
Abstract
Recent Advances:
The 2019 Nobel Prize awarded to the mechanisms for oxygen sensing and adaptation according to oxygen availability, highlighting the fundamental importance of gaseous molecules. Gaseous molecules, including reactive oxygen species (ROS), can interact with different cations generated during metabolic and redox dysregulation in cancer cells. Cross talk between calcium signaling and metabolic/redox pathways leads to network-based dyregulation in cancer.
Significance:
Recent discovery on using small molecules targeting the ion channels, redox signaling, and protein modification on metabolic enzymes can effectively inhibit cancer growth. Several FDA-approved drugs and clinical trials are ongoing to target the calcium channels, such as TRPV6 and TRPM8. Multiple small molecules from natural products target metablic and redox enzymes to exert an anticancer effect.
Critical Issues:
Small molecules targeting key ion channels, metabolic enzymes that control key aspects of metabolism, and redox proteins are promising, but their action mechanisms of the target are needed to be elucidated with advanced-omic technologies, which can give network-based and highly dimensioal data. In addition, small molecules that can directly modify the protein residues have emerged as a novel anticancer strategy.
Future Directions:
Advanced technology accelerates the detection of ions and metabolic and redox changes in clinical samples for diagnosis and informs the decision of cancer treatment. The improvement of ROS detection, ROS target identification, and computational-aid drug discovery also improves clincal outcome.Overall, network-based or holistic regulations of cancer via ion therapy and metabolic and redox intervention are promising as new anticancer strategies. Antioxid. Redox Signal. 34, 1108–1127.
Introduction
Volatile substances and gases play essential roles in regulating human physiology and pathology (125, 180, 205, 214). The recent 2019 Nobel Prize, awarded to Prof. William G. Kaelin, Prof. Peter J. Ratcliffe, and Prof. Gregg L. Semenza for their discovery of the mechanisms for oxygen sensing and adaptation according to oxygen availability (51, 79, 81, 119, 166, 197), highlights the fundamental importance of gaseous molecules for life. In particular, oxygen is the most important gas for the maintenance of life and plays an essential regulatory role in protein modifications through processes that include prolyl hydroxylation, oxidation, and ubiquitylation. For example, the transcription factor hypoxia-inducible factor-alpha (HIF-α) plays a central role in oxygen homeostasis. It can interact with the von-Hippel–Lindau (VHL) protein and trigger its ubiquitylation by O2-regulated prolyl hydroxylation on the HIF-α and VHL protein complex (81), leading to the activation of erythropoietin production to overcome hypoxia. In addition to oxygen, many other forms of superoxides, which are categorized and termed reactive oxygen species (ROS), perform multiple other protein modifications in response to changes to the redox state, which eventually lead to cell fate changes.
Many volatile substances are also generated during metabolic dysregulation, which leads to the uncontrolled transport of various cations, such as Ca2+, Na+, K+, Fe2+, and Cu2+, within and between cellular compartments (44, 144). These ions are involved in the pathogenesis of complex diseases, such as neurodegenerative diseases, cardiovascular diseases, inflammatory diseases, and cancer (12, 154, 178, 216). The abnormal flux of these ions is triggered by metabolic dysregulation and redox cycling in cancer cells. Together, these ions also lead to the modification of some key protein targets, which may eventually alter cellular signaling and cancer cell fate. Therefore, understanding the regulatory mechanisms underlying the changes in ion concentrations is important for the development of novel therapeutic intervention strategies.
Recently, multiple small molecules have been developed that can modify the function of key proteins that respond to changes in the concentration of ions and gaseous molecules. Some of these small molecules are chemically synthesized and others are derived from natural products (8, 61, 104, 179). In addition, there is an increasing demand for novel technologies for use in uncovering the mechanisms of chemical modifications and learning how they regulate the redox balance in cancer therapy. It is expected that novel technologies will lead to the discovery of more small-molecule species useful for the modification of the redox context and novel redox targets and the determination of biomarkers. In this review, we cover these issues and discuss them comprehensively.
Suppressing Cancer Growth by Modulating Ca2+-ROS Cross Talk Using Small Molecules
Ca2+− ROS cross talk modulates cell functions and cell signaling pathways
The cross talk between Ca2+ and ROS plays a major role in various signaling pathways that are crucial for physiological and pathophysiological functions. Tightly regulated Ca2+ homeostasis is important for cellular functions, such as proliferation, differentiation, cytokine secretion, and apoptosis (1, 146, 191). Ca2+ signaling is also involved in cell volume regulation (159), cell membrane potential regulation, mechanotransduction, and the modulation of the microenvironment. Physiologically, calcium is a second messenger that plays many roles in cellular metabolism, specifically in promoting ROS production. However, many studies have also reported that the redox status can affect cellular Ca2+ homeostasis through a feedback control mechanism. Of note, Ca2+ and ROS interactions can have dual effects on cellular processes (29).
Cross talk between Ca2+ and ROS in signaling pathways has been reported to control the fate of various cancer cells, sometimes in opposite ways depending on the cancer type and basal ROS level. Accumulating evidence has shown that the interaction between Ca2+ and ROS directly programs cell death by activating the mitochondrial transition pore. In contrast, Ca2+ and ROS can coordinate injury repair and prevent normal cell death, resulting in an abnormal life span of these cells and enhanced proliferation (113). Consequently, this abnormal relationship is involved in the progression of numerous diseases, such as cardiovascular diseases, inflammation, autoimmune diseases, and cancer. Therefore, the fine-tuning of the signaling cascades through Ca2+ and ROS levels to control cell fate is important.
Redox not only controls cellular Ca2+ homeostasis at the plasma membrane (PM) but also regulates endoplasmic reticulum (ER) and mitochondrial Ca2+ levels during cancer progression. Oxidants are involved in important signaling events that regulate Ca2+ homeostasis at the cell membrane through various channels and transporters (113). ROS overload and oxidative stress can inhibit cytoplasmic Ca2+ efflux while promoting Ca2+ influx, mainly through transient receptor potential (TRP) channels and store-operated calcium entry (SOCE) (145). Among the 28 TRP channels, many have been reported to exhibit altered activity in response to oxidative stress through direct and indirect channel oxidation, some serving as tumor promoters or suppressors. For example, the expression of TRP channels (Fig. 1), such as TRPA1; TRPC1, 3, 6; TRPM7, 8; and TRPV1–4, 6, has been reported to be remodeled to enhance the progression of various cancers in different stages, which have been well reviewed by Hempel and Trebak (73). In addition, TRPM2 has been identified as a potential tumor suppressor due to its ability to mediate cellular Ca2+ influx in response to oxidative stress, leading to cancer cell death (95). Cross talk between the TRP channel and ROS is also related to their interaction with nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (183).
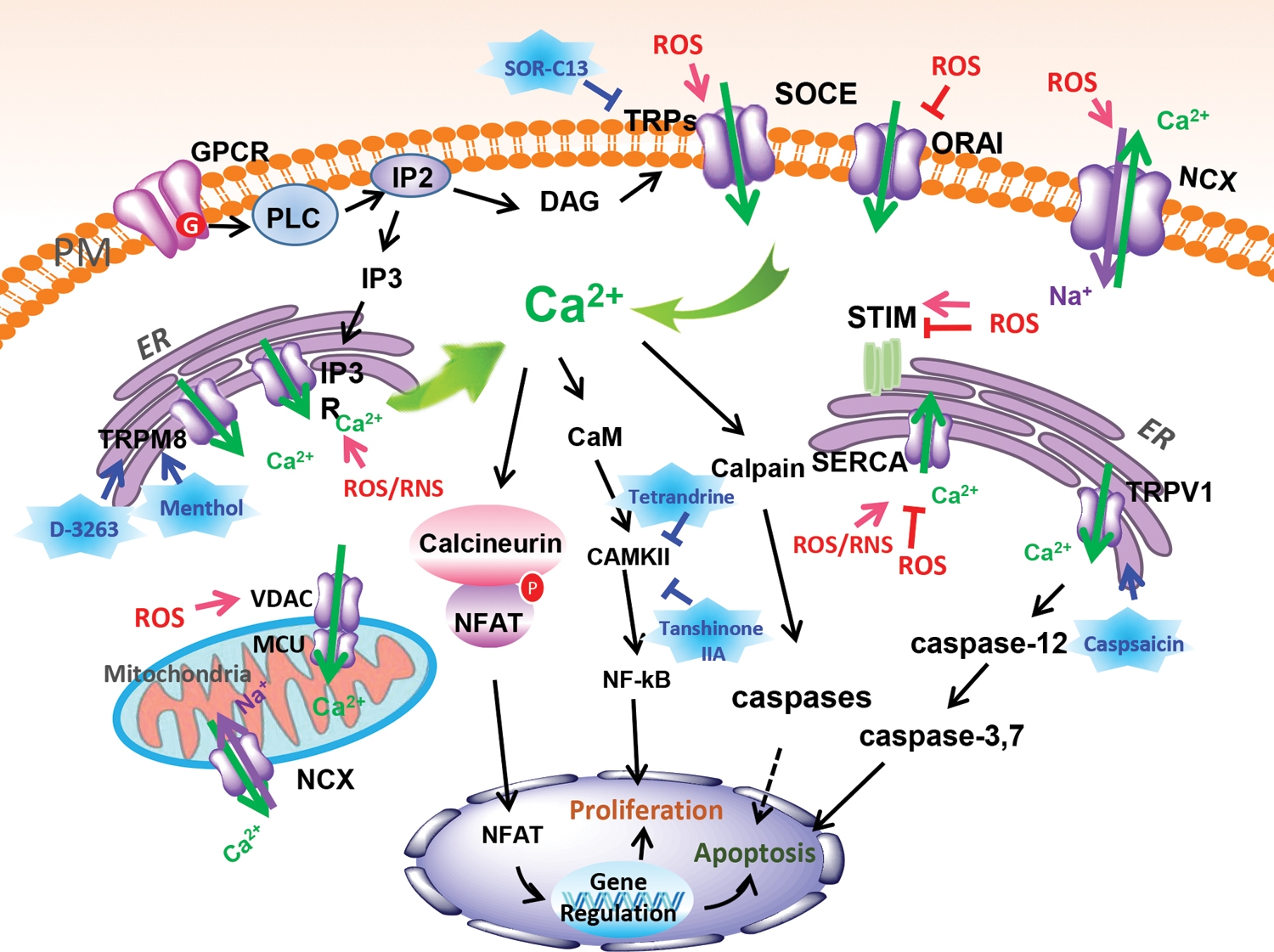
Despite the multifunctional roles of TRP channels in the progression of cancer, the major Ca2+ entry pathway in cancer cells is mediated by SOCE (23, 145, 173, 189). SOCE is regulated either through the ER and mitochondrial Ca2+ pumping or through mitochondria-derived redox signaling. SOCE involves the PM channel protein calcium release-activated calcium channel protein 1 (ORAI1), the gating of which is regulated by the ER membrane-localized protein stromal interaction molecule 1 (STIM1) or STIM2 (173, 189). These proteins interact with ORAI1 when Ca2+ is depleted from the ER and initiate Ca2+ influx through the PM. Isoforms of both ORAI and STIM are expressed differentially in different types of cancers, enabling cell proliferation and metastasis (23). For example, T cell immunoglobulin and mucin domain 1 (TIM1) expression in hepatoma carcinoma is positively regulated by HIF-1α through hypoxia, a condition that enhances SOCE and thus promotes tumor growth (101). However, when hypoxia induces acidification of the cell microenvironment, ORAI1 is uncoupled from STIM1, causing a considerable decrease in SOCE (116).
Unlike the situation at the PM, the relationship between redox reactions and Ca2+ in the ER and mitochondria is closely related to signaling pathways, such as those mediated by inositol 1,4,5-trisphosphate receptor (IP3R), mitochondrial Ca2+ uptake (MCU), sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), and voltage-dependent anion channels (VDACs) (Fig. 1) (2, 13, 33, 193). These pathways are involved in the control of MCU processes. This control is important for the generation of NADH through the tricarboxylic acid cycle, for the induction of mitochondrial respiration and the production of ATP. Accordingly, various metabolic enzymes, substrate transporters, and the mitochondrial membrane potential are regulated in a complex way. Cross talk between components in redox reactions and ER-mitochondrial Ca2+ and their associated signaling pathways is also relevant for the regulation of carcinogen-induced mutations. For example, decreased protein expression levels of IP3R3 and SERCA2b are observed in Kirsten rat sarcoma viral oncogene homologue (KRAS)-mutant colorectal cancer cells (142, 193).
Ca2+ signaling may also be involved in the regulation of the tumor microenvironment by programmed death-1/-ligand 1 (PD-1/PD-L1). Studies on the transmembrane protein TMEM16F (a Ca2+-gated ion channel) showed that mice lacking TMEM16F because of genetic ablation fail to activate T cells or internalize PD-1, resulting in immune cell exhaustion. Thus, TMEM16F contributes to cancer immune escape (94, 210). Therefore, therapies targeting the redox-Ca2+ signaling pathway may be potential candidates for treating cancer or overcoming resistance to chemotherapy and immunotherapy.
Immunotherapy by inhibitors of the immune-checkpoint blockade has received widespread attention recently because of the success of inhibitors of the PD-L1/PD-1 interaction. A variety of tumors, such as melanoma, nonsmall-cell lung cancer (NSCLC), color rector cancer, and T cell lymphomas (88, 90, 129, 203), activate this blockade to escape immune surveillance. It is likely that PD-L1 activation is not the only trick cancer cells use to ensure their survival. Rather, the complex regulation of the immune defense may offer plenty of other opportunities for cancer cells to protect themselves against immune attack. Therefore, it is very important to scrutinize all the players involved in immune regulation to find possible disruptions of their function(s) in tumors. The adaptive immune response is regulated by a variety of mechanisms, many of which involve ion channels and Ca2+. As previously detailed in the context of SOCE and ROS, Ca2+ influx is a major trigger for the activation of cellular processes, including the immune response. However, transport pathways for Ca2+ alone are not sufficient to cause changes in intracellular Ca2+. Due to the requirement of electroneutrality, the movement of Ca2+ must be accompanied either by the cotransport of negatively charged ions or by a counterflux of other positive ions. This balance is achieved through numerous types of ion channels.
Targeting Ca2+-ROS signaling to develop small-molecule anticancer agents
Targeting components of Ca2+− and redox-induced signaling, immune regulation, and their cross talk has been shown to be a powerful approach to developing small-molecule drugs for cancer treatment. FDA-approved antihypertensive agents, such as amlodipine, felodipine, manidipine, and cilnidipine, block calcium channels (124). They also are remarkably potent as an L-type calcium channel blocker, which can inhibit filopodia formation via inhibit the activation of MYO10 in pancreatic and breast cancer cell models to inhibited cell migration and invasion (124). In addition, several agents in clinical trials (82) dampen tumor cell proliferation. These include SOR-C13 and SOR-C27 (phase 1 clinical trial [NCT01578564]), a TRPV6 channel inhibitor that suppresses the Ca2+-nuclear factor of activated T cell (NFAT) pathway, resulting in cancer cell death and metastases reduction in TRPV6 high-expression cancer cells, including colon, ovarian, breast, and prostate cancers, and certain leukemia and lymphomas (15, 59, 211), and D-3263 (phase 1 clinical trial [NCT00839631]), which targets the TRPM8 channel, showing no major toxicity effect in treated prostate cancer patients (94) (Table 1). In addition, various small molecules from natural products, such as menthol (127), tetrandrine (208), and tanshinone IIA (112, 170, 199) (Table 1), are also effective in anticancer therapy targeting ROS-Ca2+ signaling pathway components (Fig. 1). For example, tanshinone IIA has been shown to downregulate TRPM7 and proinflammatory factors to alleviate acute lung injury (98).
Summary of Compounds Targeting Ion Channel, Redox Balance, and/or Metabolism in Cancer Therapy
AKT, protein kinase B; AMPK, AMP-activated protein kinase; AR, androgen receptor; CaMKK-β, calcium/calmodulin-dependent protein kinase kinase-β; CNC-bZIP, Cap ‘n’ Collar basic leucine zipper; DHEA, dehydroepiandrosterone; D-pen, D-penicillamine; EGFR, epidermal growth factor receptor; EGR-2, epidermal growth receptor 2; ER, endoplasmic reticulum; MAFG, musculoaponeurotic fibrosarcoma oncogene homologue G; MAPK, mitogen-activated protein kinase; Msr, methionine sulfoxide reductase; mTOR, mechanistic target of rapamycin; N/A, no mention; NADPH, nicotinamide adenine dinucleotide phosphate; NOX3, NADPH oxidase 3; Nrf2, nuclear factor erythroid 2-related factor 2; NSCLC, nonsmall-cell lung cancer; PI3K, phosphatidylinositol-3 kinase; PKC, protein kinase C; PKM2, pyruvate kinase isozyme M2; RASFs, rheumatoid arthritis synovial fibroblast cells; ROS, reactive oxygen species; TCA cycle, tricarboxylic acid cycle; TRPM8, transient receptor potential melastatin 8.
Moreover, tanshinone IIA induces apoptosis in NSCLC cells by inducing ROS production and decreasing the mitochondrial membrane potential (199). Furthermore, tanshinone IIA has been reported to inhibit angiogenesis through the HIF-1α/β-catenin/VEGF pathway; as a consequence, it suppresses the progression of colon cancer in both cell and xenograft models (170). Tetrandrine mechanistically induces cancer cell death through calcium/calmodulin-dependent protein kinase kinase-β (CaMKK-β) regulation and AMP-activated protein kinase (AMPK) signaling and serves as a novel protein kinase C-α inhibitor that leads to autophagy (208). These examples are only a few of many. The development of computational technology to model molecular docking and intensified efforts aimed at acquiring better insight into the role of cross talk between ROS and Ca2+ in cancer progression will certainly contribute to the development of anticancer agents.
Modulating Copper/Iron Redox Cycling to Suppress Cancer with Small Molecules
Copper/iron redox cycling in cancer cells
Many types of cancer exhibit increased intratumoral copper ion levels and/or control copper distribution (68, 162, 222). Excess copper in cancer cells is an effective catalyst for ROS production (184), acting mainly through two mechanisms of oxidative stress induction (Fig. 2A). First, it directly catalyzes the formation of ROS by a Fenton-like reaction (141). Second, excess copper is caused by the redox cycling reaction of copper with oxygen and cell-reducing substances such as mercaptans (85). In the presence of superoxide anion (O2 •−) radicals or biological reducing agents, such as ascorbic acid or glutathione (GSH), cupric ions (Cu2+) can be reduced to cuprous ions (Cu+), which catalyze the formation of reactive hydroxyl radicals (•OH) through the decomposition of hydrogen peroxide via the Fenton reaction (141), which decomposes hydrogen peroxide. It is worth noting that the net result of this redox cycle reaction not only consumes antioxidant thiols but also produces superoxide, which can be readily converted to other types of ROS. Consequently, copper-overloaded cancer cells are often at increased risk of ROS production and increased oxidative stress upon further stimulation. This important feature may be the most relevant for the design of anticancer drug molecules.
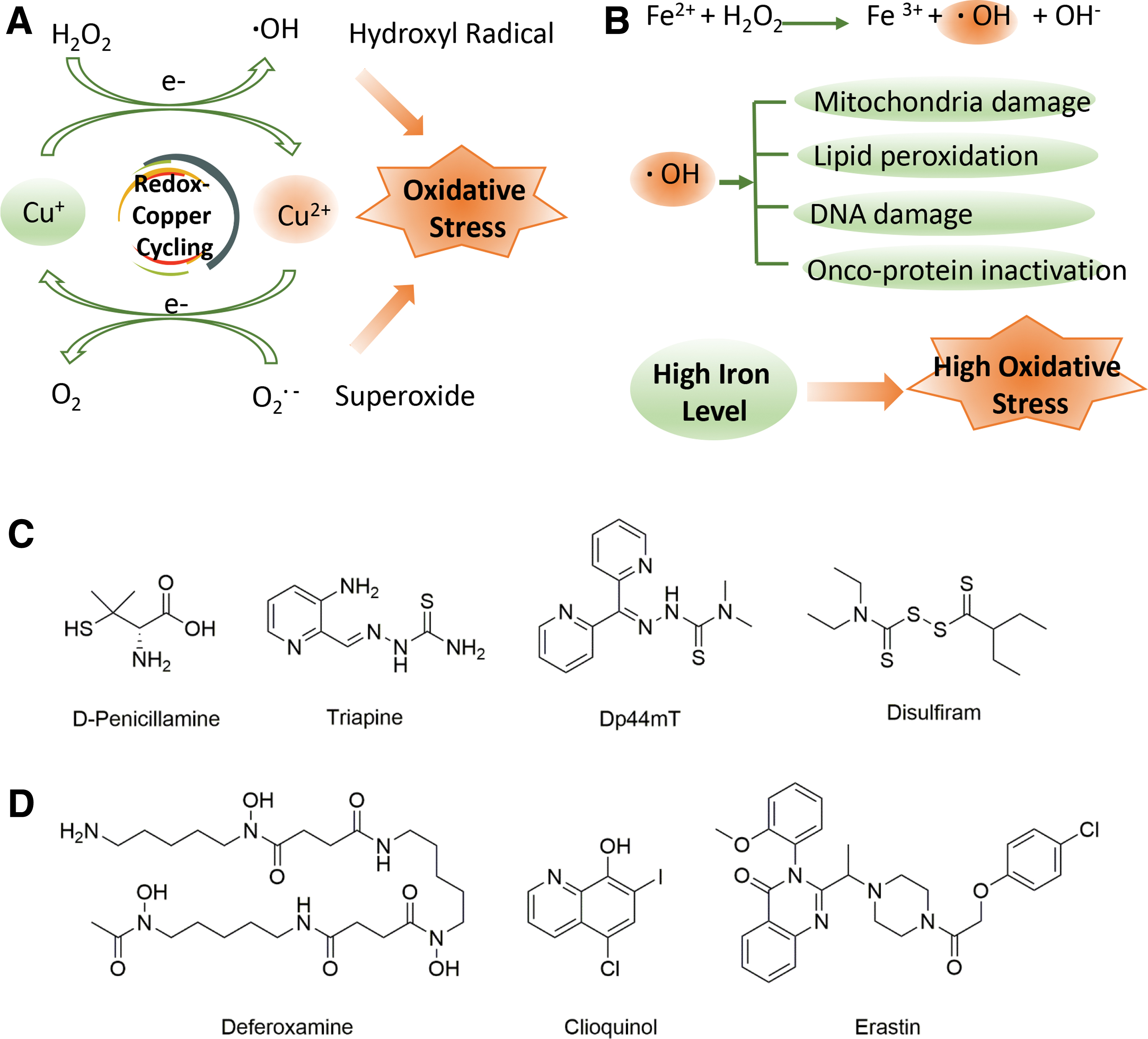
Iron (Fe) is also essential for the cellular metabolism of normal cells and cancer cells, and plays an important functional role in heme-containing enzymes (196). Ribonucleotide reductase is an iron-containing enzyme that plays a role in cellular respiration, which is important for cell cycle progression and plays a crucial role in the organism. Consequently, iron plays a key role in cell proliferation, replication, and metabolism. In cancer cells, several disruptions of iron metabolism occur during the steps of tumor initiation, progression, and metastasis (7). It is worth noting that cancer cells require large amounts of iron and show a strong dependence on their proliferation (7). Interestingly, excess iron is usually associated with toxicity because it leads to the formation of toxic hydroxyl radicals formed by the Fenton reaction (Fig. 2B) (62). The Fenton reaction results in hydroxyl radicals produced by the reaction of Fe2+ with hydrogen peroxide. This suggests a potential toxicity of free redox activity by Fe2+ (Fig. 2B) (87). In the case of iron, its catalytic utility lies in its ability to transition between the ferrous (Fe2+) and ferric (Fe3+) states, enabling it to act as an electron donor or acceptor (Fig. 2B) (87). The products of these reactions can induce cell death by initiating a reaction with biomolecules, resulting in damage to mitochondria, lipid membranes, and DNA (Fig. 2B) (87).
In addition, excess iron produces alkoxy and peroxy radicals when they react with unsaturated lipids (102). Iron-catalyzed free radicals can inhibit the oxidation of amino acids, leading to protein inactivation (Fig. 2B) (175). It is worth noting that this harmful oxidation reaction, especially when targeted to DNA, can lead to mutagenesis and thus suggests a role for iron in cancer development, progression, and metabolism (128, 176). In addition, ROS such as superoxide anion and hydrogen peroxide also act as free radicals upon their iron-induced production (44). Excessive ROS increase oxidative stress, overwhelming the cell defense system and making deadly oxidants that potentially damage DNA and other biomolecules (80). The close relationship between iron and oxidative stress has enabled researchers to develop a large number of iron-based therapeutic agents in recent years.
Targeting copper/iron redox cycling to develop therapeutic molecules for cancer
In recent years, targeting copper or iron redox cycling to develop therapeutic molecules for cancer has received much attention, and several excellent review articles have reported on copper (38, 160, 185) and iron anticancer agents (30, 50, 117). The following are some special examples of anticancer therapeutic molecules developed by targeting copper or iron redox cycling. D-penicillamine (D-pen, Fig. 2C), a metabolite of penicillin, is a good copper-chelating agent (195) that can effectively bind copper ions in tissues to form a soluble complex, which is finally excreted in the urine. Thus, it can effectively eliminate high concentrations of copper in tumor tissues (38). In addition, copper-loaded D-pen can induce human leukemia and breast cancer cells to produce a large amount of ROS, eventually leading to cancer cell apoptosis (66, 67) (Table 1).
The thiosemicarbazone compound triapine (Fig. 2C and Table 1), also known as OCX-191 or 3-AP, and Dp44Mt (Fig. 2C) can chelate copper ions to produce ROS (54, 92). These findings show that the anticancer mechanism is based on the induction of oxidative stress. Disulfiram (DSF, Fig. 2C and Table 1), an ancient antialcoholic drug, can also chelate copper ions (Cu2+) to stimulate ROS production in nodular and superficial diffuse melanoma cells, activating the apoptotic pathway (121). It has been reported that DSF complexes with copper ions can activate ROS and induce apoptosis by inhibiting nuclear factor kappa-light-chain-enhancer of activated B (NF-κB) (215). Therefore, DSF synergizes with copper ions and has a significant antitumor effect, making it promising for combination therapy.
Targeting iron redox cycling or metabolism in cancer cells is also a promising approach to cancer therapy. The iron carrier deferoxamine (DFO, Fig. 2D and Table 1) was the first iron chelator to be tested for its anticancer properties (21). Contingent on concentrations and exposure time, DFO can inhibit the growth of human neural tumor cell lines by lengthening the period of the cycle and causing the cells to arrest in G2/M blocking the proliferation of the S phase of the cell cycle in vitro (155). In preliminary clinical trials, this chelating agent has also been shown to be effective in the treatment of leukemia and neuroblastoma. Eight-hydroxyquinoline is also a member of an important class of iron chelators (48). The potential value of this class of clioquinol (5-chloro-7-iodo-8-pyrrolidine, Fig. 2D and Table 1) in anticancer strategies has been widely demonstrated. It is capable of sequestering a variety of metal ions including iron. After synergizing with FeCl2 for Raji cells for 72 h, the IC50 was measured by 3-(4,5-diethylthiazol-2-yl)-5-(3-carboxymethoxyphe) (MTS) method ranging from 1 to 10 μM, showing good cell inhibitory activity (41). In addition, ferroptosis inducers can regulate oxidative stress in the ER and cause iron-dependent cell death (42). In contrast, ferroptosis suppressor protein 1 (FSP1), also known as AIFM2, serves as a protector of human cells to form ferroptosis, which can target ubiquinone in the cell membrane to generate ubiquinol, which can inhibit peroxidation and block ferroptosis (11, 46, 177). Accumulating evidence supports the importance of ferroptosis in cancer therapy (71, 169). Erastin (Fig. 2D and Table 1) is a small molecule that triggers ferroptotic cell death. Erastin binds to and inhibits VDAC2 and VDAC3 and functionally inhibits the cystine/glutamate reversal protein system. Cells treated with erastin are deprived of cysteine and are thus unable to synthesize the antioxidant GSH (70). Therefore, the depletion of GSH by erastin indirectly inactivates GPX4, leading to the accumulation of toxic lipid ROS and the peroxidation of lipids, thereby exerting an anticancer effect (43).
In summary, targeting copper or iron redox cycling could induce oxidative stress in cancer cells and cause cancer cell DNA damage, ultimately leading to cancer cell death. Developing novel copper or iron ionophores and using the difference in copper/iron levels between normal cells and cancer cells to selectively act on cancer cells is an excellent anticancer strategy in the future. For example, the development of a cleavable and recyclable copper shuttle has achieved selective treatment of cancer (219). In addition, the photodynamically activated ROS treatment strategy of metal nanoclusters is also a promising research field in recent years (213). These strategies can be used to develop small molecules as anticancer drugs.
Modulating Pentose Phosphate Pathway Suppresses Cancer by Reducing Tumor Redox Capability
Abnormal metabolic patterns meet the requirements for redox homeostasis
Cancer cells are derived from abnormally proliferating cells. They alter their intracellular metabolic patterns to eternally grow and proliferate (69, 149, 190, 221). In 1920, Otto Warburg observed that, under conditions with sufficient oxygen, cancer cells still preferentially undergo aerobic glycolysis, but not mitochondrial oxidative phosphorylation, to produce energy (75, 201). As an explanation of this specific cell fate, it has been reported that the altered metabolism of cancer cells is not suited for maximal ATP production (20, 110) but provides the building blocks needed for sustained macromolecular synthesis (16, 202) and the maintenance of NADPH levels and cellular redox homeostasis (192).
In cancer cells, altered redox status and enhanced ROS emerge as characteristics for driving and sustaining cancer cell proliferation (137). Due to increased free radicals, originated from metabolic activity, mitochondrial malfunction, oncogenic stimulation, and diverse chemical compounds such as alcohol and tobacco (183), cancer cells exhibit increased intrinsic redox stress, inducing cancer cells to express high levels of ROS (139). Besides, it has been proven that ROS at moderate concentration can activate the cancer cell survival signaling pathway, such as mitogen-activated protein kinase (MAPK)/extracellular regulated protein kinase (ERK), c-Jun N-terminal kinase (JNK), and phosphoinositide 3-kinase (PI3K). ROS only at high concentrations can induce cancer cell apoptiosis (59). Consequently, cancer cells for their survival have higher levels of ROS antioxidants, including vitamin E, vitamin A, bilirubin, ferritin, superoxide dismutase (SOD), glutathione, and melatonin (37, 97), and scavenging enzymes, such as catalase, N-acetyl cysteine, SOD2, coenzyme Q, and glutathione peroxidase (118, 151), which protect cell from ROS-mediated activation of death-inducing pathways (24). In addition, the pentose phosphate pathway (PPP) is an alternative metabolic pathway that, starting with glucose-6-phosphate, generates precursors such as phospho-pentose for nucleotide biosynthesis and NADPH for anabolic reactions and thus contributes to the maintenance of redox homeostasis (49).
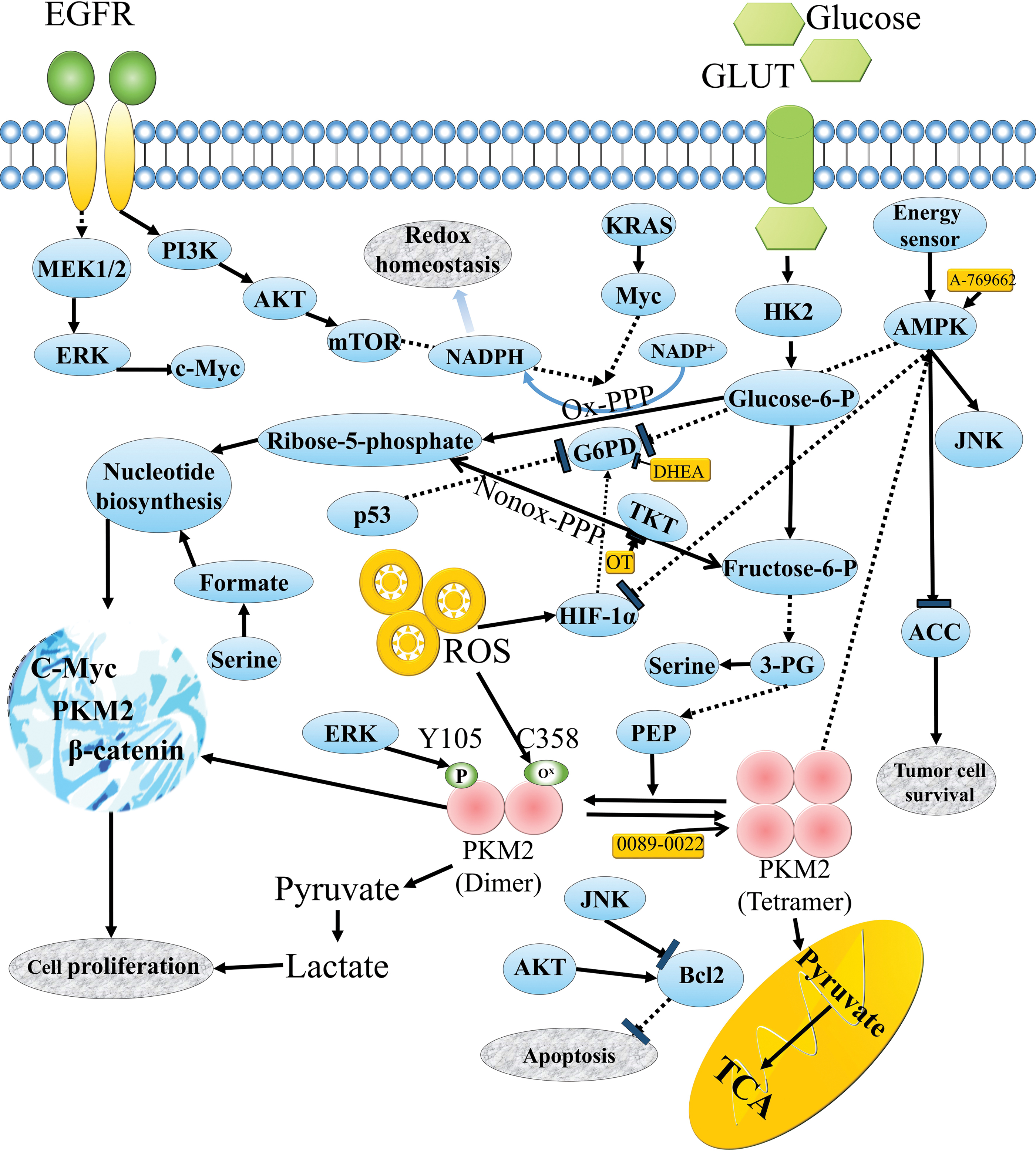
In addition to the critical role of PPP in maintaining ROS balance, numerous research groups have shown that it participates in many other oncogenesis pathways. For example, growth factors can impact the expression and activity of a key enzyme of PPP, glucose-6-phosphate dehydrogenase (G6PD), via the MAPK and PI3K signaling pathways (Fig. 3) (182). Since these signaling pathways are often hyperactivated in cancer due to the activation of oncogenes (such as KRAS, MYC, and growth factor receptors) or the inactivation or loss of function of oncosuppressors (such as p53 and PTEN), any alterations that impact these players may lead to inhibition of PPP activity (56). Moreover, it has been extensively reported that the mechanistic target of rapamycin can control cell metabolism, including the glycolytic pathway and PPP, to regulate cancer cell proliferation and anabolism (78, 118). Thus, according to the well-established theory that excessive ROS cause damage and cell death, cancer cells are more dependent on antioxidant enzymes to cope with extracellular stress than normal cells (24). Accordingly, inhibition of the key antioxidant enzymes is expected to cause ROS-mediated damage in cancer cells (34).
Targeting metabolic enzymes to reveal a novel anticancer treatment
The PPP is divided into an oxidative and a nonoxidative branch, two independently operating metabolic modules (18). First, oxidative PPP is an irreversible metabolic pathway driven by G6PD, the first and rate-limiting enzyme acting as a control element (28). While this pathway is involved in two cellular processes essentially related to anabolism and redox homeostasis (synthesis of ribose and NADPH) (123, 204), reactions of the nonoxidative branch are reversible, allowing carbon recirculation (in the formation of ATP, amino acids, or lactate) (151), with transketolase (TKT) and transaldolase being the two key enzymes (63, 86). Consequently, G6PD and TKT are potential targets for modulating PPP.
In cancer cells, higher levels of G6P can also be achieved by enhancing the amount of glucose uptake and glycolysis. For instance, glucose phosphorylation is conducted by the glycolytic rate-limiting enzyme hexokinase 2 (HK2), which is the main isoform expressed by cancer cells (134). In addition, the pyruvate kinase isozyme M2 (PKM2) dimer, expressed by many cancers, provides an advantage to cancer cell proliferation by facilitating anabolic metabolism, as it only takes two further steps for its product to complete the synthesis of lactate (37). PKM2 can be controlled by posttranslational modifications (e.g., oxidation, phosphorylation, and acetylation) that impair the PKM2 tetramer-mediated metabolic function of favoring PKM2 dimer formation (as ROS oxidize PKM2 at Cys358) (40, 84a). Thus, the PKM2 dimer leads to an accumulation of upstream glycolytic intermediates and their subsequent turning into anabolic pathways such as PPP, glycerol, and serine/glycine synthesis (109). Moreover, AMPK is a critical cellular fuel sensor for maintaining metabolic homeostasis, such as managing glucose uptake, fatty acid uptake, and oxidation, lipolysis, cholesterol synthesis, and protein synthesis. AMPK is generally downregulated in many cancers (156).
Accordingly, the strategies for targeting redox capability could be subdivided into two types: blocking key enzymes in PPP or targeting glycolytic regulating enzymes. For instance, G6PD and another key enzyme of the PPP nonoxidative branch, TKT, have been proposed as potential therapeutic targets in cancer (5), and inhibitors of the two enzymes have been designed and assessed. The inhibitor of G6PD, dehydroepiandrosterone (10), and an inhibitor of TKT, oxythiamine (4), have both been investigated to induce G1 phase cycle arrest by decreasing nucleic acid ribose synthesis, while further suppressing tumor growth both in vitro and in vivo (Table 1). These inhibitors have been described as potential antitumor agents (133).
In addition, both HK2 inhibitors and PKM2 tetramer activators are potential targets for anticancer treatment (161). For example, 2-deoxy-D-glucose (2DG) is a competitive analogue of glucose and can be phosphorylated to produce 2-deoxyglucose-6-phosphate, which is an inhibitor of HK-induced cell death (138). 0089-0022 (Table 1) is one of the direct PKM2 activators reported to induce cell apoptosis through inhibition of protein kinase B (AKT) signaling pathways in both epidermal growth factor receptor (EGFR) and KRAS mutant NSCLC cells. At the same time, it has less cytotoxic effects on normal lung cells (99). For the energy sensor AMPK, enhanced induction of cell death through direct AMPK activators, such as A-769662 (Table 1), has been reported compared with indirect activators, such as metformin. The results demonstrate that A-769662 can induce tumor cell apoptosis through the activation of AMPK (Fig. 3) (150).
Overall, these results provide a glimpse into the promising future of modulating key enzymes of PPP and its upstream rate-limiting enzymes to disrupt tumor redox capability for the suppression of tumor growth.
Modulating Protein Oxidation Using Small Molecules As a Novel Cancer Therapeutic Strategy
Excess ROS generation leads to protein oxidation and alters biological processes
As an important posttranslational modification, oxidation effectively modulates the function of target proteins in cancer (200). For example, oxidation of active site cysteine 797 to sulfenic acid in EGFR, which is a cancer driver mutation gene, will enhance kinase activity (187), whereas direct oxidation of a reactive cysteine in the activation loop of cAMP-dependent protein kinase (PKA) will result in the inactivation of the kinase (77). By oxidation of proteins, increased ROS levels in cancer cells facilitate the process of carcinogenesis through various signaling pathways, such as increasing the activity of oncoproteins (39), inducing an influx of Ca2+ (152, 181), and promoting cell metabolism (72, 130). Therefore, understanding the regulation of the oxidation of proteins will remarkably contribute to the treatment of cancer.
Oxidative modifications of proteins can generally be classified into two categories: irreversible and reversible oxidation. The irreversible modification was frequently related to the permanent loss of protein function, which led to protein degradation or toxic aggregation (206). However, during reversible oxidation, proteins act as buffer systems to maintain redox homeostasis and provide protection from irreversible oxidation and protein dysfunction (19, 136, 212). The dominant factor that controls the extent of protein oxidation is the intensity of the ROS action.
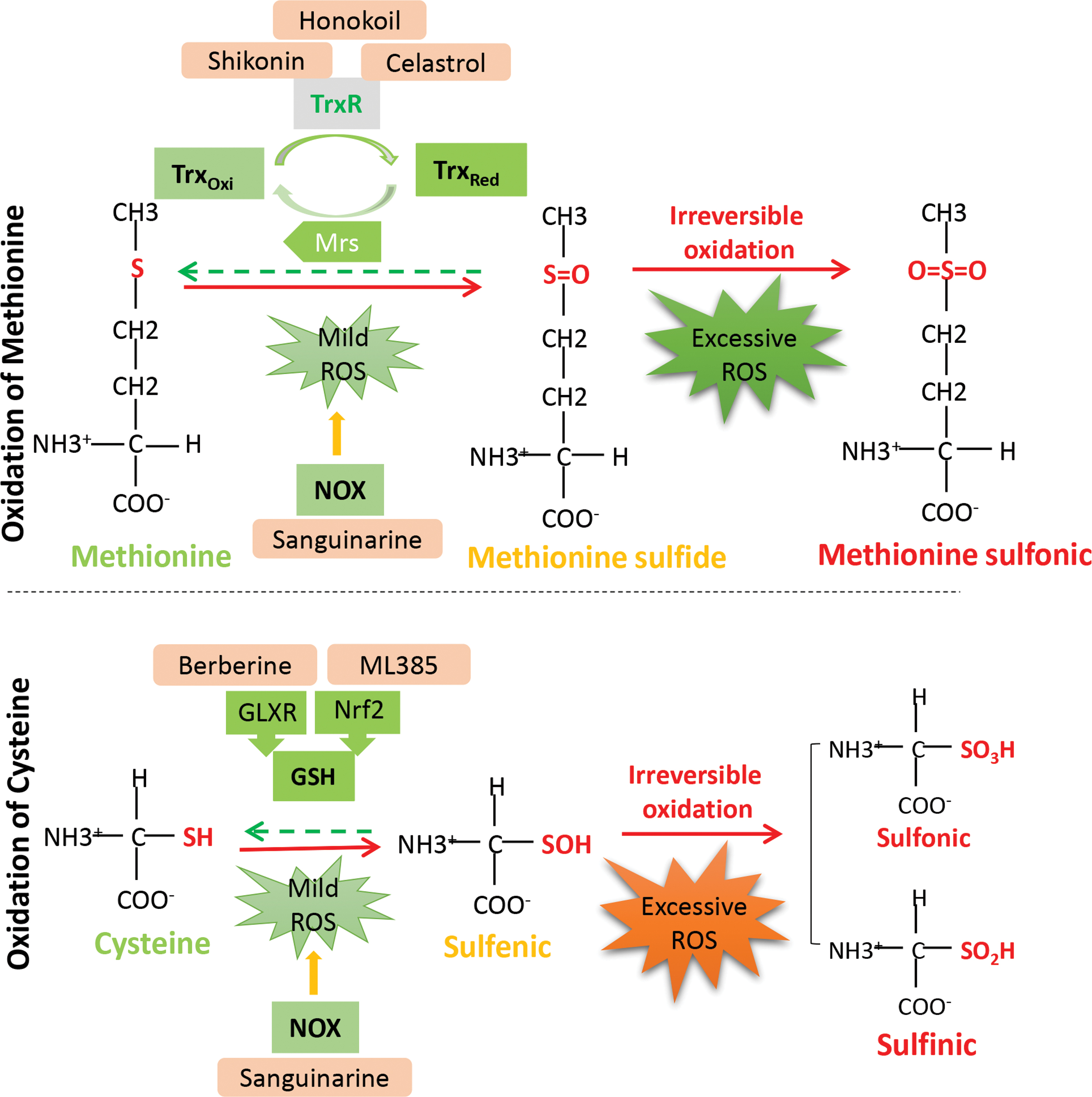
Cancer cells are characterized by increased ROS levels, which can continuously induce reversible oxidation of proteins and accordingly maintain the activity of oncogenes (60, 120, 132). However, ROS are a double-edged sword. Increased ROS also provide an opportunity to eliminate cancer cells. In fact, sulfur-containing amino acids (cysteine and methionine) are the most easily oxidized and are therefore the most relevant to tumor progression (31). When exposed to mild oxidants, these amino acids react with oxidants to prevent DNA mutation and tumor initiation. Oxidation of cysteine leads to the formation of a sulfenic acid intermediate (SOH), which can be reduced and reversed by the antioxidants thioredoxin (Trx), glutaredoxin, or glutathione (91, 158). However, further oxidation results in the irreversible formation of the sulfinic (SO2H) and sulfonic (SO3H) forms, which cause permanent inactivation and degradation of proteins (Fig. 4) (103). Similarly, methionine can be oxidized to methionine sulfoxide and reduced by methionine sulfoxide reductase through TXR (89). However, methionine sulfoxide can be further hyperoxidized to methionine sulfone and become irreversible (89). Therefore, further elevating ROS to highly toxic levels to disable the key antioxidant systems is a promising approach for anticancer therapy and even for overcoming chemotherapy resistance. Results can be achieved by stimulating ROS generation, inhibiting antioxidant defense and a combination of both approaches.
Small-molecule intervention on protein oxidation as novel therapeutic strategy
To enhance the intracellular ROS level and further oxidize proteins in cancer, enzymes related to ROS generation and elimination or its downstream effectors have been widely used for anticancer drug discovery. Up to now, the anticancer effect of agents with ROS regulating ability has been validated in numerous preclinical studies, including lung, gastric cancer, and leukemia models, and their efficacy was evaluated by a mouse xenograft model. Mitochondria and NADPH oxidase (NOX), the major generators of endogenous ROS, have been assessed by numerous preclinical studies, which demonstrated their anticancer efficacy by regulating their activity (53). For instance, by suppressing the mitochondrial respiration chain to induce the accumulation of cellular ROS levels, agents such as metformin, berberine, and celastrol effectively inhibited the proliferation of tumors (3, 26, 52) (Table 1). In addition, sanguinarine, a NOX3 activator, was demonstrated to selectively induce hyperoxidation and degradation of an oncoprotein (EGFR carrying the T790M mutation) while sparing wild-type EGFR. Alternatively, targeting ROS scavengers was found to be another way to enhance protein oxidation (97) (Table 1). Nuclear factor erythroid 2-related factor 2 (Nrf2), the Trx/thioredoxin reductase (TrxR) system protein, sirtuins (SIRTs), histone chaperones, and many other proteins serve as important targets for impairing redox homeostasis in cancer by rendering cancer cells vulnerable to oxidative stress (Fig. 4) (35, 157, 168). Various small inhibitors of these targets, such as shikonin, Honokiol, and ML385, have also been reported to possess potential anticancer capacity (Fig. 4) (100, 111, 172) (Table 1).
Taken together, these data suggest that although a surge in ROS induces protein oxidation in cancer cells and promotes the progression of tumors, it is also cancer's Achilles heel, providing options for its treatment. Modulating the oxidation of key proteins involved in redox balance will significantly facilitate the development of new anticancer strategies.
Novel Technologies for the Detection of ROS and Research on Drug Mechanisms
Although increasing evidence has shown that redox plays an important role in the physiological processes of cells, the role of specific redox markers remains difficult to identify. Therefore, advanced multidisciplinary techniques are required to detect redox markers and to address their roles.
Molecular probe-based tools for redox detection
Oxidation of cysteine to S-nitrosothiols (RSNOs) (57), cysteine sulfenic or cysteine sulfenic acid (RSOH) (55), and sulfinic acids (RSO2H) (131), persulfides (RSSH) (107) and disulfides (RSSR) (135) provides a few prominent examples of oxidative posttranslational modification products, which play key roles in signal transduction and protein function. Using chemical probes to detect and quantify these molecules and to determine which proteins are subjected to these modifications will help to understand their physiological functions.
Currently, a series of detection methods for RSNO have been reported, such as the biotin switch technique (58, 83), assays based on gold nanoparticles (84), phenylmercury probes (47), organophosphine probes (198), and benzenesulfinate probes (153). RSOH plays a key role in redox regulation by acting as an early oxidation product of the reaction of cysteine with ROS and reactive nitrogen species. Because a large number of chemical probes contain releasable 1,3-diketones that can react directly with sulfenic acid, the detection of sulfurous acid modifications has become increasingly common. The most commonly used sulfinic acid traps are dimethyl ketone and its derivatives, such as DYn-2 (136). Dimedone stably derives protein sulfenates, which can be monitored colorimetrically or with purified protein in vitro using mass spectrometry (MS) (126). However, antibodies have been developed that overwhelmingly detect protein sulfenates derivatized by dimedone. It is easier to monitor protein sulfenation in cells and tissues (65, 171). Moreover, it can be detected by using a modified biotin switch technique (163) using 1,3-diketone chemical probes (9).
A higher oxidation state of sulfur in RSO2H, however, leads to reduced reactivity compared with that of the molecule in the lower oxidation state, making it difficult to design chemical probes that react specifically with the modified residue. Despite this complication, many assays have been developed (106, 108, 114, 209). Oxidation of cysteine to generate persulfide, the oxidation state, is thought to occur through various reactions with hydrogen sulfide. Cavallini et al. reported early evidence of persulfide formation in proteins upon the addition of sodium sulfide to disulfide-containing proteins, such as insulin and RNase (22). Since the persulfides are strong nucleophiles, including native cysteine thiol, developing selective methods for their detection is a challenge. Despite such diverse problems, a number of novel methods have been developed (32, 45, 93, 122, 164, 167, 218). Due to the similar reactivities of persulfides and thiols, however, there is still a long way to go in designing chemical tools that specifically react with persulfides.
RSSR are important for maintaining the structure of proteins and participate in various catalytic and signaling processes. The presence of intermolecular disulfide bonds can often be inferred through SDS-PAGE analysis, which has been used to study the formation of disulfides among moieties in both the cytoplasm and myocardial cells (17, 36, 174). Moreover, a series of detection methods have been reported (6, 115). In view of the roles of these oxidative modifications in signal transduction, the development of specific probes with higher selectivity will have high practical value.
MS-based methods for protein identification
MS has been used for the identification and characterization of proteins for many years. It is capable of detecting changes in the mass-to-charge ratio (m/z) of ionized analytes after oxidative modification of proteins. Therefore, MS is a powerful method for detecting oxidative posttranslational modifications (207). In recent years, MS methods for the analysis of native and oxidized proteins have experienced substantial progress. They can be classified in terms of “top-down” and “bottom-up” analysis. The latter is by far the most commonly used method because it has been widely used in proteomic research to study protein oxidation. Over the years, many bottom-up methods have been developed to enable large-scale identification and quantification of modifications based on protein oxidation. Since the levels of oxidative modifications are relatively low in complex samples (<5%), special methods and enrichment strategies have been used to label oxidized proteins. These methods include indirect methods that can detect cysteine oxidation after losing reactivity with a thiol modifier (such as N-ethylmaleimide or iodoacetamide) (25, 89). Traditionally, N-ethylmaleimide and iodoacetamide are used for the initial alkylation of reduced thiols. However, they react with both -SH and -SOH cysteine in proteins, and so, this method is not suitable for the determination of -SOH (27). In addition, another compound that can be used to indirectly detect thiol oxidation is ABD-F, which recognizes only free cysteines that are in the thiolate state. Biotin switches are the most commonly used tag-switch tool used in redox proteomics. Since their introduction, many modifications have been made to achieve more efficient sample enrichment and docking during MS analysis (186).
Ascorbate is widely used to selectively reduce protein nitrosylation (-SNO) in the indirect tag-switch method (64, 217). The direct method relies on a selective reaction of a chemical probe (typically a biotin tag or a clickable alkyne or azide tag) with a specific oxidative modification. Selective chemical probes compatible with MS analysis have been reportedly used to label proteins carrying -SO2H (108), -SNO (114), or -SOH (147) residues. In addition, the latest time-of-flight secondary ion mass spectrometry (ToF-SIMS) technology is used to explore the electro process at the surface of samples. Qian et al. achieved an electrochemically reversible redox reaction of NAD+/NADH derivatives through the functionalization of a gold electrode with a specially designed NAD+ derivative and characterized the electrochemical redox products by ToF-SIMS (148). Lee et al. developed and validated a rapid and sensitive method for the study of the cytotoxicity of zinc oxide nanoparticles using human skin equivalent HaCaT cells in a model system. The method is based on a combination of ToF-SIMS and fluorescence imaging using confocal laser scanning microscopy (96). Collectively, MS-based methods can be used to identify the site(s) of modification, to quantify site occupancy, and to determine relative changes in oxidation levels between experimental conditions.
Computation-based technologies for the elucidation of redox mechanisms
The transfer and transport of electrons through biological matter are some of the key steps of redox reactions. Currently, with the development of computational molecular science and high-performance computing, atom-scale simulation methods allow scientists to understand and interpret these processes in terms of microscopic protein structures and dynamics. A series of energy gap calculations for the electron transfer of redox reactions has been successfully performed using classical molecular dynamics (MD) studies and the density-functional theory of novel MD for simulations (165, 194). Moreover, computational simulation techniques can also be used to illustrate the effects of redox on protein structure. For example, Heppner et al. revealed the structural impact of sulfenylation of cysteines, indicating that Cys-277-SOH causes solvent exposure of Tyr-416 to promote its (auto)phosphorylation and that Cys-185-SOH destabilizes pTyr-527 binding to the SH2 domain (74). Bonanata et al. reported on MD simulations of human serum albumin (HSA) on a submicrosecond timescale, showing that sulfur exposure to the solvent is limited and fluctuates when in the thiol form. However, the exposure increases when HSA is in the thiolate form, which is stabilized by a persistent hydrogen-bond network involving Tyr84 and bridging water molecules connected to Asp38 and Gln33 in the backbone. Insight into the mechanism of Cys34 oxidation by H2O2 is provided by ONIOM (QM: MM) modeling, which includes quantum water molecules (14). MD simulation of reduced versus oxidized EGFR, reinforced by experimental testing, indicated that sulfenylation of Cys797 allows for new electrostatic interactions with the catalytic loop of the protein (187). These studies show that computer-based MD simulation techniques will remarkably contribute to understanding microscopic redox processes in the context of cancer.
Concluding Remarks and Future Perspectives
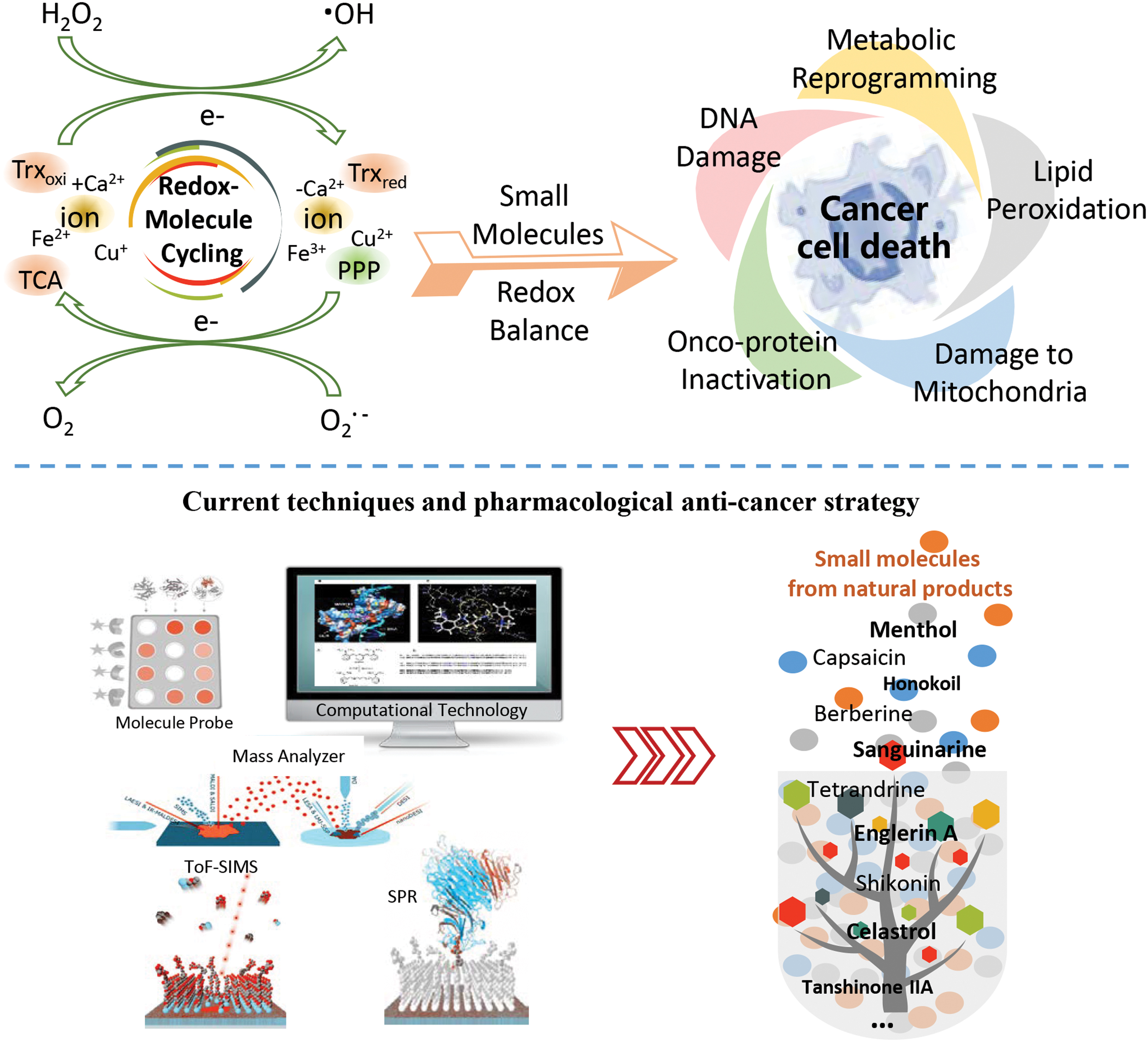
In perspective, many questions and technical challenges remain such as: Does cell redox homeostasis play a similar role compared with the well-known mechanism of glycolysis, local hypoxia, and lactic acid formation in tumor? Can multidisciplinary technology be applied to improve the detection of redox markers? Is it possible to quickly determine the oxidative level of cysteine and methionine in the human body? How to optimally match the bioactivity profiles of single drugs for maximal therapeutic effects, based on knowledge about the relative specificity of target proteins or gene profiles? Interdisciplinary collaborations between biologists, chemists, and physicists toward the development of biochemical and optical platforms to define distinct redox signaling events hold promise for resolving long-standing, burning questions in redox biology and becoming the key for breakthrough. With the fast pace development of advanced technologies, ROS and ion detection sensitivity, drug discovery throughput, and the new drug target and biomarker discovery abilities could be enhanced. Since cancer cells and normal cells habor distinct metabolic and redox characteristics, it provides insight to develop small molecules that can selectively target cancer cells but spare normal cells. The use of MS and computer-based MD simulation techniques can help to profile the specific protein modification and oxidation residues, which can greatly broaden the spectrum for understanding the molecular mechanism of action of ROS and ion intervention on protein. As such, we expect an increasing number of redox targets and biomarkers to be discovered in the future, which will facilitate the discovery of new small molecules for redox intervention (Fig. 5).
Footnotes
Authors' Contributions
L.L., E.N., X.J.Y., and E.L.H.L conceived the review. E.L.H.L., H.L.L., X.X.F., Y.W.W., R.Z.L, and J.M.Z. wrote the article. L.L., E.N., X.J.Y., T.J.H., and E.L.H.L. revised the final proof the article. All authors reviewed the article.
Funding Information
Funded by the Macau Science and Technology Development Fund (File no. 0096/2018/A3, 0055/2020/A, 0003/2019/AKP) and the Joint Research Fund for Overseas Chinese, Hong Kong and Macao Young Scientists of the National Natural Science Foundation of China (Grant No. 81828013), and the Macao Young Scholars Program (AM201926).