
Abstract
Significance:
The systematic investigation of oxidative modification of proteins by reactive oxygen species started in 1980. Later, it was shown that reactive nitrogen species could also modify proteins. Some protein oxidative modifications promote loss of protein function, cleavage or aggregation, and some result in proteo-toxicity and cellular homeostasis disruption.
Recent Advances:
Previously, protein oxidation was associated exclusively to damage. However, not all oxidative modifications are necessarily associated with damage, as with Met and Cys protein residue oxidation. In these cases, redox state changes can alter protein structure, catalytic function, and signaling processes in response to metabolic and/or environmental alterations. This review aims to integrate the present knowledge on redox modifications of proteins with their fate and role in redox signaling and human pathological conditions.
Critical Issues:
It is hypothesized that protein oxidation participates in the development and progression of many pathological conditions. However, no quantitative data have been correlated with specific oxidized proteins or the progression or severity of pathological conditions. Hence, the comprehension of the mechanisms underlying these modifications, their importance in human pathologies, and the fate of the modified proteins is of clinical relevance.
Future Directions:
We discuss new tools to cope with protein oxidation and suggest new approaches for integrating knowledge about protein oxidation and redox processes with human pathophysiological conditions. Antioxid. Redox Signal. 35, 1016–1080.
Table of Contents
I. Introduction 1018 II. Oxidative Modification of Proteins Compromising Their Structure, Function, and Fate 1018 A. Protein oxidation to carbonyl groups 1018 1. Is protein carbonylation a regulatory mechanism? 1020 2. Specificity/susceptibility 1020 3. What could explain specificity? 1020 4. Pathologies associated with protein carbonylation 1020 B. Carbonyl-driven protein modification: common chemical mechanisms of protein glycation and modification through by-products of lipid peroxidation 1021 1. Protein glycation 1023 2. Adducts formed with by-products of lipid peroxidation 1023 a. Aldehydic protein modification 1024 (1) Protein-HNE adducts 1024 (2) Protein-HNE adducts as markers of pathophysiological processes 1025 (3) Protein-HNE adducts and signaling 1025 (4) MDA- and acrolein-protein adducts 1026 (5) Oxysterol protein adducts 1027 (6) Protein adducts with products of PUFA cyclization 1027 (7) Methods for detecting aldehydic protein adducts 1028 (8) Aldehydic modification of serum proteins 1029 C. Amino acid covalent crosslinking 1030 1. Detection and analysis of oxidative protein/protein crosslinks 1031 2. Roles in pathophysiology 1032 D. Protein nitro- and nitroso-derivatives 1033 1. Tyrosine nitration 1033 a. Biological mechanism 1033 b. Biological consequences 1035 c. Denitration 1035 d. Detection of nitrotyrosine (free and protein-bound nitrotyrosine) 1035 e. Proteomics 1035 (1) Examples of nitrated proteins 1035 (a) Manganese superoxide dismutase 1035 (b) Apolipoprotein A-I 1035 (c) Fibrinogen 1035 (d) α-Synuclein 1035 2. Cysteine nitrosation 1035 a. Biological mechanisms (nitrosation vs. nitrosylation) 1035 b. Denitrosation 1036 c. Biological consequences 1036 d. Detection of nitrosothiols 1036 e. Proteomics 1036 (1) Examples of nitrosated proteins 1036 (a) Thioredoxin 1036 (b) Guanylyl cyclase 1 1037 (c) Glyceraldehyde 3-phosphate dehydrogenase 1037 (d) Peroxiredoxin 2 1037 E. Other protein oxidative modifications 1037 III. Role of Protein Sulfur-Amino Acids in Redox Processes and Their Oxidative Modifications 1037 A. Thiol-centered redox mechanisms in catalysis and signaling 1038 1. Thiol/disulfide switches 1038 2. Thiol-sulfenic acid switches 1039 3. Thiol-sulfinic/sulfonic acid switches 1040 4. Relationships between H2O2 signaling and tyrosine phosphorylation 1041 B. Protein S-glutathionylation 1043 1. Reversal mechanism 1044 2. Methods to detect protein S-glutathionylation 1044 3. S-glutathionylation as a modulatory mechanism of protein function 1044 4. Physiopathological processes associated with protein S-glutathionylation 1044 C. Protein persulfidation 1045 D. Oxidation of protein methionine residues 1045 IV. Fate of Oxidized Proteins 1046 A. Protein aggregation 1046 1. Mechanisms involved in protein aggregation induced by protein oxidation 1047 2. Cellular consequences of aggregate accumulation 1047 3. Human pathologies associated with oxidized protein aggregation 1048 B. Proteolysis 1048 1. Proteasome 1048 2. Lon protease 1049 3. Autophagy 1050 V. Conclusions 1051 A. Oxidatively modified proteins as biomarkers of pathological conditions 1051 B. Prevention of protein oxidation 1052 1. Antioxidants 1053 2. Elimination of oxidized proteins and aggregation prevention 1053 C. Emerging approaches to integrate current knowledge on oxidative processes, protein oxidation, and human pathologies 1054
I. Introduction
As early as the 1920s, biochemists started investigating protein photo-oxidation (258, 283) in total blood or plasma induced by light incidence (visible and ultraviolet [UV]) in the presence of iron compounds. They were concerned with the amount of oxygen consumed. Despite these studies being in accordance with present knowledge, the link between protein oxidation and O2 metabolism took much longer to unravel.
Free radical biochemistry dates back to World War II when the astonishment provoked by radiation-induced mutations and poisoning stimulated the field. Indeed, researchers dedicated a tremendous amount of effort to understanding radiation chemistry. Later, in 1954, Gerschman et al. suggested a common mechanism to account for biological damage induced by X-ray irradiation and hyperoxygenation (225). Then, in 1962, Denham Harman hypothesized a role for oxygen-derived free radicals in aging and cancer (257). These two publications paved the way for future reactive oxygen species (ROS) investigations.
In the first half of the 20th century, Otto Heinrich Warburg and Leonor Michaelis proposed that O2 would produce one-electron intermediates during respiration (24). Accordingly, in 1969, McCord and Fridovich discovered that a known protein, erythrocuprein, was able to efficiently catalyze the dismutation of the superoxide anion radical (O2 •−), a by-product of oxygen metabolism (418). This protein later became known as superoxide dismutase (SOD) and proved that oxygen radicals are formed during cell metabolism, reinforcing the claims of the pioneers Warburg, Michaelis, Gershman, and Harman. Moreover, the SOD discovery refocused attention on proteins as generators, consumers, and targets of oxyradicals, consequently attracting protein biochemists who began to study and investigate the relationship between proteins and ROS.
Earl Stadtman and his group were the ones who started systematic studies on the chemical mechanisms of protein oxidation. In fact, he dedicated at least 30 years of his 60-year scientific life to that subject. Besides contributing to the chemical mechanisms underlying the ROS-mediated oxidation of protein amino acid side-chains, his group revealed significant correlations between protein oxidation and pathological conditions, particularly in the aging process. His contribution to the study of thiol-based peroxidase enzymes was also remarkable (see below, Section III.A).
Another hallmark in the field of redox biology was the discovery of the physiological enzymatic synthesis of the free radical nitric oxide (NO•) (439). It was from this finding that redox biochemistry acquired irreversible importance in biology. In addition, the cross-reaction between NO• and O2 •− generating another nitrogen-based reactive species (ONOO−) opened up a new topic of investigation, protein modification by reactive nitrogen species (RNS), which has gained an important status in biochemistry and cellular biology.
Due to their abundance and high reaction rate constants, proteins are major targets for radicals and two-electron oxidants in biological systems (265). Although oxidative damage can occur throughout the whole protein molecule, certain amino acid side-chains are more susceptible because of their reduction potential and spatial localization (151, 265). Sulfur (Cys and Met) and aromatic (Trp, Tyr, Phe, and His) amino acids have the lowest reduction potentials and react fast with oxidants.
The motivation for the present review was the amazing number of publications on the subject. For example, a search on the PubMed platform using the keyword protein oxidation in the Title/Abstract returned more than 115,000 articles, including 12,000 reviews, published in the last 40 years. Considering only the last decade and specific oxidative modifications (sulfur amino acids; carbonyl formation; adducts of lipid peroxidation products; protein crosslinking; and glycation), an underestimated number of 28,000 articles and 3500 reviews stood out. Therefore, the objective of the present review is to integrate the most critical information available on protein oxidation and highlight the role of these modifications in human pathological conditions. We also discuss the role of the redox protein thiol-based catalysis and cellular signaling.
II. Oxidative Modification of Proteins Compromising Their Structure, Function, and Fate
A. Protein oxidation to carbonyl groups
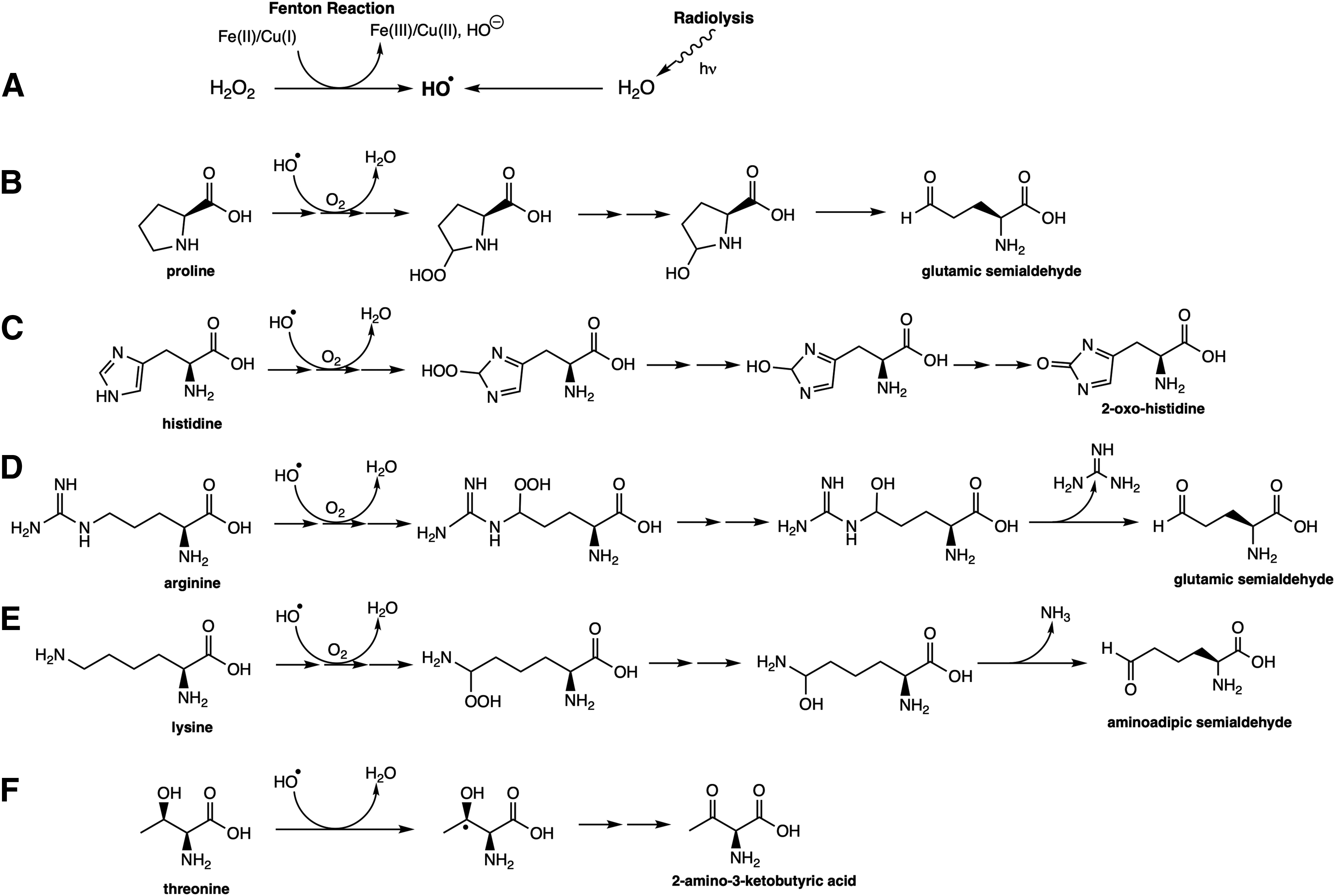
The first demonstration of the formation of a carbonyl group in proteins was made by Garrison et al. utilizing radiation (220, 221). However, systematic investigations focusing on oxidant-mediated protein oxidation were initiated with studies on glutamine synthetase (GS), which converts glutamine into a nitrogen source for nucleic acid and amino acid biosynthesis. In the first study, Stadtman's group (373) demonstrated that GS, a metalloprotein, oxidizes when submitted to oxidative systems (rabbit liver microsomal fraction incubated in the presence of ascorbate, Fe2+, and Mn2+), and catalase and metal chelators inhibit this process. This work also anticipated the susceptibility of oxidized proteins to proteolytic degradation. Later on, it was demonstrated that GS oxidation was highly site-specific (one His and one Arg residue, both close to the metal-binding site), which was in contrast to the protein oxidation observed upon radiation (130, 190). A further set of in vitro experiments led to understanding the metal-catalyzed oxidation (MCO) of proteins, rendering carbonyl groups and other derivatives, depending on the residue involved (608). As demonstrated by Stadtman's group, specific amino acids are prone to the formation of carbonyl groups in proteins and, as later confirmed, the final products are also specific (547). For example, Lys residues are oxidized to aminoadipic semialdehydes, Arg and Pro residues to glutamic semialdehydes, His to 2-pyrrolidine, and Thr to 2-amino-3-ketobutyric acid (Fig. 1). The MCO depends on a Fenton reaction that generates the hydroxyl radical. The low concentrations of free Fe (II) and Cu (I) or peroxides at physiological conditions suggest that the MCO of protein amino acid residues is limited to those amino acids with a high affinity for metals (Arg, Lys, and Pro). In agreement, Desmylaer et al. (167) demonstrated that yeast cells lacking the iron storage protein YFH1p presented higher protein carbonylation levels, which could be reverted by expressing human ferritin L expression. However, MCO is not the only mechanism to trigger protein carbonyl formation. Radiation, as pointed above (221), protein peroxyl radical as the intermediate species (151, 265) and derivatives of reactive halogenous species (266), can also generate a primary carbonyl moiety in proteins.
The literature frequently refers to protein carbonylation as the products of Michael's addition (Fig. 2), where proteins are modified by 4-hydroxy-2-nonenal (HNE), an aldehyde product of lipid peroxidation that preserves a carbonyl moiety centered in the aldehyde. However, in the present review, those products are not considered carbonyl derivatives. Protein modification by lipid peroxidation by-products is discussed in another section (Section II.B.2).
The detection of carbonyl proteins is the most common method utilized by investigators for estimating oxidative stress. Despite being widely utilized as a parameter of oxidative stress, the functional and structural consequences of carbonyl formation are not completely clear. Questions such as: “How many residues in a protein have to be carbonylated to promote the functional and structural modifications required for functionally inactivating the protein?” or “What are the determining factors for the degradation or aggregation of carbonylated proteins?” still have not been answered. Although the linking of protein carbonyl formation to pathological conditions or as a marker of aging has been proposed (discussed below), very few reports support this idea in a way that demonstrates that carbonylation by itself would underline the protein damage and fate.
The identification of protein carbonyl groups by derivatization with dinitrophenylhydrazine followed by spectrophotometric analysis or immunoblotting (374) is the most utilized method for detecting protein carbonyls. More recently, mass spectrometry (MS) has been utilized to identify carbonylated proteins and carbonylated amino acid residues in a given protein (21, 90, 131, 263). A few years ago, a multicenter study demonstrated the importance of protein carbonyl determination standardization (25).
1. Is protein carbonylation a regulatory mechanism?
At first glance, it is unlikely that protein carbonylation has a regulatory role since it has been considered an irreversible modification. However, data in the literature indicate a reversible mechanism of protein carbonylation, as evidenced by a decreased pool of carbonylated proteins, which cannot be attributed to protein degradation or de novo protein synthesis (579, 715). However, these data need to be confirmed with further investigations.
2. Specificity/susceptibility
Whether there are proteins more prone to carbonylation has been a matter of investigation. In several studies working with aged organisms, some proteins, in different organs of the same organism and different organisms, were found repetitively carbonylated (90). Among those proteins, 20% are involved in glucose metabolism, the tricarboxylic acid cycle, and the electron transport chain. Also, heat shock proteins (HSPs) and elongation factors of protein synthesis were carbonylated during aging, from bacteria to humans (90). Elegant studies from Nyström's group showed that increased mistranslation in Escherichia coli achieved utilizing specific mutations and drugs was paralleled by increased protein carbonylation (181). Moreover, mutants harboring hyperaccurate ribosomes exhibited drastically attenuated protein oxidation during growth arrest (34). As interpreted by the authors, verified protein carbonylation in aging cells might be a consequence of reduced transcriptional and translational fidelity independent of increased oxidant formation.
The crosstalk between protein carbonylation and the methionine sulfoxide reductase system has been proposed (448). There are data showing that methionine sulfoxide formation precedes carbonylation as organisms (yeast and mice) lacking the methionine sulfoxide reductase A enzyme (MsrA) accumulate carbonylated proteins. The hypothesis is that the structural change in the protein carrying the methionine oxidation increases the vulnerability of the protein to carbonylation. While this mechanism sounds attractive, no data demonstrate that the increased occurrence of carbonyl proteins (e.g., in aging and neurodegenerative diseases [NDs]) is associated with decreased methionine sulfoxide repair in humans.
3. What could explain specificity?
Protein location in specific subcellular compartments and their abundance would be essential parameters to preview protein carbonylation specificity. Nevertheless, classical work by the Sohal and Levine groups demonstrated that in the flying muscles of an aged population of Drosophila melanogaster, aconitase was the only carbonylated protein in the mitochondria (146). On the contrary, abundant proteins such as cytochrome c remained unchanged, as observed previously by Yan et al. (727). So, what could explain specificity? Cabiscol's group (90) identified a total of 179 proteins from different organisms that were increasingly carbonylated during aging and grouped them according to either their physiological function or location. They found that 9% and 2% of the total proteins listed were cytoplasmic and mitochondrial, respectively. Concerning their physiological function, 11% were HSPs, 11% were involved in amino acid and protein metabolism, and more than 20% in energy production. Another important group was the nucleotide-binding proteins because of the high probability of metal binding to the nucleotide (90).
In an attempt to determine if there are protein sites more prone to carbonylation, Temple et al. (635) showed that only two Lys residues, among 59, were carbonylated in the human serum albumin when the protein was challenged in vitro with ascorbate/Fe3+. However, when challenged with hypochlorous acid (HOCl), five Lys residues were modified. These data suggested that carbonylation is selective to some structural features and dependent on the oxidative species. In agreement, Maisonneuve et al. (399) reported that protein carbonylation should be a predictable process since Arg-Lys-Pro-Tyr-rich sequences are the main carbonylatable sites. Amazingly, the abundance of these sites is related to protein function since a high percentage of proteins containing carbonylatable sites were found to be involved in translation, ribosomal structure, energy production, and nucleotide transport.
The increased application of MS analysis and proteomic approaches for identifying oxidative modifications in proteins is expected to reveal additional information on the susceptibility and specificity of protein carbonylation.
4. Pathologies associated with protein carbonylation
Although the association of protein carbonylation with degenerative processes was proposed based on its prevalence during aging, very few reports are presented in the literature regarding human aging (4, 228, 459). Increased protein carbonylation in aging might be a marker as its production depends on the decreased antioxidant defense and decreased proteolysis. There have been discussions about whether protein carbonylation is the cause of senescence or merely an aging diagnostic marker. These possibilities came from some studies showing that protein carbonylation is (i) increased in species with short life expectancies [e.g., crawler; (597)]; (ii) increased in nonculturable cell populations of E. coli when compared with culturable cells (168); and (iii) decreased in the mitochondria of mice submitted to caloric restriction (356).
In the present decade, there are many reviews relating increased and/or specific protein carbonylation to a broad spectrum of human pathologies, including diabetes and obesity (270, 558), skeletal muscle dysfunction (38), obstructive pulmonary disease (765), cardiovascular diseases (641, 675), chronic kidney disease (658), NDs (239), and hepatocellular carcinoma (228). The majority of the determinations of protein carbonyl levels in those studies were performed in the plasma using different carbonyl detection techniques. Some of these studies established a correlation between the pathology and the pool of carbonylated proteins. For example, Bollineni et al. (65) examined the plasma of lean and obese individuals with or without type 2 diabetes (five individuals per group). They identified 18 carbonylated proteins in the plasma of the diabetic group. Those proteins were not resident plasma proteins but from other sources. Although the study demonstrated that protein oxidation underlies this pathological condition, the presence of the same nonoxidized protein in the plasma had already been linked to type 2 diabetes, obesity, and metabolic diseases. Therefore, their oxidation would not be a marker for these conditions unless it was a determinant for their occurrence in the plasma. Zinellu et al. (765) reviewed studies centered on determining plasma protein carbonyls in patients with chronic obstructive pulmonary disease (COPD). They concluded that one study, out of 16, found a positive correlation between increased carbonyl proteins and COPD. Some of the studies found that sometimes, but not always, plasma carbonyl proteins proceed in parallel with disease progression. An extended review by Tucker et al. (658) on chronic kidney diseases reports that protein carbonyls increase with disease progression. Since protein carbonyls increase in parallel with the severity of the diseases, it is plausible that protein carbonyls could be used as markers for tracking disease progression. In addition, the levels of carbonyl proteins decrease after renal transplantation and l-carnitine supplementation. However, none of these studies identified the proteins prone to carbonyl modification.
In summary, carbonylated proteins are present in pathological processes, but no specific proteins are associated with those conditions.
B. Carbonyl-driven protein modification: common chemical mechanisms of protein glycation and modification through by-products of lipid peroxidation
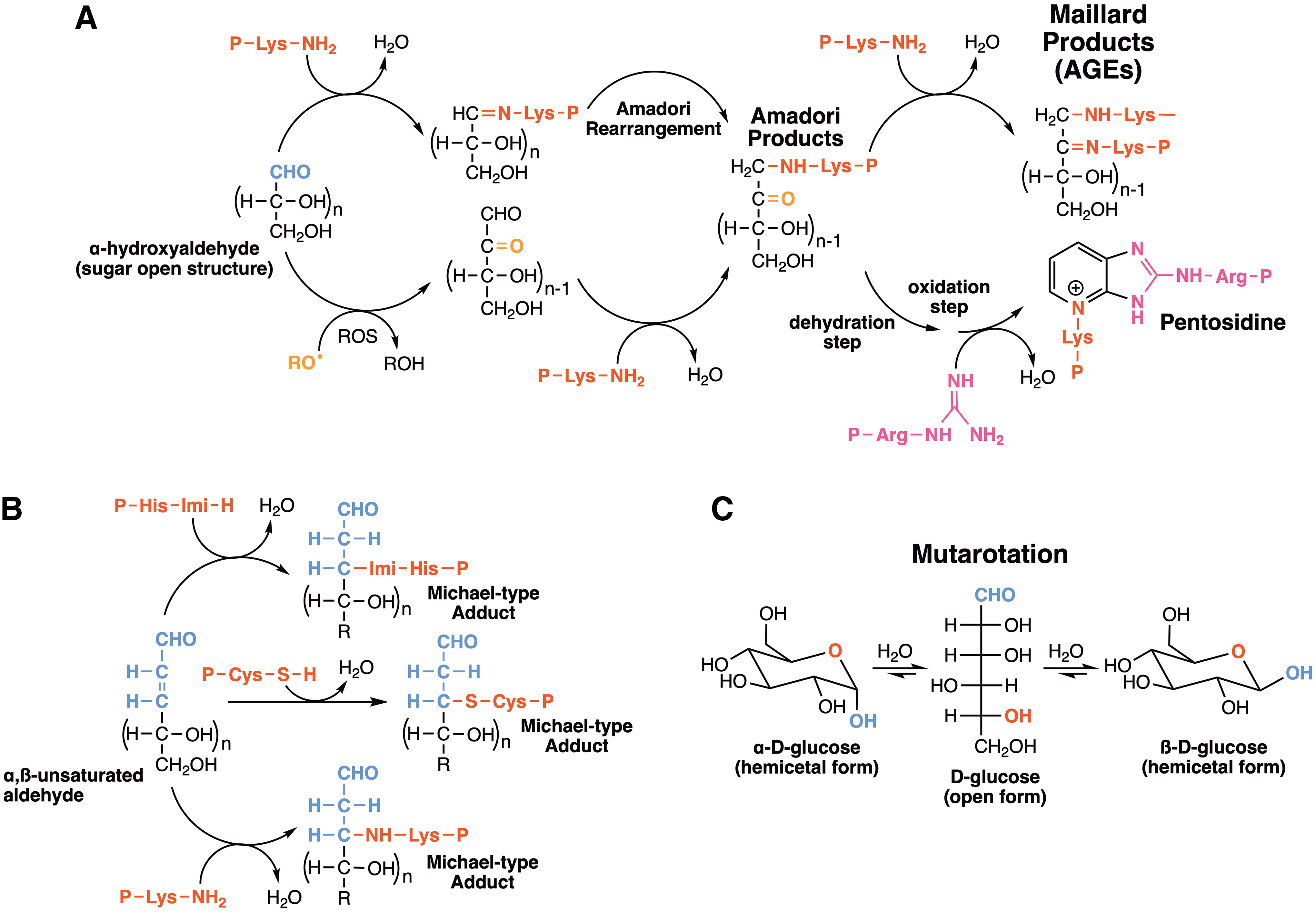
Reactive carbonyl species—aldehydes and ketones—derived from the aerobic oxidation of lipids and oxidized and nonoxidized carbohydrates in cells are reportedly capable of conjugating with amino and thiol groups of proteins. The starting point of the target chemical change is the generation of adducts, bearing –C = N– (imine, Schiff base) groups or C–S–C (thioether) bonds, by nucleophilic attack of amino or thiol groups of proteins to carbonyl moieties originated from either intact or oxidized carbohydrates and lipids. Furthermore, similar 1,2-additions of protein basic amino acids (Lys, Arg, His) to carbonyls give rise to stable products, including the so-called Amadori and Maillard products derived from α-hydroxyaldehydes, α-dicarbonyls, and aldoses, namely advanced glycation end-products (AGEs) (591, 609, 610). In addition, highly reactive methylglyoxal (α-oxopropanal), originated as a by-product of glucose and fructose catabolism, is a putative precursor of AGEs in diabetes, cardiovascular diseases, and neuropathies (414, 530).
Conversely, protein sulfhydryl groups are prone to 1,2-addition to either aldehyde fragments, derived from free radical-mediated oxidation of proteins, or to malondialdehyde (MDA), α-hydroxy aldehydes, and α,β-unsaturated aldehydes generated by lipid peroxidation yielding Michael-like adducts (1,4-addition), collectively called advanced lipoxidation end-products (ALEs) (490, 680) (Fig. 2).
Aldehydes are more reactive to nucleophilic addition than the corresponding ketones. This increased activity is due to the alkyl groups of ketones having higher steric hindrance in the transition of sp2-carbonyl to sp-saturated carbon. Moreover, since sp2-carbonyls are more electronegative than sp-alkyl groups, alkyl groups of ketones have a higher inductive electron-donating effect than the hydrogens of aldehydes. Short-chain alkanals, β-alkenals, α-oxoaldehydes and other α-dicarbonyls, α-hydroxyketones, β-ketoacids and -esters, β-diketones, and α-aminoketones whose MCOs generate α-dicarbonyls are among the most reactive and better-studied aldehydes and ketones and must be highlighted (Fig. 3). These compounds undergo biochemically relevant nucleophilic addition with sulfhydryl and amino groups of proteins and nucleobases.
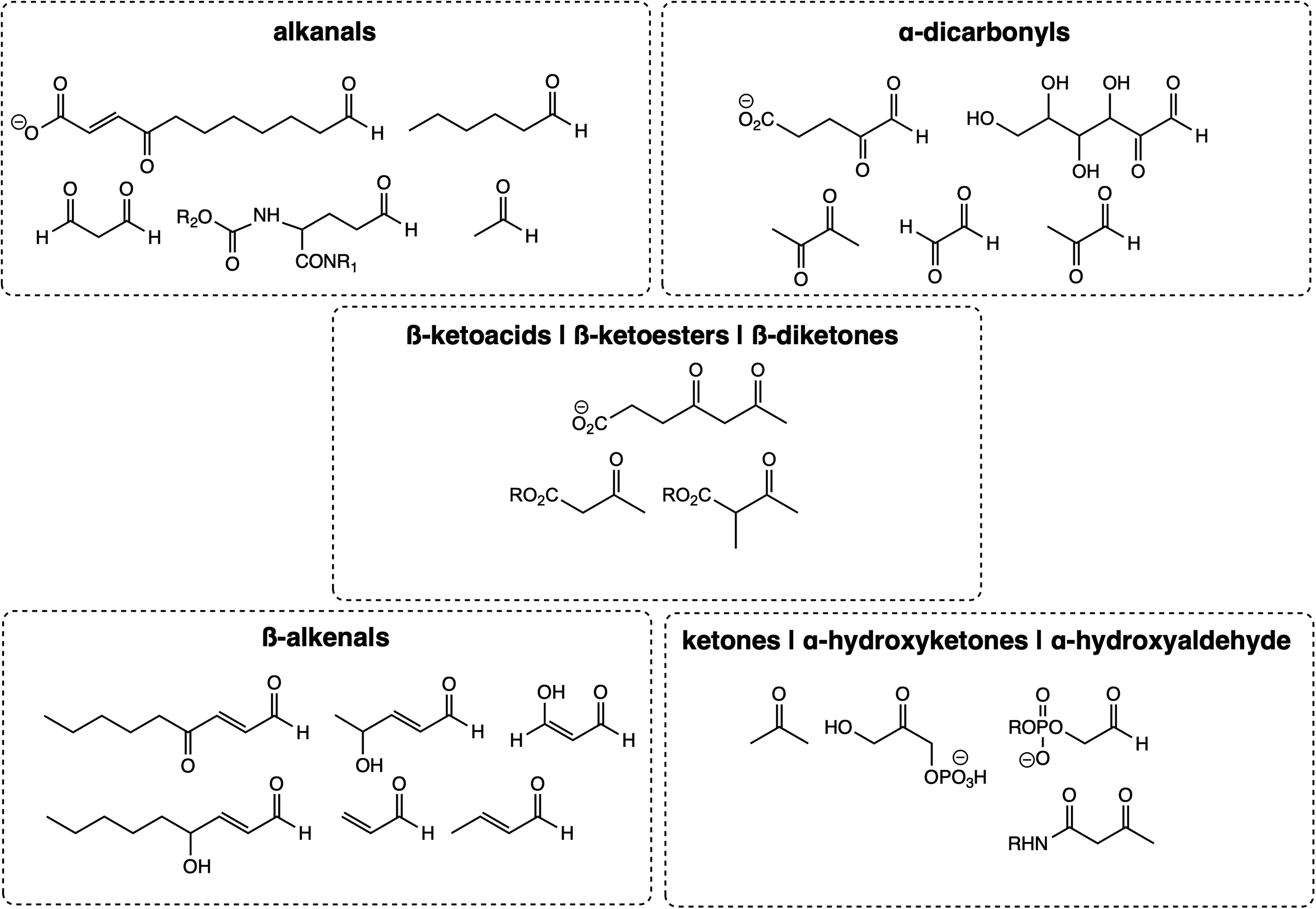
To construct a ruler to predict the reactivity of in vivo carbonyl metabolites with surface protein amino acids, that is, their electrophilicity, rate constants of the nucleophilic addition of peroxynitrite in phosphate buffer pH 7.2–7.4 at room temperature obtained in vitro may provide exciting and eventually helpful comparative parameters (411 –413). Table 1 lists the reaction kinetics of several sugar and fatty acid oxidized carbonyl products with proteins and nucleobases in ascending order. Accordingly, acetone (a ketone body) is the poorest electrophile due to the inductive static effects of two methyl groups and steric hindrance.
Bimolecular Rate Constants (k2) of Peroxynitrite Nucleophilic Attack on Different Carbonyl Metabolites in Phosphate Buffer a
100–125 mM.
For comparison: the k2 value for peroxynitrite addition to CO2 is in the range 3–6 × 104 M −1·s−1 (162), which prevails over the reaction with carbonyl additions, except for the highly reactive α-oxoaldehydes under high intracellular CO2 cellular concentrations, for example, plasma concentrations (162).
RT, room temperature.
The increasing reactivity of the acetaldehyde (ethanal), propanal, and isobutanal is listed here to corroborate the higher electrophilicity of aldehydes compared with ketones and also to attest to the increasing carbonyl deactivation owing to their substituent bulkiness. Acrolein (ACR) is an α,β-unsaturated aldehyde prone to Michael-type 1,4-additions of nucleophiles; the ene conjugation with the carbonyl group exacerbates its reactivity. Finally, the α-dicarbonyls diacetyl (dimethylglyoxal, 2,3-butanedione), methylglyoxal, and glyoxal itself (ethanediol), for the arguments stated above, are, respectively, one, two, and three orders of magnitude higher than that of alkanals; the electron-withdrawing effect of a vicinal carbonyl group in the glyoxals increases the partial positive charge of the carbonyl side, thus facilitating a nucleophilic attack of a protein or an amino group of DNA.
As expected, buried amino acid residues would be much less accessible to the attack by sugars or low-molecular-weight electrophiles (Table 1). Accordingly, Gao and Wang (216) found that the electrophile modified only four solvent-accessed Arg residues out of six using methylglyoxal-challenged hemoglobin. The authors suggested that methylglyoxal-triggered site-specific modifications of hemoglobin could be exploited as a biomarker for clinical applications.
1. Protein glycation
Protein glycation is intrinsically associated with high glucose levels in the blood and tissues. At first glance, protein glycation is not necessarily associated with oxidative mechanisms, although oxidatively modified sugars retain similar reactivity to render Amadori end-products with protein amino groups (Fig. 2). Nonenzymatic protein glycation occurs when sugars condense with protein amino groups forming a Schiff's base, which rearranges to form an Amadori product (Fig. 2). Amadori products further react with amino groups of other protein molecules to generate Maillard products, also known as AGEs. Among the reported AGEs, pentosidines that originate from Arg and Lys, hydroimidazolones from Arg, and other biomarkers derived from Arg, Lys, and Cys are of bioanalytical interest and can be quantitatively measured (529). Pentosidines are fluorescent Lys-Arg intermolecular protein crosslinks and sequentially oxidized, thus referred to as a glycoxidation process. They accumulate in bone and connective tissues (573) and are used as urine and blood biomarkers of age-related diseases such as osteoporosis and diabetes (560, 575) (Fig. 2). AGEs were first described in vivo as a modification of long-lived proteins, such as hemoglobin, lysozyme, collagen, elastase, and alkaline phosphatase (486). Indeed, glycated hemoglobin is a suitable marker for high glucose levels. AGEs are also formed from exogenous sources, such as cigarette smoking (477) and food (200).
The relationship between protein glycation and pathologies associated with protein-based oxidative stress relies on the fact that protein glycation is involved in protein aggregation and delayed oxidized protein degradation since AGEs may block the entry of the proteasomal core (486). AGEs are recognized by a cell surface type I receptor belonging to the immunoglobulin superfamily (471), which implies a set of metabolic interferences, including interactions with oxidative and nitrosoactive stress (486). Nevertheless, it is out of the scope of the present review to cover the rich literature on AGEs, as it is not a direct consequence of protein modification through by-products of oxidative mechanisms. However, their participation in oxidative stress should not be ruled out. A search in the PubMed platform (April/2020) using Protein glycation as the keyword returned 15,000 total publications that included 2800 reviews. For more information, we advise readers to search for specific literature on AGE-associated pathologies.
2. Adducts formed with by-products of lipid peroxidation
Intracellular intermediates of lipid peroxidation, such as lipid hydroperoxides, can be formed either by enzymatic or nonenzymatic mechanisms (105, 230, 479). Lipid hydroperoxides are formed by reactions involving lipoxygenases, cyclooxygenases, and cytochrome P450. They are efficiently reduced to the corresponding alcohols by the antioxidant enzymes, glutathione peroxidases (Gpxs), and peroxiredoxins (Prxs). While certain types of lipid oxidation products exert beneficial functions, acting as ligands and modulators of cellular signaling processes, the overproduction of lipid hydroperoxides and/or their inefficient reduction can promote deleterious effects by increasing the production of reactive lipid by-products.
The uncontrolled production of lipid hydroperoxides is associated with the induction of cell death (e.g., ferroptosis) (202), inflammation, and several pathologies, such as cardiovascular and NDs. Notably, lipid hydroperoxides can react with metal ions or other one-electron oxidants to produce peroxyl and/or alkoxyl radicals in a Fenton-like reaction (739). These radical intermediates can further propagate lipid peroxidation, yielding various secondary reactive lipid species known as lipid electrophiles.
Esterbauers's group characterized several short-chain aldehydes, including HNE, MDA, and ACR (186). Chemical structures of major lipid electrophiles are shown in Figure 4. Fatty acid-derived aldehydes can be grouped by size into short-chain (<6C), medium-chain (6–12C), and long-chain aldehydes (>14C, e.g., hexadecenal, cyclopentenone PGs, and isoprostanes [IsoPs], levulinaldehydes). In addition, lipid electrophiles can be categorized into truncated oxidized phospholipids (oxPL) and sterol-derived aldehydes (β-hydroxy-5-oxo-5,6-secocholestan-6-al, Seco-A and its aldol product, Seco-B).
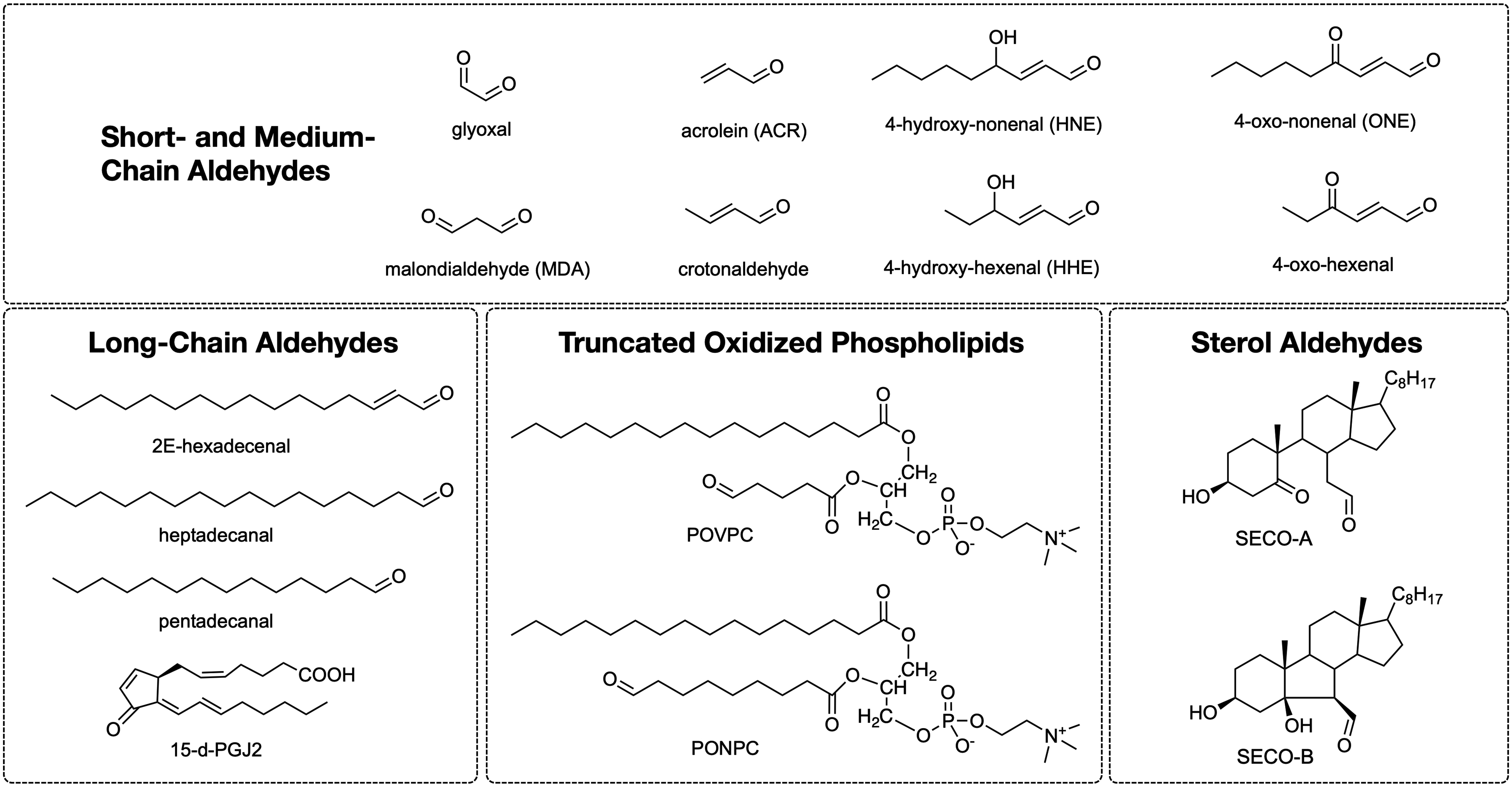
The short-chain and medium-chain aldehydes include several alkanals and β-alkenals (α, β-unsaturated aldehydes) produced by polyunsaturated fatty acid (PUFA) lipid peroxidation. Moreover, various long-chain aldehydes (IsoPs and isolevuglandin [IsoLG]) are produced by fatty acid cyclization reactions (427, 565), as well as from ether glycerolipids (plasmalogens) and sphingosine-1-phosphate, by enzymatic and nonenzymatic oxidation mechanisms (554). These lipid electrophiles react with nucleophilic groups in biomolecules such as proteins to produce lipoxidation adducts, which is reviewed in the next sections.
a. Aldehydic protein modification
Although the modification of proteins by aldehydes and its interference with protein function have been reported since the 1940s (database: PubMed; keywords: aldehydes AND proteins), evidence that aldehydic products of lipid peroxidation could affect protein function was first reported by studies with rat liver microsomal fractions (45, 195). The first detected protein adduct with lipid peroxidation by-products was described in liver microsomal proteins (46) and upon the formation of low-density lipoprotein (LDL)-HNE adducts (317). Since then, there have been thousands of reports in the literature on this subject. In fact, there has been an almost 2.5-fold increase in the number of publications in the present decade, with many investigators currently dedicated to unraveling the meaning of protein adducts with aldehydic by-products of lipid peroxidation in physiological and pathological processes (224, 747).
(1) Protein-HNE adducts
HNE is probably the most studied lipid by-product [highlighted in a special issue dedicated to HNE (519)]. It is highly reactive toward nucleophilic residues in proteins. As shown in Figures 2 and 5, the mechanisms by which HNE modifies proteins are almost exclusively centered on Michael addition to Lys, Cys, and His protein residues. Besides the chemical possibility of forming Schiff's bases where the HNE aldehyde group reacts with the ɛ-amine group of protein Lys residues, it is estimated that more than 99% of HNE-modified proteins result from Michael addition. As mentioned above, since a carbonyl group in the HNE tail is preserved in proteins modified by the Michael addition, these products are usually classified as carbonyl-proteins.
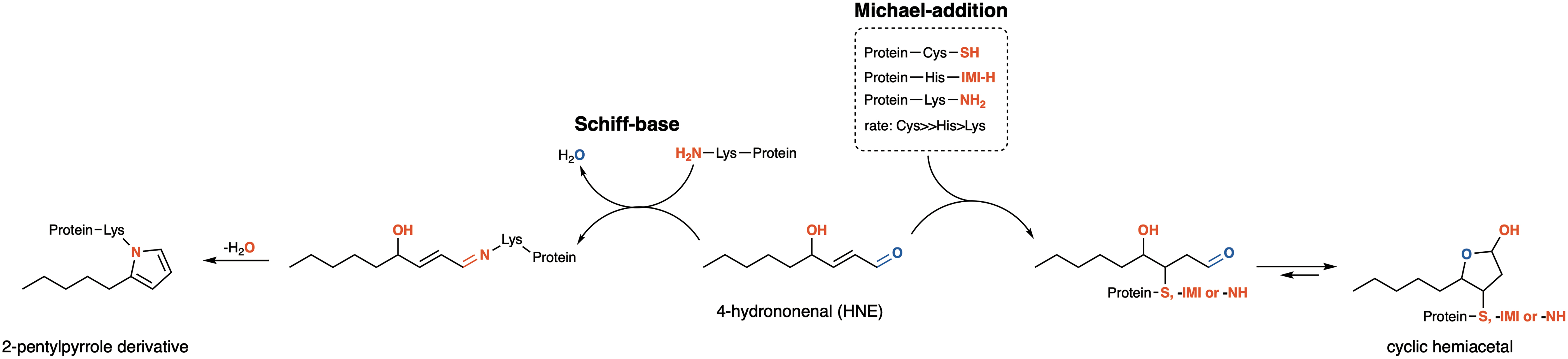
In recent years, proteomic approaches have been used to uncover HNE-modified proteins (732, 755). Data reported in these studies are based on cells incubated with HNE (732) and extracted cellular proteins incubated with 4-HNE (755). The primary importance of these studies was (i) to identify the proteins that are potentially prone to HNE modification, (ii) to specify the protein motif/sequences that are more likely to be modified residues, (iii) to investigate the stability of the modified protein, and (iv) to discover the most common residues undergoing modification. The main conclusions from these studies are that (i) 400 to 600 proteins were found modified by HNE, (ii) the modification was only through the Michael addition mechanism, (iii) Cys and His residues were the most common modification sites, and (iv) in one study, aspartic acid was found to be enriched around both modified His and Cys residues (755), while in another study, Lys was found in the vicinity of Cys-modified residues (732).
Regarding the stability of the modified protein, when cells incubated with HNE were allowed to recover for 4 h, 87% of the quantifiable HNE adducts were reduced by twofold (732). However, the study found that among four modified-Cys residues in the FAM120A protein, one of them was still modified after the 4-h recovery, suggesting an unknown intracellular repair mechanism. Indeed, proteasomal inhibition did not affect the turnover of the HNE-modified proteins. The modified proteins found in those studies are localized in almost all intracellular compartments and the extracellular exosome. Moreover, they are functionally related to RNA processing, protein ubiquitination, and cell cycle, among others. On the contrary, when six liver cell lines without any treatment were analyzed for the pool of HNE-modified proteins, only a few proteins were found modified, 14 at Cys residues, 14 at His, and 5 at Lys (755).
(2) Protein-HNE adducts as markers of pathophysiological processes
Only 8% of the HNE produced inside cells bind to proteins (586). The proteins potentially modified by HNE found in human samples are mainly transcription factors of pathways involved in the antioxidant response, inflammation, cell cycle, protein synthesis, and apoptosis (104, 224). The concept of a biomarker as an indicator of diseases or their progress cannot yet be assigned to HNE-modified proteins. For this, many studies of epidemiological nature are necessary.
Nonetheless, there are many reports in the literature pointing in that direction. One important observation is that while protein-HNE adducts are relatively vulnerable to degradation, they are more stable than HNE itself. Moreover, proteins in the extracellular compartments are much less exposed to proteases. Notably, in recent years, many reviews highlighting protein-HNE adducts in human pathological processes have been published. Table 2 summarizes human diseases where protein-HNE adducts are reported either as a possible biomarker or a contributing factor in the development of the investigated pathological processes.
Human Pathological Conditions Associated with Protein-HNE Adducts
HNE, 4-hydroxy-2-nonenal; AD, Alzheimer's disease.
(3) Protein-HNE adducts and signaling
One crucial question about oxidatively modified proteins is whether they function as signaling or regulatory biomolecules. Herein, we define a regulatory role as a process where the protein is modified so that its function is changed to modulate or cope with the process it is engaged in, discussed in Section IV. On the contrary, a signaling process implies the participation of the modified protein in promoting a cellular response to a given stimulus (e.g., antioxidant response, gene transcription, and others). In the case of protein-HNE adducts, the most widely reported effect is the participation of these adducts in cell signaling involving the activation of kinases and transcription factors related to redox homeostasis (Nrf2, NF-κB, AP-1, PPAR) (28, 389, 751).
One of the most explored mechanisms is the modulation of the mitogen-activated protein kinase (MAPK) family, including the well-known ERK, JNK, and p38-MAPK subfamilies. The mechanism by which members of the MAPK signaling cascade are activated is not well known. Data show that protective or pathological functions are dependent on HNE concentrations used to promote cellular responses either to cope with oxidative stress or to induce apoptosis. Physiological HNE concentration is estimated to be in the range of 0.28–0.68 μM in normal human plasma, while concentrations up to ∼5 μM can be found in rat hepatocytes (751). Most of the signaling effects have been observed in studies using low physiological doses of HNE (<1 μM), whereas high supraphysiological doses (>10–20 μM) have been shown to induce processes such as autophagy, senescence, and cell cycle arrest (28, 751).
Other signaling cascades affected by protein-HNE adducts involve protein kinase C isoforms (PKCs), a family of serine/threonine kinases phosphorylating various targets and playing an essential role in cell proliferation, differentiation, and tumorigenesis processes (28). The selectivity of HNE with the various PKC isoforms (conventional: α, βI, βII, γ; novel: δ, ɛ, η, θ; and atypical: ζ, λ/τ) depends on HNE levels, which is indicative of differential effects based on the reactivity of the isoforms for HNE (114). Conventional and novel PKCs are lipid-sensitive (modulated by diacylglycerol levels) and calcium-dependent isoforms. HNE can activate PKC indirectly by inducing phospholipase C activation, which cleaves phosphatidylinositol-4,5-bisphosphate to generate inositol triphosphate and diacylglycerol. In contrast, HNE PKC inhibitory effects are thought to be mediated by direct adduction with the protein. However, the underlying mechanism of HNE selectivity toward different PKC isoforms remains unknown.
Among transcription factors directly or indirectly affected by HNE are those related to genes of the antioxidant response, protein folding, inflammation, and cell cycle (389). At low HNE concentrations, protein-HNE adduct formation modulates gene transcription, allowing cells to recover from moderate oxidative stress. An example is the Keap1-HNE adduct, which prevents the Nrf2-Keap1 complex from forming, thereby allowing the translocation of Nrf2 to the nucleus to induce the transcription of antioxidant and detoxification enzymes (heme oxygenase-1, Prx, thioredoxin [Trx], Gpx, glutathione S-transferase [GST], etc.). The regulation of Nrf2 signaling involves multiple signaling molecules, including Keap1, PKCs, and p21, among others. Moreover, Keap1 is equipped with multiple cysteine-based sensors that are modulated by various types of endogenous/exogenous stressors, including H2O2 and electrophiles such as HNE (625). Details on the sensing mechanisms of the Nrf2-Keap1 system toward different electrophiles and oxidants are still under investigation.
HNE is metabolized through enzymatic detoxification systems, namely, Phase I and Phase II metabolic pathways. As a result of both metabolic pathways, the percentage of HNE found on proteins was reported to be around 5% of the total HNE in rat liver and brain (26, 433, 751). HNE is enzymatically modified through glutathione (GSH) conjugation as a substrate of GSTs (Phase II). Hence, GSTs function as significant determinants of cellular levels of HNE and play a role in the regulation of the HNE-protein adduct formation. Another important class of enzymes in HNE detoxification is the family of aldehyde dehydrogenases (ALDHs), which converts HNE to 4-hydroxy-2-nonenoic acid. The ALDHs have been implicated in a series of pathological conditions as protective players against HNE toxicity (433). Experimental approaches in animal models, by ALDH2 gene deletion or ALDH2 drug activation, have confirmed and given clues about the involvement of protein-HNE adducts in many pathologies, such as neurodegeneration, heart diseases, atherosclerosis and, nonalcoholic fatty liver diseases (433). NADH-dependent alcohol dehydrogenase and NAD(P)H-dependent aldo-keto reductase have also been shown to detoxify HNE by reducing it to 1,4-dihydroxy-2-nonene (751).
(4) MDA- and acrolein-protein adducts
MDA is generated as a by-product of nonenzymatic lipid peroxidation and a side product that arises during thromboxane A2 biosynthesis (28). MDA concentrations in human plasma have been found in the range of 0.36 to ∼15 μM (599). MDA reactivity is based on its electrophilic nature, thereby reacting with nucleophiles such as the protein amine residues (Lys, Arg, His) to generate Schiff's base adducts (28). MDA reacts in vivo with primary protein amines to form the N-(2-propenal) Lys or Lys-Lys crosslinks (663). These adducts are also referred to as advanced lipoxidation end-products (ALEs; Section II.B; Fig. 2). MDA can also react with DNA and proteins, and in some cases, mediate the formation of DNA-protein crosslinks (682). The mouse apoB-100 fraction of oxidized LDL was one of the first protein-MDA adducts detected (488). These adducts are found in almost all the pathologies associated with HNE-protein adduct formation (Table 1) (28).
ACR is an unsaturated aldehyde and the strongest electrophile of all reactive aldehydes and consequently presenting the highest reactivity with nucleophilic residues in proteins (Cys, His, and Lys residues) (186). The FDP-Lys adduct (3-formyl-3,4-dehydropiperidino-lysine) is a stable adduct that was first detected in oxidized LDL (664). Studies by Esterbauer showed that ACR reacts 110–150 times faster with GSH than HNE (186). Conjugation to thiols can occur spontaneously or be catalyzed by GST isoenzymes (27). GSH conjugation and conversion to mercapturic acid metabolites (2-carboxymethylmercapturic acid and 3-hydroxypropylmercapturic acid, as a major urinary product) in the liver and kidney are the main routes for ACR elimination through urine. ACR is produced: (i) as an end product of lipoperoxidation, (ii) by myeloperoxidase (MPO) from threonine at sites of inflammation, (iii) from protamine oxidation, and also (iv) as a metabolite of the anticancer drug cyclophosphamide (613). Human exposure to ACR not only occurs through metabolism and endogenous lipid peroxidation but also by the oral ingestion of food (e.g., cooking oil) and water, as well as through respiratory (cigarette smoke, automobile exhaust) and dermal routes (432).
Increased protein-ACR adduct concentrations have been reported in many human pathological conditions, such as cardiovascular diseases, diabetes, spinal cord injury, alcoholic liver disease, photo-damaged skin, and others. Furthermore, protein-ACR adducts are suggested to play a role in the development of many pathological conditions (83, 432). However, the causality and mechanisms by which ACR adducts induce each pathological condition remain to be determined. The pathological association of ACR with various diseases has been based on detecting higher levels of ACR adducts in sera of patients (83). At the experimental level, ACR has shown to induce pro- or anti-inflammatory effects, depending on the ACR dose and exposure duration (83, 432). ACR (up to 30 μM) caused suppression of innate macrophage responses, and this effect was causally correlated to ACR adduction and inhibition of JNK2 and NF-κB (286). Moreover, ACR treatment has shown to induce endoplasmic reticulum (ER) stress, impair protein biosynthesis and gene transcription, modulate membrane permeability, and increase apoptosis (432). As described in a myocardial ischemic injury model, ACR forms adducts with the mitochondrial PKCɛ leading to mitochondrial dysfunction (691). Besides, ACR high reactivity with thiol proteins impairs critical antioxidant systems based on the protein thiol catalysis by causing GSH depletion (432).
(5) Oxysterol protein adducts
Oxysterols are oxygenated derivatives of steroids that are formed by enzymatic and nonenzymatic pathways. The most abundant steroid in mammalian cells is cholesterol, playing essential roles in membrane structure and signaling. The enzymatic pathway is responsible for cholesterol hydroxylation and its conversion to bile acids and steroidal hormones (458). Since it is unsaturated, cholesterol is oxidized nonenzymatically by free radicals, singlet oxygen, and ozone. These reactions produce oxysterol containing hydroperoxy, hydroxy, ketone, epoxide, and aldehydic groups (171, 595, 764). Importantly, secosterol aldehydes (SecoA and SecoB) are electrophilic oxysterols produced in the reaction of cholesterol with ozone (704), singlet oxygen (643, 667), and free radicals (764). Secosterol aldehydes have been detected in biological samples, including the human brain (66) and atherosclerotic tissues (703), and have been implicated in the pathogenesis of cardiovascular and NDs. Concentrations of secosterol aldehydes in systemic circulation were found to be ∼30 nM under basal conditions and ∼200 nM in patients with inflammatory artery disease (705).
Secosterol aldehydes can modify several proteins leading to misfolding and aggregation. They have been shown to accelerate the in vitro amyloidogenesis of β-amyloid peptides (Aβ 1–40 and 1–42), the α-synuclein fibrillation (66), and SOD1 aggregation (143, 144). These modifications were hypothesized to favor the formation of neurotoxic protein aggregates linked to Alzheimer's, Parkinson's, and amyotrophic lateral sclerosis. Aβ modification occurred by Schiff base formation with basic amino acid residues, specifically Lys 16 and 28, and the N-terminal group of Asp 1 (670). Similar adduct formation has been observed with bovine myelin basic protein (bMBP) (138). Conformational changes and agglomeration of this protein have been attributed to the covalent attachment of SecoB, leading to increased exposure of the peptide domain V86-T98, which is related to the immunological reaction, and decreased exposure of the F42 and F4 proteolytic regions, which lead to the cleavage of bMBP (138). Secosterol aldehydes also form Schiff base adducts with Lys residues located in loop VII (electrostatic loop) and nearby the dimer interface in apoSOD1, enhancing its propensity to aggregate (143). Notably, apoSOD1 aggregation was dramatically increased by the hydrophobic sterol aldehydes and not by less hydrophobic aldehydes, HNE or 4-hydroxy-2-hexenal (HHE), indicating that aldehyde hydrophobicity critically affects protein aggregation (144).
A study evaluating the reactivity of secosterol aldehydes in human atherosclerotic tissue observed that this aldehyde induces the formation of apolipoprotein C-II (apoC-II) amyloid fibers in vitro (615). Notably, macrophages secrete apoC-II in the atherosclerotic process, and its fibrillation is directly related to plaque formation in the disease (615). Of note, secosterol aldehydes also inhibited nitric oxide synthase (NOS), which may contribute to the development of vascular and NDs (355). The inhibition mechanism seems to be related to the blockade of the enzyme binding site with its cofactor calmodulin through the formation of Schiff bases with Lys residues present in this region (355). Furthermore, the role of secosterol aldehydes in cancer-related mechanisms was investigated by Nieva et al. (478). They have shown that secosterol aldehydes, HNE and HHE, form adducts with Lys residues on wild-type p53 protein. However, similarly to the apoSOD1 study (143, 144), only the hydrophobic secosterol aldehydes induced p53 amyloidogenesis, while the more polar aldehydes, HNE and HHE, were not able to induce protein aggregation (478).
Overall, studies reviewed in this section highlight the potential role of electrophilic oxysterols in protein modification. Being highly hydrophobic, sterol aldehydes derived from cholesterol dramatically affect the conformational stability of the protein to which they are bound. Further in vivo studies are required to clarify the importance of oxysterol-induced protein modifications and their potential role in cancer, and cardiovascular and NDs.
(6) Protein adducts with products of PUFA cyclization
Arachidonoyl-containing lipids are oxidized by free radical-catalyzed peroxidation producing a series of prostaglandin F2-like compounds known as IsoP (447), which are important modulators of inflammatory signaling (555). Although some classes of IsoPs undergo Michael addition reactions with protein thiol groups, no protein-IsoP adducts have been identified. Presently, the only evidence of protein-IsoP adducts comes from the direct inhibition of IKK in a cellular model, where the biosynthesis of PGD2 was induced, which subsequently increased IsoP, a metabolite of PGD2 levels. As a result, NF-κB was activated, and the proinflammatory response ensued (557).
Levuglandin (LG) and IsoLG derivatives are acyclic γ-ketoaldehydes derivatives (levulinaldehydes) produced from the spontaneous rearrangement of arachidonate endoperoxide intermediates, prostaglandin H2 and H2-IsoP, generated through cyclooxygenase and free radical pathways, respectively. Of note, the cyclooxygenase pathway produces two levulinaldehyde stereoisomers, the LGE2 and LGD2, while the free radical pathway generates several isoLG stereo and regio-isomers (563, 565). The levulinaldehydes produced by the free radical pathway are also referred to as “isoketals (IsoK)” to distinguish them from the enzymatic products. However, chemically they are the same group of compounds, and the IsoLG nomenclature is used to designate both of them. Of note, IsoLGs have never been isolated from biological sources, most likely because of their high reactivity toward biomolecules (half-life of ∼2 min). Notably, IsoLGs have been reported to react approximately two orders of magnitude faster than HNE or MDA (76).
Immune assays have detected protein-IsoLG adducts in human samples, such as plasma, brain, and meningeal vessels (566). Protein–IsoLG adducts have also been detected by liquid chromatography tandem mass spectrometry (LC-MS/MS) (154, 737). Using this approach, IsoLF-Lys adducts in CYP27A1, a sterol C27-hydroxylase, have been identified in the human retina. Protein-IsoLG adducts have been implicated in several pathological conditions, such as alcoholic liver disease, Alzheimer's disease (AD), age-related macular degeneration, atherosclerosis, cardiac arrhythmias, cancer, end-stage renal disease, glaucoma, multiple sclerosis, and thrombosis (565). As discussed below, protein-IsoLG-modified adducts are supposed to be important proteasomal inhibitors (565). The so-called neuroketals are formed analogously to IsoLG through the oxidation of docosahexanoic acid, a PUFA highly enriched in the brain (51).
IsoLGs have a common γ-ketoaldehyde core linked to two different hydrocarbon chains. The γ-ketoaldehyde core reacts with the ɛ-amine group of protein Lys residues and amino groups in nucleic acids and aminophospholipids (565). The reaction mechanism proceeds through the formation of an imine adduct (Schiff's base), which irreversibly cyclizes to a pyrrole adduct (76). In the presence of oxygen, the pyrrole adduct is converted over time into highly stable lactam adducts, as well as hydroxylactam adducts (76). Alternatively, the pyrrole adduct can also react with other nucleophiles such as thiols or other pyrroles to produce protein/protein and protein-DNA crosslinking (55). Two different structures for the protein/protein crosslinks have been proposed, the bis-aminal and the pyrrole/pyrrole crosslinks.
Membrane proteins are vulnerable targets for IsoLG modification, leading to the formation of phospholipid IsoLG-protein complexes, which can impair the function of ion channels, enzymes, and receptors (75). Sirtuin, a deacetylase enzyme located very close to membrane lipids, is prone to IsoLG modification, reducing its activity and increasing the overall acetylated protein levels. The imbalance of protein deacetylation affects protein/protein interaction as acetylation alters electrostatic interactions and hydrogen bond networks (476), ultimately interfering with signal transduction. The degradation of protein-IsoLG adducts by the proteasome was compromised because adducted proteins are not suitable substrates, and the tentative degradation results in proteasomal inhibition (155).
In conclusion, increased levels of protein-IsoLG adducts are observed in many human diseases, such as atherosclerosis, myocardial infarction, hypertension, and AD (224). However, whether IsoLG adducts contribute to the pathogenesis of these diseases remains to be determined. Evidence supporting a potential role for these adducts on the development of pathological conditions comes from data where cell treatment with exogenous IsoLG induces a variety of relevant responses, including increased macrophage uptake of LDL, activation of platelet aggregation, inhibition of sodium and potassium channels, and inhibition of the proteasome (153). Notably, the presence of IsoLG adducts is increased in patients with atherosclerosis (564). Moreover, IsoLG-protein adducts were found to be elevated in HDL derived from patients with hypercholesterolemia, shedding light on the potential role of HDL-IsoLG adducts in cardiovascular disease (417).
(7) Methods for detecting aldehydic protein adducts
MDA was the first by-product of lipid peroxidation to be measured in biological samples as a free aldehyde (342). Aldehydic protein-adducts formed with MDA, HNE, and ACR, and other aldehydes have been evaluated using different techniques, including immune labeling/immunostaining (747) and mass spectrometric methods (MS). Various polyclonal and monoclonal antibodies have been raised against aldehyde-protein adducts and are now commercially available (324, 326, 489, 649, 665, 685). These antibodies have been successfully applied for the detection, quantification, and tissue distribution of lipid aldehydes, such as HNE, in oxidatively modified LDL particles, ischemic heart, and neurodegenerative disorders [reviewed by (604)].
Despite the usefulness of immunoassays, this technique does not yield information about the precise identity of the modified proteins or structural details about adduction sites. Thus, high-resolution tandem mass spectrometry (MS/MS) analysis has emerged as the gold standard method for characterizing protein posttranslational modifications (PTMs) (10, 673). Proteomic methods commonly used to characterize protein-aldehyde adducts can be divided into “gel-based” and “gel-free” approaches (397). In “gel-based” assays, proteins are separated by one- or bidimensional gel electrophoresis before digestion by proteases, and in the “gel-free” approach, proteins are digested directly in solution. The peptide mixture is then analyzed by MS and identified by peptide mass fingerprinting (PMF) or by MS/MS. In PMF, intact peptide masses are searched against a database containing in silico digested proteins. In MS/MS analysis, peptides are fragmented, and the collection of fragment ions is used for peptide sequencing. The MS/MS proteomic analysis is often coupled to nanoflow liquid chromatography (LC-MS/MS), which greatly increases the coverage and precision necessary for detection and identification of modified proteins
Butterfield et al. have pioneered gel-based redox proteomics to identify oxidatively modified proteins in NDs (88). The gel-free LC-MS/MS strategy is the most commonly used approach for identifying aldehyde-protein adducts (29). The identification of adducted or oxidized proteins is based on the analysis of peptide mass shifts that can be searched manually or using automated computational tools (756).
Detection and identification of modified proteins can be especially challenging in complex mixtures containing thousands of proteins. The analytical complexity is increased due to the great diversity of targets and the extremely low concentrations of the lipid electrophiles (pmol-nmol/mg protein) and their respective protein adducts (572). For this reason, enrichment strategies have been used before MS analysis (124, 678, 689, 732, 755). Such chemoproteomic methods include aldehyde analogues bearing terminal azide or alkyne functionalities, such as azido- or alkynyl-HNE, subsequently captured by click reactions and enriched over avidin-based matrices (302). Indeed, combinations of click chemistry, streptavidin-based enrichment, and MS analysis have been successfully applied to identify sets of target proteins for alkynyl-HNE (678) and alkynyl-ONE (622) in cell lines. Similar strategies based on alkynyl-labeled sterols (631, 709) have been used to characterize the oxysterol adductome. Using alkynyl-HNE and isotopically tagged (light 12C6- and 13C6-heavy labeled) photocleavable azido-biotin reagents, Yang et al. identified and quantified ∼98 alkylation sites (86 Cys and 12 His residues) in intact RKO cells (732). Similarly, Zhang identified and quantified 2257 HNE-modified peptides mapping 1121 proteins in six liver cell lines using an aminooxy labeling strategy (755).
In conclusion, quantitative analysis of protein modifications induced by lipid electrophiles and/or other oxidizing agents is still challenging. Some accurate and high-throughput identification/quantitation methods based on MS analysis (2) and the use of specific tags and isotopically labeled compounds is now available (124, 678, 689, 732, 755). However, there are still limitations regarding the synthesis and availability of these specific reagents, cellular or in vivo delivery routes, the sensitivity of the assay, and other factors. Indeed, further developments of highly specific, sensitive, and quantitative protein PTM analysis methods might help solve many outstanding questions regarding protein modification mechanisms.
(8) Aldehydic modification of serum proteins
Several serum proteins have been shown to form protein-aldehydic adducts (747). This section describes the structural characterization of blood protein modifications induced by aldehyde by-products derived from lipid peroxidation and discusses their potential use as biomarkers. Other oxidative modifications involving the direct reaction of proteins with hypohalous species (HOCl), singlet oxygen, and RNS (ONOO−, NO2 •) are not discussed here. However, they can be found in other excellent reviews (152, 171, 196, 264, 505, 616).
Since the proposal of the oxidation hypothesis of atherogenesis, numerous studies provided ample evidence supporting the involvement of oxidized LDL in atherosclerosis (234, 470, 612, 616). Biochemical mechanisms involved in LDL oxidation have been proposed. However, the in vivo mechanisms and exact composition of oxidized LDL particles responsible for atherosclerosis initiation and progression remain unclear (612). Oxidants responsible for LDL oxidation have been the subject of extensive studies and debate. Various in vitro and in vivo experiments have implicated MPO and MPO-derived oxidants (HOCl), lipoxygenase, NADPH oxidases, NOS, metal ions, and heme proteins in the mechanism of atherogenic oxidized LDL particle generation (97, 269, 401, 636). Common features observed under these oxidative insults include the induction of lipid peroxidation, antioxidant depletion, and the modification of apoB-100, the main protein present in LDL particles (375).
Early studies by Esterbauer and collaborators showed that HNE and MDA were the primary aldehydes formed during LDL oxidation (186, 317). Antibodies raised against HNE or MDA-modified LDL recognized oxidized forms of LDL and epitopes in atherosclerotic lesions (250, 317, 666). Moreover, MS analyses have provided important qualitative and quantitative details on sites of modifications induced by electrophilic compounds derived from fatty acid, phospholipid, and cholesterol oxidation on apoproteins (3). Lys and His were the primary residues modified by the aldehydes during copper-catalyzed LDL apoB-100 oxidation (64, 584, 666). Quantitative analysis of hydrolyzed aldehyde-amino acid adducts in an oxidized LDL sample by an adductomic method developed by Uchida's group demonstrated the presence of 6 mol/mol of HNE-His Michael adducts and 6 mol/mol of N ɛ-(8-carboxyoctanoyl)-Lys adducts (584). The latter is suggested to be formed by the reaction of 9-oxononanoylphosphatidylcholine (also called PONPC) with Lys residues of LDL apoB-100 (584).
Apart from modifications induced by short-chain aldehydes, studies point toward the relevance of lipoprotein(a) [Lp(a)] modifications induced by oxPL in cardiovascular diseases (61, 672). Owing to genetic, epidemiological, and clinical studies indicating an association between elevated Lp(a) and the risk of developing cardiovascular disease, attention has been recently focused on this lipoprotein particle (62). Lp(a) is a lipoprotein very similar to LDL in terms of lipid composition, which contains an apo(a) glycoprotein covalently linked to apoB-100 via a single disulfide bond. Immunohistochemical analysis using EO6, a monoclonal antibody that specifically recognizes oxPL-modified proteins, demonstrated an accumulation of oxidized Lp(a) in human atherosclerotic lesions (297) and plasma of patients with cardiovascular diseases (657). MS analysis of the Lp(a) fraction isolated from patients indicated the presence of different oxPL species (672). OxPLs are covalently linked to apo(a), specifically through Lys residues located at a Kringle domain called KIV10 (367). Remarkably, studies over the past 15 years conducted mostly by the Tsimikas' group (317, 365, 628, 655, 670) have provided consistent experimental evidence demonstrating the importance of oxPL as a proinflammatory risk factor associated with the epidemiology of cardiovascular disease and for Lp(a) as a major carrier of oxidized lipids in the plasma. Moreover, epidemiological and clinical data indicate that elevated plasma concentrations Lp(a) in arterial lesions are probably a causal risk factor for the development of cardiovascular diseases (61). The mechanism by which Lp(a) induces pathological events is not fully known.
Oxidized lipids and lipoproteins can exert protective or adverse effects depending on the type of reactive lipid species formed, location, tissue, cell type, and protein-adducts formed (426). Aldehydic-protein adducts, protein crowding, and hypoxic/anoxic environments may modulate immune responses (352, 431). Covalent adducts formed by lipid aldehydes (e.g., oxidized truncated phospholipid- and MDA-adducts with protein) on the surface of oxidized lipoproteins can act as epitopes, known as oxidation-specific epitopes (OSEs) (57). These epitopes constitute damage-associated molecular patterns that are recognized by pattern recognition receptors (e.g., scavenger receptors and toll-like receptors), enabling the immune system to mediate their clearance (57). The requirement of OSEs for oxidized LDL or apoptotic cell clearance was demonstrated by experiments showing that monoclonal antibodies that bound to the OSEs inhibited binding and degradation of oxidized LDL by up to 91% (284). Thus, OSEs present in oxidized LDL or apoptotic cells, either as free oxidized lipid or as lipid-protein adducts, act as essential ligands required for their uptake and phagocytosis. Importantly, under pathological conditions, the clearance capacity of available phagocytes is overwhelmed, and the accumulated OSEs in damaged cells or lipoproteins trigger a condition of chronic inflammation (57).
In addition to LDL modifications, studies have also reported oxidative modifications of high-density lipoproteins (HDL). Notably, it has been shown that HDL is a primary carrier of circulating plasma lipid hydroperoxides (74). Apolipoprotein A-I (apoA-I) is the major protein of HDL, comprising ∼75% of the protein content, modified by the lipid-derived aldehydes—ACR and MDA (580, 581, 651). Aldehyde-induced alterations in HDL components have been proposed to produce dysfunctional HDL particles that lack cardioprotective properties. ACR and MDA adduction to apoA1, one of the significant HDL apoproteins, has been shown to potently alter its capacity to remove cholesterol from macrophages by impairing two critical steps in the ABAC1 pathway (580, 581). MS analysis revealed that both aldehydes primarily modified Lys residues. More recently, apoA1 was reported to be extensively oxidatively modified within the human aorta (173). In particular, apoA1 served as a selective target for oxidative modifications by MPO-generated chlorinating and nitrating oxidants within the artery wall (49, 581, 758). Quantitative analysis in human atheroma showed that ∼20% of apoA1 within the lesion is oxidized specifically at Trp72, forming monohydroxylated Trp product (2-OH-Trp). (290). Remarkably, oxidized apoA1 was shown to be dysfunctional and highly enriched in atherosclerotic lesions (290). Moreover, Trp oxidation in apoA1 significantly inhibits the ABCA1-dependent cholesterol efflux acceptor activity (290, 746).
Furthermore, albumin, a highly abundant serum protein (5–55 mg/mL, ∼0.6 mM), reacts with many electrophilic metabolites (559). Albumin-HNE is increased in patients with type 2 diabetes and alcoholic cirrhotic patients (449, 650). Sites of Albumin-HNE adduction have been characterized ex vivo by reacting albumin with HNE and then submitting the modified protein to protease digestion and peptide sequencing by MS (11, 93, 626). Some discrepant results are reported with regard to the preferred sites of modifications. These differences are attributed to the fact that commercially available albumins often contain mixed disulfides at Cys34 and present considerable variability in terms of fatty acids and other ligands that can alter its conformation and reactivity with electrophiles (559). Serum albumin modifications, especially at Cys34 (pKa 6.55), are hypothesized to serve as a potential biomarker for monitoring human exposure to exogenous and endogenous electrophiles (461, 559) as well as a biomarker of oxidative stress (379). Improved immunoassays combined with MS-based analytical approaches have been used to monitor in vivo albumin-aldehyde adduction (93, 538). Analytical strategies include an untargeted LC-MS/MS adductomic pipeline for the global characterization of albumin Cys34 oxidation and conjugation to electrophiles (241). Interestingly, a study conducted with urate electrophiles showed an increase in albumin-urate adducts in the plasma and synovial fluid from individuals with gout and rheumatoid arthritis (660).
Together with albumin, hemoglobin-aldehyde adducts represent promising blood biomarkers. Serum proteins are herein emphasized because, based on our understanding, investigating the presence of oxidatively modified proteins in the serum might be a promising approach to establish biomarkers for pathophysiological conditions and the progress of pathologies based on epidemiological studies (see discussion below). Hemoglobin-aldehyde adducts have been studied using several electrophiles (96), including acetaldehyde (614), 4-oxo-2-nonenal (740), and 2-octenal (741).
Regarding the fate of protein-aldehydic adducts, they are easy substrates for proteasomal degradation (244). The proteasome is also a notable intracellular target of modification by aldehydes (e.g., HNE), resulting in its inhibition (203). It has been shown that mildly crosslinked HNE-modified proteins are preferentially degraded by the proteasome, especially by the 20S proteasome (20SPT). However, extensively modified proteins can contribute to the accumulation of modified proteins, such as in AD, in which HNE-modification of β-amyloid peptide generates a progressively more selective and efficient inhibition of the human 20SPT chymotrypsin-like activity (585). Proteasome activity inhibition by Aβ1–40 was increased from ∼2% (without HNE) to 25% with 5 μM HNE and ∼40% with 10 μM HNE (585). This inhibition was correlated with the increased crosslinking and formation of amyloid-beta oligomers induced by HNE (585). Interestingly, molecular analysis has shown that oligomeric forms of proteins involved in NDs (Aβ, α-synuclein, and mutant huntingtin) adopt a three-dimensional (3D) conformation that inhibits the 20SPT through allosteric impairment of the substrate gate in the 20S core particle, thereby blocking protein degradation (638). In addition to proteasomal degradation, accumulating evidence suggests that lysosome (410) and autophagy (277) may play essential roles in HNE-protein adduct degradation.
C. Amino acid covalent crosslinking
Protein crosslinks refer to the formation of covalent bonds between two amino acid side chains within a single protein subunit (intramolecular) or between two subunits of the same protein or different proteins (intermolecular). The covalent bond between two amino acid residues (or between two amino acids) can be catalyzed by enzymes or occur through spontaneous chemical reactions. The formation of crosslinks does not always occur through oxidative processes, but when it does, the oxidation may occur by two- or one-electron mechanisms (252). For instance, both of these oxidative mechanisms may produce the disulfide bond (-S-S-) from Cys residues, resulting in different biological responses. The disulfide crosslink is the most investigated protein/protein crosslink and has structural (115) and signaling functions (see Section III). In these cases, specific enzymes catalyze crosslink formation, such as protein disulfide isomerases (PDIs) (357, 708) and Prxs (474, 550). Other specific enzymes mediate the reduction of the disulfide bond back to the original Cys residues. Hence, disulfides are biologically reversible crosslinks. An abundance of oxidative protein modifications may occur under pathophysiological conditions, making disulfide crosslinks inaccessible to biological reductants, contributing to protein aggregation (210, 603). Conversely, the reduction of the disulfide bond in some proteins may also lead to misfolding and aggregation (733).
In addition to the disulfide crosslink, many other posttranslational protein modifications known in biological systems are protein/protein crosslinks. Most of them are irreversible because, up to now, there are no described enzymes capable of reversing the crosslink to the unmodified protein residue form. A number of these posttranslational crosslinks form through oxidation reactions or from oxidized biotargets and may be functional, but can also be dysfunctional.
Among the functional, we cite the crosslinks produced by the action of lysyl oxidase (LOX) and peroxidasin enzymes, which are essential for the formation and maintenance of the 3D structure of extracellular proteins (409). Peroxidasin uses H2O2 and halide ions to form hypohalous acids, preferentially hypobromous acid (HOBr), which promotes intermediate sulfimine (S = N) crosslinking between the sulfur of an Met residue with the nitrogen of an Lys/hydroxyLys residue (54). LOX and similar enzymes catalyze the oxidative deamidation of Lys or hydroxyLys to produce reactive aldehydes (30, 545), which form crosslinks by spontaneous reaction with other LOX-derived aldehydes (aldol condensation) or with other Lys/hydroxyLys residues (Schiff base formation) (Section II.B). Another example is the dityrosine (Tyr-Tyr) crosslink produced by peroxidase-catalyzed oxidation of Tyr residues. These crosslinks have crucial roles in providing stability and elasticity to many structural proteins of invertebrates. For example, these crosslinks are highly abundant in the fertilization envelope of sea urchin eggs (273), the adhesive glues of mollusks (687), the cuticles of insects (570), and the oocyst walls of parasites (396, 607).
Protein/protein crosslinks generated from reactive carbonyl metabolites or reactions with reducing sugars and their metabolites are likely dysfunctional. In these cases, if the carbonyl reagent only possesses this reactive center toward a protein amino acid residue, a reagent-protein adduct is formed. However, if the reagent possesses another reactive center, it may react with another protein residue, producing intramolecular or intermolecular protein/protein crosslinks (Sections II.A and II.B). Similarly, reactive carbonyl metabolites derived from the oxidation of Tyr (quinones) (30, 187) and Trp (kynurenine and N′-formylkynurenine) (182, 725) residues generate crosslinks that are likely dysfunctional in mammals (645, 662). However, Tyr-derived quinone products are abundant in structural proteins of invertebrates (30, 85).
There are other enzymatic and nonenzymatic protein/protein crosslinks produced by nonoxidative reactions. Transglutaminase enzymes catalyze the formation of the Gln-(C = O)NH-Lys crosslink (isopeptide bond), which is crucial in the blood coagulation cascade (517), but apparently also relevant for protein aggregation in NDs (690, 706) and cataract (385). Other crosslinks produced by nonoxidative mechanisms, such as Lys-Asp (694) and thioether crosslinks (695), are also present in lenses with cataracts.
The discovery that inter- or intramolecular crosslinked proteins are poor substrates for the proteasome, leading to the inhibition of proteasomal activity and contributing to protein aggregation (245), increased the interest in these posttranslational protein modifications. Protein aggregation is a hallmark of age-related diseases, such as NDs, atherosclerosis, and cataract, the occurrence of which is augmented with the increasingly aged human population (543). Despite the increasing interest in protein/protein crosslinks, their detection and analysis remain challenging tasks, as recently reviewed (252) and briefly summarized below.
1. Detection and analysis of oxidative protein/protein crosslinks
For many years, the predominant detection methods, such as sodium dodecyl sulfate–polyacrylamide gel electrophoresis and light scattering, only revealed gross protein modifications such as dimerization and/or oligomerization. Later, more specific methods involving antibodies or total protein hydrolysis followed by gas chromatography coupled to MS or LC-MS were developed. Both of these methodologies and antibody development require knowledge of the amino acid residues involved in the crosslink and the nature of the covalent bond. In this context, the long-known Tyr-Tyr crosslink that possesses strong fluorescence (232) became, after the disulfide, the most detected oxidative crosslink in biological samples. However, the proteins in these samples that contain Tyr-Tyr crosslinks and the specific residues that participate in the linkage remain mostly uncharacterized (17, 452).
The scenario started to change with the development of strategies to analyze enzymatic protein hydrolysates by LC-MS/MS strategies. These methodologies not only confirmed the formation of Tyr-Tyr crosslinks in proteins submitted to different oxidants or light in the presence of photosensitizers but also enabled the identification of the Tyr residues involved in the linkage. They also enabled the characterization of novel specific crosslinks in oxidized proteins, such as Trp-Trp (419, 503, 582), Trp-Tyr (209, 370), as well as Tyr-Lys and His-Lys (409). Although Trp-Tyr crosslinks were characterized before in the active sites of enzymes with peroxidase activity, these characterizations were dependent on X-ray crystallography (52, 724). Concerning the mechanisms for the formation of these novel crosslinks, the production of Tyr-Lys and His-Lys crosslinks by oxidation reactions remains under investigation (409). Conversely, there is a consensus in the literature that the formation of Trp-Trp and Trp-Tyr crosslinks occurs through radical-mediated mechanisms, as is the case with Tyr-Tyr crosslinks. The one-electron oxidation of Trp and/or Tyr residues leads to the corresponding protein-derived radicals (protein-Trp•/protein-Tyr•), which rapidly (k ∼ 5 × 108 M −1·s−1) recombine with itself or with the other to produce the crosslink (Trp-Trp, Tyr-Tyr or Trp-Tyr) (252, 502, 503). The yields of these products are likely to be low under most physiological conditions since high yields of protein-Trp• and -Tyr• radicals are required to favor their recombination reactions. In addition, these radicals react relatively slowly with O2 (k ∼ 105 M −1·s−1) (189, 292) and rapidly with O2 •− (k ∼ 109 M −1·s−1) (99, 145, 435), which are biologically ubiquitous. Still, protein crowding and hypoxic/anoxic environments may favor the formation of protein crosslinks by radical recombination (502).
Up to this point, most of the performed LC-MS/MS studies to investigate posttranslational crosslink modifications were limited to purified proteins oxidized in vitro. However, recent improvements, such as performing enzymatic hydrolysis of oxidized proteins in water labeled with 18O, optimization of MS/MS fragmentation methods, and the use of software packages to search for crosslinks opened up new avenues to study crosslinks present in oxidized proteins from biological samples (409). As a proof of concept, these authors used the developed strategies to analyze a protein extract from the gram-positive lactic acid bacterium Lactococcus lactis exposed to peroxyl radicals generated from the decomposition of 2,2-azobis (2-amidinopropane) dihydrochloride (AAPH). They were able to characterize 24 Tyr-Tyr, 4 Tyr-Trp, and 3 Trp-Trp specific crosslinks in specific proteins of the extracts (409). More recently, we reported the presence of Trp-Trp and Trp-Tyr crosslinks in crystallin proteins of human lenses with advanced nuclear cataract (502) (Fig. 6A). Therefore, we can anticipate that as instrumentation, software, and the understanding of oxidant chemistry advance, it will be possible to characterize crosslinks in biological samples more frequently and to discover novel protein crosslinks (252).
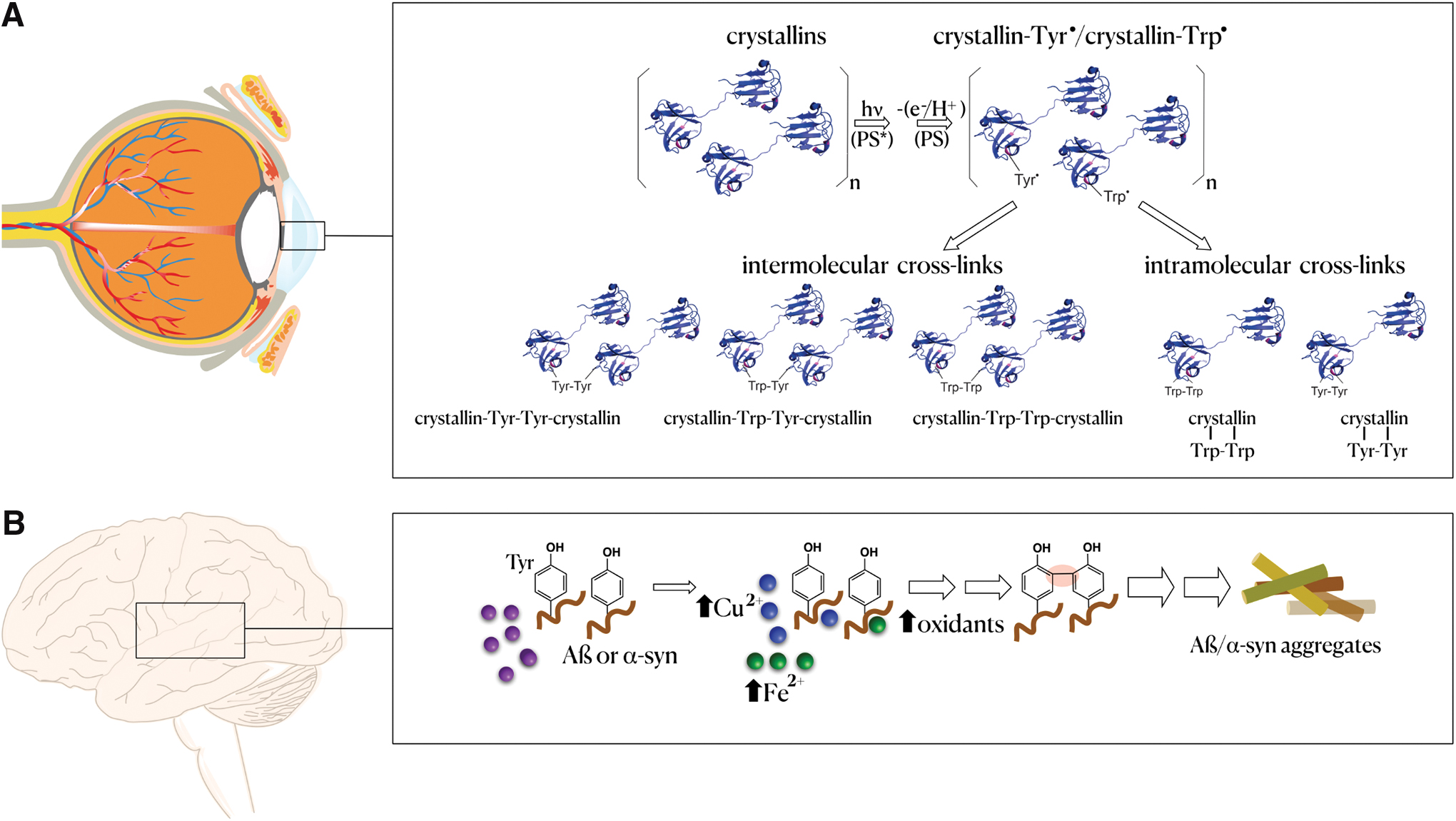
2. Roles in pathophysiology
As stated above, the characterization of many oxidative crosslinks is recent, and there is a lack of data on their occurrence in biological samples. The exception is the Tyr-Tyr crosslink identified from the oxidized amino acid and protein-Tyr residues after total protein hydrolysis (enzymatic or chemical) (232). Therefore, the detection and quantification of Tyr-Tyr in biological samples are a useful biomarker of oxidative imbalance. Furthermore, increased Tyr-Tyr levels suggest risk for some diseases when accumulated in specific tissues (176, 232).
Indeed, several pathologies present increased Tyr-Tyr levels. For example, elevated levels have been reported in the plasma of patients with hyperlipidemia, chronic renal failure, and uremia undergoing hemodialysis (323, 378, 721), myocardium tissue after acute infarction, lenses of patients with cataracts, and urine of diabetic patients. The levels of Tyr-Tyr increase during aging (194, 207, 702) and acute inflammation (53). During the inflammatory response, the activity of the heme-containing enzyme MPO localized in phagosome membranes of immune cells, mainly neutrophils, produces large quantities of HOCl from hydrogen peroxide and chloride ions (20). HOCl promotes the formation of Tyr-Tyr (129, 273, 428), which can also be formed by peroxynitrite (391, 568) and by peroxidases/H2O2, including MPO in the absence of chloride (272). In some clinical conditions, MPO plasma levels rise above those of the healthy population (33, 78, 285). Higher Tyr-Tyr levels in patients undergoing hemodialysis correlated with increased MPO activity in response to blood contact with dialysis membranes (337).
Several studies also indicate an association between NDs and Tyr-Tyr crosslinks. The presence of these crosslinks in the β-amyloid and the α-synuclein from postmortem brain samples from AD and Parkinson's disease (PD) patients, respectively, suggests that Tyr-Tyr crosslinks may have an important role in the assembly of the β-amyloid fibrils in the neuropil and of α-synuclein assemblies in the Lewy bodies. The amyloid fibrils are key characteristics of the aggregates in these NDs (12, 13). The high concentrations of copper and iron ions found in amyloid plaques and substantia nigra are likely catalysts for in situ formation of protein-Tyr•, which are Tyr-Tyr crosslink precursors (86, 110, 170, 551, 567) (Fig. 6B). Copper ions greatly enhance Tyr-Tyr levels present in the β-amyloid (12, 13). Similarly, Tyr-Tyr crosslinks form in vitro via metal ion-catalyzed oxidation. In addition, Tyr-Tyr crosslinks are biomarkers of oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD (12, 508, 509). Of relevance, the levels of free metal ions and oxidants increase in the central nervous system with aging, facilitating protein oxidation, crosslink formation, and protein aggregation. Interestingly, possible or novel therapeutic approaches for AD (23, 325) and PD (169) rely on metal ion-targeting drugs. Indeed, of the 800 compounds used, tested, or proposed for PD treatment between 2014 and 2019, 250 present metal ion-chelating properties (646). Figure 6B summarizes the above information, displaying a hypothetical pathway for the role of metal ions in Tyr-Tyr crosslink formation and protein aggregation in the brains of AD or PD patients.
In conclusion, the characterization of many protein crosslinks formed by oxidative mechanisms occurred only recently, and many others remain uncharacterized (252). Advances in the strategies to detect and quantify protein crosslinks in biological samples are required, particularly in protein aggregates, which are possibly relevant to age-related diseases. With these and other advances, detailed investigations into the role of protein crosslinks in the pathogenic mechanism of diseases will be feasible.
D. Protein nitro- and nitroso-derivatives
The PTM nitration (Fig. 7A) and nitrosation (Fig. 7B, also called S-nitrosation) are often confused even though the associated chemical modifications are different (addition of a nitro, -NO2 group vs. addition of a nitroso, -NO group), and occur on different amino acid residues (nitration of tyrosines vs. nitrosation of cysteines). However, both protein modifications are mediated by NO•-derived oxidants, with biological tyrosine nitration occurring via an NO2 •-dependent radical process and cysteine nitrosation occurring via an NO•-dependent process.
In vivo, neither protein tyrosine nitration nor protein cysteine nitrosation occurs through enzyme-catalyzed processes. While denitrosation (removal of the –NO group from a modified cysteine) is enzyme-accelerated, no enzymatic denitrase activity (removal of the –NO2 group from a modified tyrosine) has been unambiguously identified or characterized yet. Biological tyrosine nitration is often associated with protein dysfunction, considered a marker of unbalanced cellular redox status, and correlated with a pathological condition. On the contrary, cysteine nitrosation is a reversible process, usually involved in cellular regulatory mechanisms and associated with oxidative physiopathological conditions (discussed below, section Examples of nitrated or nitrosated proteins). In a proteomic analysis, not many but specific proteins were found nitrated. On the contrary, numerous proteins were found to be S-nitrosated under physiological and pathological conditions. In this sense, the precise role of each S-nitroso protein, as well as each nitrotyrosine protein, requires further investigation.
1. Tyrosine nitration
The PTM of tyrosine by nitration consists of the addition of a nitro (−NO2) group to the aromatic ring of a Tyr residue to form 3-nitrotyrosine (3-NT).
a. Biological mechanism
The biological formation of 3-NT is a radical mechanism that first involves the formation of a tyrosyl radical, which is followed by the addition of nitrogen dioxide (NO2 •). NO2 • radicals derived from peroxynitrite (OH• and NO2 •) as well as the radicals derived from peroxynitrite reaction with CO2 (carbonate radical CO3 •− and NO2 •) can perform this reaction (532). Initially, 3-NT was considered a footprint of peroxynitrite formation in vivo, but peroxynitrite is not the only source of biological nitration since NO2 • can also do the job. For example, nitrite as a second substrate for heme peroxidases (MPO, H2O2, and NO2 −) or autoxidation of NO• (533).
The yield of biological Tyr nitration is low, and it is a selective process since only a few Tyr residues are capable of being nitrated (1, 196). Even though a specific consensus sequence for Tyr nitration has not been identified, Tyr residues in the proximity of negatively charged residues (Glu, Asp) and loop regions containing turn-inducing residues (Pro, Gly) are more prone to nitration (296, 600). Also, Tyr residues in transmembrane domains (750) or metalloproteins (602) seem to be preferentially nitrated, which could be associated with the biological nitrating agent responsible for this PTM, that is, the acceleration of NO• autoxidation in membranes and hydrophobic protein domains to produce •NO2 (434, 436), or the metal-induced catalysis of peroxynitrite-mediated nitration (602). An in-silico method to identify potential Tyr nitration sites was developed using a training data set of nitrated proteins selected from the database dbPTM SysPTM2.0, using previously identified nitrotyrosine and non-nitrotyrosine sites (260).
b. Biological consequences
When a Tyr is modified to 3-NT, the 274 nm absorbance maximum shifts to 360 nm at acidic pH (ɛ360 = 2790 M −1 cm−1) or 440 nm at alkaline pH (ɛ440 = 4400 M −1 cm−1). The incorporation of the nitro group at position 3 lowers the pKa of the adjacent phenol group (pKa tyrosine ∼10.3 and pKa 3-NT ∼7.5 depending on the protein environment); thus, Tyr nitration provokes a change in the global pI of the modified protein.
It is plausible that protein Tyr nitration can induce changes in the protein structure that could affect protein function. In the case of proteins with a critical Tyr, nitration of that residue leads to protein dysfunction. For example, the nitration of Tyr385 in prostaglandin synthase PGH results in enzyme inactivation (237). The nitration of manganese superoxide dismutase (MnSOD) Tyr34, an amino acid located in the channel where the superoxide substrate binds, also leads to inactivation (726). However, there are reports of activation or increased activity following nitration. In the case of α-synuclein monomers, nitration promotes aggregation, and nitrated synuclein was found in Lewy bodies of PD patients (278, 762).
Similarly, the nitration of fibrinogen seeds the fibrin aggregation and activates clot formation (671). Moreover, nitration of Prx2 diminishes its inactivation by hyperoxidation, resulting in an effective increase of peroxidase activity (537). Interestingly, there is also evidence showing a gain of function after nitration, as with cytochrome c, which gains a new peroxidase activity that leads to a nonfunctional apoptosome (101, 218). This gain of function is biologically relevant considering the low yields of this PTM in vivo; thus, the appearance of a new activity, although only in a few molecules, could transform a nearby substrate and have a cellular impact or affect ligand binding in signaling transduction (196).
The phosphorylation of Tyr can also be affected by nitration, which can have a significant impact on signaling pathways (1, 442). The presence of a bulky nitro group adjacent to the phenol can block its phosphorylation; thus, nitration of a protein participating in a signaling pathway could disrupt the phosphorylation/dephosphorylation cascade. On the contrary, the more anionic nitro-Tyr could mimic phospho-Tyr and promote the downstream signaling process. In addition, nitration of kinases or phosphatases can alter the efficiency of phosphorylation/dephosphorylation cascades (343).
Another biological consequence of protein Tyr nitration is the generation of antibodies against nitrated proteins; a well-documented example is the increase in the titer of antinitrotyrosine antibodies in the synovial fluid or serum of patients with autoimmune diseases (331) as well as in the circulation of patients with coronary artery disease (CAD) (640).
A basal level of protein Tyr nitration is detected under physiological conditions. However, these levels increase under conditions of inflammation and oxidative stress when the production of NO-derived oxidants is stimulated. Despite not being an enzyme-catalyzed process, biological nitration has specificity, and selected proteins are nitrated on specific Tyr residues (1, 533). The nitration of specific proteins in pathological conditions such as cardiovascular or NDs has been reported (1, 533). In general, the nitration of Tyr leads to protein dysfunction that could be involved in the progression of the disease (usually not the only nor even the main factor), but the molecular mechanisms still need to be clarified.
c. Denitration
There is ample evidence that once the protein is modified by nitration, it is promptly degraded by the proteasome (1, 243). Although some evidence of in vivo denitration processes has been provided (594), an enzyme with denitrase activity has not been isolated yet.
d. Detection of nitrotyrosine (free and protein-bound nitrotyrosine)
Specific antibodies against 3-NT have been developed to detect this PTM in tissues and quantitate its presence in biological fluids via enzyme-linked immunosorbent assay (640). Previously, different HPLC detection methods (from absorbance to electrochemical) were used for detecting this PTM, but now, the most reliable method is LC-MS/MS (40, 437, 738).
e. Proteomics
As mentioned before, the amount of nitrated proteins found in vivo is low and specific proteins are the preferential targets of nitration. The “Tyr-nitroproteome” revealed less than a hundred nitrated proteins, even under pathological conditions associated with oxidative stress (1, 196, 243).
(1) Examples of nitrated proteins
(a) Manganese superoxide dismutase
A remarkable example of a loss of enzyme activity exclusively due to nitration of a specific Tyr residue is the mitochondrial MnSOD. The exposure of MnSOD to peroxynitrite resulted in nitration of its Tyr34, in an Mn-catalyzed process, and enzyme inactivation, with no other residue being oxidatively modified (394, 541, 726). This condition was associated with organ transplant rejection (394, 395).
(b) Apolipoprotein A-I
apoA-I, the major protein component of HDL, is a selective target for MPO-catalyzed nitration (758). Of the seven Tyr residues in apoA-I, Tyr192, and Tyr166, the MPO-preferred sites were found to be nitrated in human atherosclerotic tissues (172, 759). The extent of apoA-I nitration correlates with the functional impairment of reverse cholesterol transport (758, 759). An important consequence of protein nitration in cardiovascular disease is the induction of humoral responses documented by the increased circulating immunoglobulins that recognize 3-NT in CAD patients and LA–apoA-I−/− mice as a model for atherosclerosis (495, 639, 640).
(c) Fibrinogen
Increased levels of nitrated fibrinogen were found in the plasma from patients with clinically documented CAD (494, 671). MS identified Tyr292 and Tyr422, at the C-term of the β-chain of fibrinogen, as the main sites of nitration in vivo (494).
(d) α-Synuclein
Synucleinopathies, including PD, AD, and dementia with Lewy bodies, are characterized by the presence of amyloid inclusions in neurons, which are rich in the aggregated α-synuclein protein. Nitrated synuclein was found in Lewy bodies, and nitration of α-synuclein monomers promoted aggregation, oligomerization, and fibrillation (109, 278, 601).
2. Cysteine nitrosation
The PTM of cysteine by nitrosation (also called S-nitrosation) consists of the covalent addition of a nitrosonium (−NO+) to the sulfur atom of a deprotonated Cys residue (P-CysS−), forming an S-nitrosothiol (P-S-NO).
a. Biological mechanisms (nitrosation vs. nitrosylation)
There is ample evidence of protein S-nitrosation in vivo, and it is undoubtedly linked to the formation of NO• under physiological or pathological conditions. Nevertheless, the biological mechanism of S-nitrosothiol formation is still under debate. The term “nitrosation” or “nitrosylation” depends on the mechanism of formation of the nitrosothiol, and in most cases, this is not always certain. It is clear that there is no direct reaction of NO• with the thiol or thiolate, but an oxidation step is necessary. Thus, to react with radical NO• either the thiol is oxidized to a thiyl radical, or the thiolate reacts with higher nitrogen oxides, NO2 • or N2O3, products of NO• autoxidation (Equations 1–3) (79):
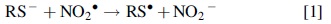
The autoxidation of NO•, even though it is accelerated in hydrophobic environments such as membranes or even proteins (436, 437), is too slow to explain the biological formation of nitrosothiols. The intermediacy of nitrosyl/iron complexes, dinitrosyl/iron complexes, and cytochrome c (bound to GSH) has been proposed (67, 79, 700), but further studies are needed to elucidate the molecular mechanisms of S-nitrosothiol biological formation. It is important to note that S-nitrosothiols can react with other thiols to form a new S-nitrosothiol, a mechanism known as transnitrosation (279). Considering the high intracellular concentration of GSH, once S-nitrosoglutathione (GSNO) is formed, it can nitrosate other thiols. For example, the transnitrosation from GSNO to cysteine to yield S-nitrosocysteine occurs with k = 140 M −1·s−1 (279).
Modeling S-nitrosation on proteins has not predicted any consensus site to underline a preferred Cys residue to be modified; however, the proximity of the reactants is necessary for transnitrosation reactions, that is, subcellular proximity to NOS, and/or availability of S-nitrosothiols such as GSNO (384). Also, solvent-exposed Cys residues favor the reaction of the thiolate (408). Recently, an in-silico method was published to predict potential S-nitrosation sites called PreSNO (261).
b. Denitrosation
Cys nitrosation is a reversible PTM, and so far, two enzymes that accelerate denitrosation in vivo have been identified: Trx and GSNO reductase. Interestingly, Trx can catalyze both transnitrosation and denitrosation (577). An alcohol dehydrogenase class III was found to efficiently catalyze the reduction of GSNO and has, therefore, been called GSNO reductase (308). Considering the high intracellular concentrations of GSH that can transnitrosate protein S-nitrosothiols and form GSNO, it is considered an efficient biological denitrosation system. The GSNO reductase-knockout mice presented markedly increased levels of S-nitroso proteins and increased mortality in endotoxic shock that was attenuated by inducible nitric oxide synthase (iNOS) inhibitors. In contrast, these mice were protected from experimental myocardial infarction due, at least in part, to S-nitrosation-mediated stabilization of hypoxia-inducible factor HIF-1α (380).
More recently, a denitrosylase activity was ascribed to sulfiredoxin (Srx), acting on nitrosated-Prx2 in dopaminergic neurons (Prx) (623). It is worth mentioning that nitrosated Prx2 was found in the Lewy bodies of PD patients (188). Srx was initially described as an ATP-dependent specific reductant of sulfinylated peroxidatic cysteine of 2-Cys Prx such as Prx2 (314). Later, other Cys-sulfinylated substrates were identified (731), and the denitrosylase activity of the S-nitrosylated peroxidatic cysteine of Prx, also at the expense of ATP (188).
The catalytic decomposition of nitrosated proteins makes a clear difference between the two PTMs, Tyr-nitration versus Cys-nitrosation. Once formed, the PTM participating in a signaling cascade needs to be rapidly eliminated to stop the signal. Thus, an S-nitrosated protein is more likely to be involved in signaling and regulatory events, and Y-nitrated proteins probably accumulate under oxidative stress pathological conditions.
c. Biological consequences
There is a change in the UV spectrum of a nitrosated protein with a maximum absorbance of around 335 nm (GSNO ɛ = 922 M −1 cm−1) (79). The global pI could significantly change since the potential negative charge of the thiolate is no longer there. A change in pI could affect protein/protein interactions (it will largely depend on the pKa of the Cys residue and its location within the protein structure), subcellular protein localization, and ubiquitylation-dependent protein degradation (276).
A protein with a critical Cys residue, once it is nitrosated, will indeed display a diminished activity; however, it is a reversible modification, and the activity can be recovered after denitrosation. Like glutathionylation, reversible cysteine nitrosation could be viewed as a protection mechanism to avoid irreversible hyperoxidation of the Cys residue (140).
Cysteine residues can form bonds to coordinate the metal center of proteins, as with Zn tetrathiolates, interactions that usually play a structural role in the protein. Thus, nitrosation of two or more of those Cys residues will probably result in metal release and compromise protein stability (73).
S-nitrosation is involved in a wide range of physiological pathways, including cellular metabolism, apoptosis, membrane trafficking, protein phosphorylation, transcription factor activation, and redox homeostasis (276). Dysregulation of S-nitrosation is associated with a growing list of pathophysiological conditions, including pulmonary hypertension, asthma, myocardial ischemia, PD, and cancer, among others (201).
Recently, the role of S-nitrosation as a ubiquitous, stable PTM that directly regulates many proteins has been challenged (249, 713). These results indicate that S-nitrosation predominantly serves as a transient intermediate in the formation of a disulfide (intra- or intermolecular, or with GSH).
d. Detection of nitrosothiols
Methods to detect and quantify nitrosothiols have been recently reviewed in Möller et al. (437). Chemiluminescence, with nM sensitivity, is considered the gold standard method for quantifying S-nitrosothiols, and it is appropriate for most biological applications (174). In addition, the Saville method has a sensitivity in the μM range and is based on Hg2+-mediated decomposition of the S-nitrosothiol to nitrite that is then measured spectrophotometrically by Griess (174). Antibodies against S-nitrosocysteine have also been raised (174). The introduction of the “biotin switch” in 2001 (303) allowed the modified Cys within a protein to be mapped, consequently displacing these previous techniques and contributing to the development of the S-nitrosoproteome. Nevertheless, the original method was repeatedly criticized, and several modifications were introduced (692). Other approaches to identify S-nitrosated proteins and the site of the modification include the use of different affinity tags: organomercurials (534), derivatized phosphines (752), and isotopic labeling mixed with biotin switch and LC-MS/MS (763).
e. Proteomics
According to the dbSNO (
(1) Examples of nitrosated proteins
(a) Thioredoxin
S-nitrosation of Cys73 of Trx has been associated with apoptosis regulation, particularly via transnitrosation of caspase 3 (429, 621). Trx has been shown to regulate the amount of S-nitrosothiols in cells (251). On the one hand, Trx behaves as a transnitrosylating agent, thus generating proteins modified by S-nitrosation. On the other hand, Trx has been described as a denitrosylating agent, thus accelerating the disappearance of the S-nitroso modification on proteins. The Trx denitrosylase and the transnitrosylase activity has been shown to depend on the active site cysteine residues Cys32 and Cys35 for the reduction of the targeted protein thiol (47, 327).
(b) Guanylyl cyclase 1
The production of NO• in the endothelium (by endothelial nitric oxide synthase [eNOS]) stimulates the catalytic activity of the NO-sensitive guanylyl cyclase guanylyl cyclase (GC1) favoring vasorelaxation and inhibition of platelet aggregation. This activation is via the reaction of NO• with the Fe-heme of the enzyme (295). However, NO• can also perform signaling through the cGMP-independent pathway by the S-nitrosation of Cys residues. In particular, the S-nitrosation of GC1 correlates with the desensitization of this enzyme to NO•, decreasing NO-dependent cGMP production. In addition, Trx could be responsible for denitrosating GC1 to regain its sensitivity to NO• (287).
(c) Glyceraldehyde 3-phosphate dehydrogenase
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), in addition to its well-known glycolytic function, GAPDH participates in nuclear events. S-nitrosation of GAPDH Cys150 inhibits its glycolytic function and promotes its translocation to the nucleus (457). S-nitrosation of GAPDH triggers binding to Siah1 (an E3 ubiquitin ligase), nuclear translocation, and apoptosis (256). In this sense, S-nitrosation of GAPDH was proposed as a molecular mechanism of cytotoxicity, and it is considered responsible for the PTM of nuclear proteins via transnitrosylation.
(d) Peroxiredoxin 2
Prx2, a ubiquitous Cys-dependent peroxidase present at relatively high concentration in cells (10–250 μM in erythrocytes), is inactivated by S-nitrosation of both the catalytic and resolving cysteine residues (Cys51 and Cys172, respectively), sensitizing dopaminergic neurons to H2O2-dependent cell death (188). Increased nitrosative stress, and Prx2 S-nitrosation, might contribute to the loss of dopaminergic neurons in PD (188).
In a recent report, Visiedo et al. (679) detected higher levels of S-nitrosated Prx1 and other antioxidant enzymes in the placentas of women with gestational diabetes mellitus, a condition in which inflammation of the placenta is increased during pregnancy, as well as elevated iNOS expression.
E. Other protein oxidative modifications
Heme peroxidases, such as MPO, eosinophil peroxidase, and lactoperoxidase, generate hypohalous acids with variable reactivity and oxidizing capacity (265). All of them can modify proteins. The mammalian immune system, upon activation, triggers a response against invasive species by MPO, generating the most reactive and powerful hypohalous species, HOCl, from hydrogen peroxide and physiological chloride (339). Despite HOCl playing a prominent role in immune defense, it is also responsible for the oxidative modification of proteins in host cells, a process that is significant in inflammatory diseases and also reported to occur during aging (100, 136). Major protein targets for HOCl are His, Arg, Lys α-amino group, the sulfur-amino acids Cys and Met, and the aromatic amino acids Trp and Tyr (265). The modification of these amino acids by HOCl has been extensively studied and reviewed (266). The detection of proteins modified by HOCl was recently described in detail through MS analysis (481). These modifications are relevant during the innate immune response in inflammatory and cardiovascular diseases. The interested readers should consult the above-cited references.
Another important protein oxidative modification is the formation of protein hydroperoxides (POOHs) (151). POOHs are formed from the reaction of carbon-centered protein radicals at a high O2 concentration generating peroxyl radicals (POO•). The peroxyl radicals generate POOH by hydrogen-atom abstraction through reaction with other H-bond species (about 70% of the initial protein-peroxyl radical). Mechanisms of formation, methods for detection, and stability and reactivity of POOHs, including studies with free amino acids and peptides, are well documented and competently reviewed (151). Still, POOHs' relevance in human pathophysiological conditions remains based on a few direct pieces of evidence. POOHs are short-lived species, and their direct detection in intact tissues remains challenging despite substantial evidence showing that alcohols and primary carbonyls are major products of POOHs. Notably, increased levels of protein alcohols were detected in human samples of atherosclerotic lesions (206) and cataractous and aged lenses (207). POOHs are also intermediates of protein oxidation by HO• generating carbonyl derivatives (Fig. 1).
Many oxidative products of specific amino acid side chains are not addressed in the present review. A recent and competent review refers to all of them (265). Noteworthy, the oxidation of free and aromatic protein amino acids (Phe, Tyr, and Trp) is particularly relevant because they play essential roles in the central nervous system as precursors in the synthesis of neurotransmitters, in the immune system, and in therapeutic proteins such as monoclonal antibodies (571). However, it is beyond the scope of the present review to explore such specificities.
Also poorly discussed in the present review are the products of the oxidation of amino acid side chains by singlet oxygen (1O2). Since the usual 1O2 sources (e.g., UV light) differ from those of intracellular oxidants, human skin and eyes are special targets of 1O2-mediated processes. The amino acids Cys, His, Met, Tyr, and Trp are the more sensitive to oxidation by 1O2. Detection of the formed products inside cells and organisms remains a challenge, particularly due to the instability and complex nature of products formed by reactions involving both 1O2 (Type II-mechanism) and radical intermediates generated by Type I-mechanism in photosensitized reactions (499). The complexity is further increased by the secondary reactions that are propagated after light exposure (151). A comprehensive review of the reactions of 1O2 with proteins and other cellular components was published recently (171). More details on the photoinduced protein oxidation products can be found in other reviews (209, 499). A comprehensive review of the reactions of 1O2 with proteins and other cellular components was published recently (171).
III. Role of Protein Sulfur-Amino Acids in Redox Processes and Their Oxidative Modifications
The emergence of life on the earth is linked to the high presence of H2S and NO• in the atmosphere and iron-rich (Fe2+) oceans that existed around 3.8 billion years ago (133). Sulfide is alledged to have primacy in the origin of life. In combination with transition metals, one- and two-electron reactions would have formed various catalytic species to synthesize organic compounds, including the sulfur-containing amino acids (Met and Cys).
Due to the presence of thiol (R-SH) groups in its side chain, Cys is the most redox-active residue in most proteins. Exceptions are proteins that contain selenium Cys (25 proteins in human proteome), as this residue is more reactive than sulfur Cys (262, 450). The sulfur atom is large and polarizable, and as a consequence, a thiol group is electron-rich and nucleophilic. The free amino acid Cys presents low reactivity for redox reactions, but in specific protein environments, Cys residues can gain redox activity (408, 711). The thiol reactivity can be enhanced by stabilizing its deprotonated form, called the thiolate anion (RS−). In some proteins, the pKa value of the Cys residues can be as low as 3.5 (177), whereas the pKa for the free Cys residue is around 8.0. The structural stabilization of the transition state in the active site of enzymes is a significant factor in enhancing enzymatic activity (254). More about the chemistry and reactivity of thiol groups is beyond the scope of this review, and there are other reviews on this matter (522, 539).
Methionine residues have an essential role in protein protection against oxidative modifications as well as in aggregation. As shown in the following sections, the oxidative modification of sulfur-containing amino acids has either a critical catalytic or regulatory protein function, different from the oxidative modifications discussed above.
A. Thiol-centered redox mechanisms in catalysis and signaling
1. Thiol/disulfide switches
Initially, we describe some thiol/disulfide switches (Fig. 8) in proteins as mechanisms underlying catalysis and signaling. The two-electron oxidation of thiols in proteins to disulfides is a common theme in biology initially associated with structural roles as permanent crosslinks that stabilize the protein structure (115). These structural disulfides adopt the most favorable conformations when the dihedral angle is 90°C, and the distance between the two sulfur atoms is 2.05 Å (6, 475, 550a).
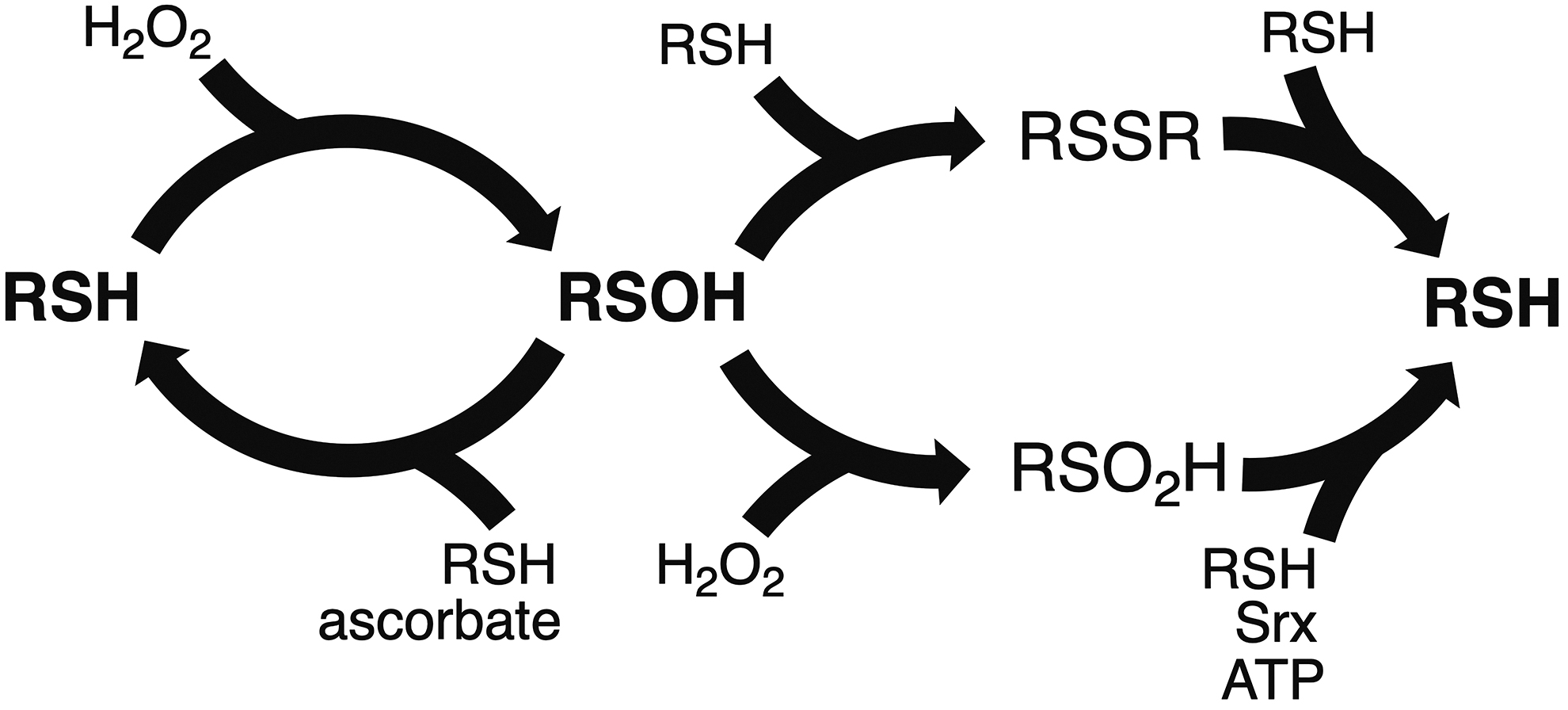
Nonetheless, disulfides are also recognized to play functional roles through redox processes (115, 267). In this situation, disulfide bonds are continuously formed and disrupted. Therefore, a highly stable disulfide is not suitable, and this covalent bond then assumes less stable structural conformations that are more easily reduced back to dithiols. Indeed, these disulfides are strained in stereochemically disfavored conformations that present intrinsic torsional energy (267). These “functional” disulfides are sometimes referred to as “forbidden” (267).
A specific type of functional disulfide is the allosteric one. To understand this concept, we need to consider that an allosteric effector is a compound that changes the activity of a protein by binding in a region that is distant from the active site (440). Allostery is possible due to dynamics in the protein structure. Therefore, an allosteric disulfide represents a covalent bond that, once formed, triggers a conformational change that affects protein activity (115).
These dithiol/disulfide pairs may also function as redox switches during the catalysis of enzymes, such as ribonucleotide reductase, a protein involved in the synthesis of deoxyribonucleotides, the DNA building blocks. The dithiol of ribonucleotide reductase can reduce ribonucleotides into deoxyribonucleotides by a complex sequence of electron transfer reactions that result in the formation of a disulfide (185).
2-Cys Prxs are other Cys-based enzymes in this dithiol/disulfide category that play central roles in hydrogen peroxide metabolism. In this case, a highly reactive thiol present in the so-called peroxidatic Cys (CP) reacts with H2O2 giving rise to sulfenic acid (R-Cys –SOH) intermediates that then undergo condensation with a resolving Cys (CR) to form a disulfide (Fig. 9).
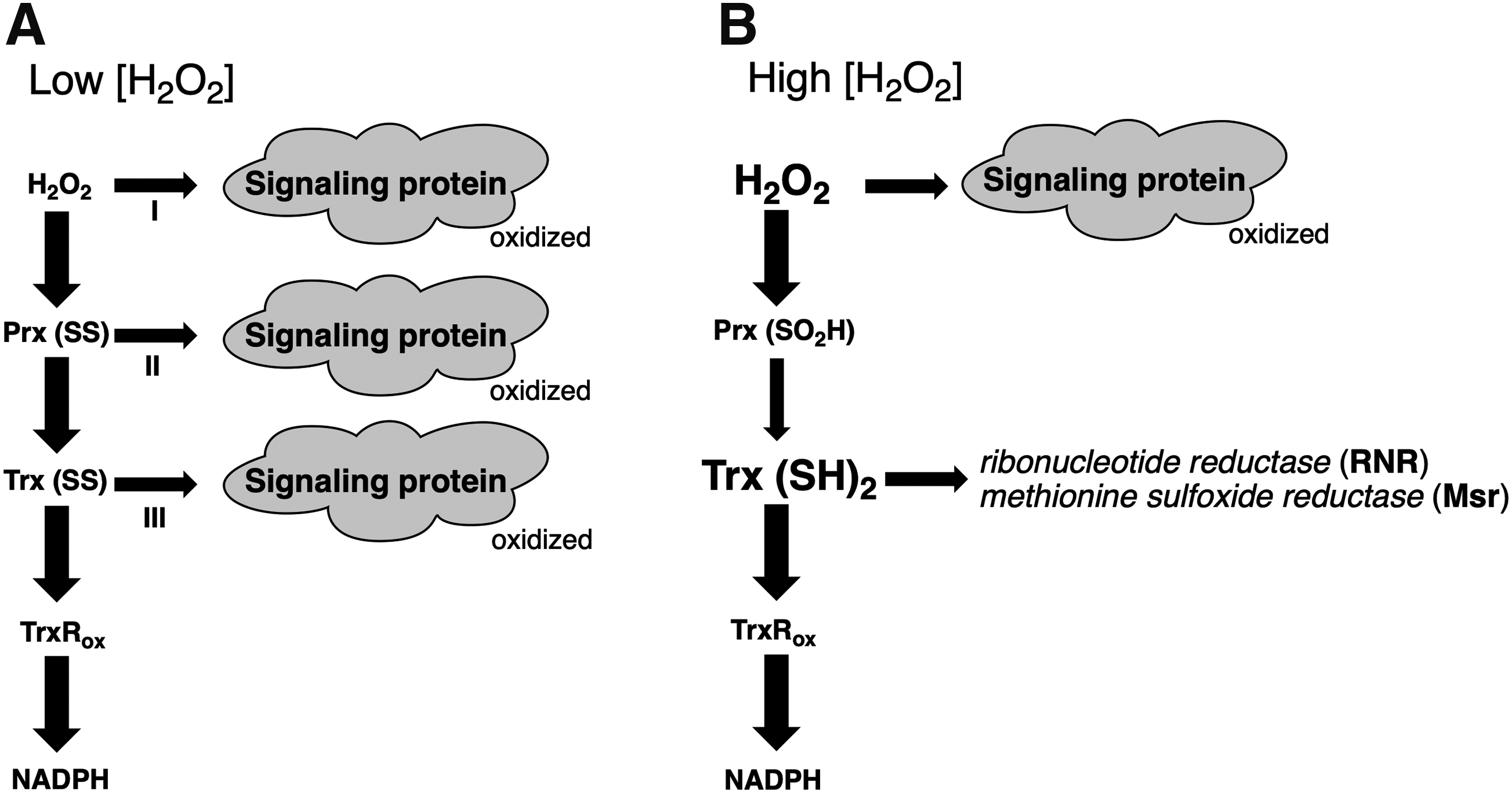
The regeneration of the reduced forms of ribonucleotide reductase and 2-Cys Prx involves an SN2 nucleophilic displacement reaction, called the thiol/disulfide exchange reaction. Uncatalyzed thiol/disulfide exchanges proceed at slow rates, but some oxidoreductases, such as Trx, glutaredoxin (Grx), and PDI, can accelerate these reactions by six to seven orders of magnitude (475). Since thiol/disulfide oxidoreductases display specificity to their substrates, they are involved in the redox regulation of signaling pathways (475).
Indeed, some transcription factors are redox-regulated by dithiol/disulfide switches that involve the participation of Trx or Grx enzymes. For instance, OxyR is a bacterial transcription factor that, upon oxidation of critical Cys residues by H2O2, forms a disulfide bond (Fig. 9A, arrow I), resulting in the induction of gene expression (760). Following the oxidative insult, Grx reduces the disulfide bond back to the dithiol state (760); consequently, the transcription of target genes ceases.
OhrR is another redox-regulated protein, but, in contrast to OxyR, it is a repressor rather than an activator of transcription (208). In the reduced state, OhrR binds to the promoters of target genes, inhibiting their expression. Upon oxidation of critical Cys residues by organic hydroperoxides, OhrR undergoes structural movements that provoke its release from the DNA, exposing the promoters of the target gene to the transcriptional machinery. Similar to OxyR, the oxidation of OhrR results in the formation of a disulfide that, in this case, is reduced explicitly by Trx instead of Grx (590).
Thiol/disulfide switches are also involved as a posttranscriptional mechanism that controls intracellular iron levels. Iron is required for several processes, such as DNA synthesis, respiration, and heme biosynthesis (123). However, high levels of iron may promote undesirable oxidation of vital cellular components (123). Therefore, the levels of this metal in cells are strictly regulated. Iron regulatory proteins 1 and 2 (IRP1 and IRP2) are the key iron sensors in mammalian cells that activate posttranscriptionally the synthesis of proteins involved in iron metabolism. IRP1 and IRP2 are cytosolic proteins that bind RNA stem-loops known as iron-responsive elements located in untranslated regions of mRNAs. IRP2 is the predominant RNA-binding protein in vivo (358), and unlike IRP1, IRP2 protein does not coordinate a [4Fe-4S] cluster and does not display aconitase activity (242). Under iron-replete conditions, IRP2 is degraded by a process involving polyubiquitylation and proteasomal degradation that requires a 73-amino acid sequence that is not present in IRP1 (298, 299). NO+ donors and exogenous heme increase IRP2 degradation in a process that depends on Cys residues present in the 73-amino acid segment (71). Notably, the redox state of two other Cys residues (Cys512 and Cys516) can regulate the mRNA-binding activity of IRP2 through a thiol/disulfide switch (767).
2. Thiol/sulfenic acid switches
Besides disulfides, redox regulation of thiol groups in proteins can also proceed through other intermediates such as nitrosothiols (Section II.D). We now focus our attention on sulfenic acid (R-Cys-SOH) derivatives that are the direct product of the two-electron oxidation of thiols (Fig. 9). The reaction between sulfenic acids and thiols gives rise to disulfides. Therefore, oxidants such as H2O2 generate disulfides in proteins with the intermediacy of sulfenic acids. In some cases, however, the sulfenic acids are protected from thiols by the protein environment.
Sulfenic acids are unstable compounds that can react not only with other thiols but also with other nucleophiles. The half-lives of sulfenic acids can also be limited by oxidation to sulfinic (-SO2H) and sulfonic (-SO3H) acids or self-condensation to yield thiosulfinates (9). In addition, sulfenic acids can also reversibly react with backbone amides, generating sulfenamides (248). However, sulfenic acids can gain stability in specific environments, where steric hindrance effects due to bulky substituents can protect them from organic compounds (108) or protein microenvironments (745). In these proteins, thiol/sulfenic acid switches can function in catalytic or signaling processes.
Therefore, as sulfenic acids are present as a dynamic and transient oxidation product of thiols, these species are generally analyzed by dimedone (5,5-dimethyl-1,3-cyclohexanedione)-based labeling reagents that trap these intermediates before they undergo other reactions (14, 473). The reaction of dimedone with sulfenic acid is slightly selective but very slow (340, 583). As an alternative, strained alkynes and alkenes have been developed (523) that selectively trap sulfenic acids with rates more than 100 times faster than reaction with dimedone (524). In the same line, benzothiazine (205)- and trans-cycloocten-5-ol (574)-based probes were described. Noteworhty, electrostatic or steric effects can likely modulate the rate of any probe reactivity toward a protein sulfenic acid (583). Perhaps the description of a thiol/sulfenic acid switch in GAPDH was the first with functional implications. Notably, the conversion of the glyceraldehyde phosphate dehydrogenase activity into acylphosphatase activity of GAPDH was paralleled with the formation of a sulfenic acid on a critical Cys residue (Cys 149) (742).
Several other studies also described thiol/sulfenic acid switches in catalysis and redox signaling. Nevertheless, the development of chemoselective probes has allowed for the investigation of sulfenic acids in cellular contexts. For instance, when H2O2 oxidizes Cys-797 of epidermal growth factor receptor (EGFR) in human epidermoid carcinoma A41 cells, the kinase activity of this protein is enhanced (501). These processes are consistent with the mechanism described in Figure 9A, arrow I.
Although the formation of sulfenic acids by the oxidation of thiols is well studied, the reverse process (reduction of sulfenic acids to thiols) is still poorly characterized. Indeed, the identity of the reducing agent is controversial. As reviewed by Kettenhofen and Wood (328), trivalent arsenicals are considered specific reductants for sulfenic acids, but they are not physiological compounds (328). In biological systems, Trx and GSH are considered general reductants of sulfenic acids (132). In both cases, two sulfhydryl groups are required for the reduction of one sulfenic acid in a two-step process. In the first step, a mixed disulfide between the protein and the thiol is generated. When GSH is the thiol involved, the protein is glutathionylated. These reactions are generally fast, attaining values in the 105 M −1·s−1 range for nonprotein molecules (462).
On the contrary, the corresponding rate constant for sulfenic acid in human albumins is in the 3–100 M −1·s−1 range (659). In the second step, a second sulfhydryl moiety is required to resolve the mixed disulfide. In the case of GSH, an oxidoreductase such as Grx is generally required to catalyze the reaction.
However, the identity of sulfenic acid reductants varies from protein to protein. Furthermore, these reductions of sulfenic acids in proteins display a high degree of specificity. For instance, mammalian Prdx6 (1-Cys Prx enzyme) is not reducible by GSH or Trx enzymes (199), although Trx and GSH are considered general reductants for sulfenic acids (132). Regeneration of the reduced form of Prdx6 can be achieved by its heterodimerization with πGST, which can then be reduced by GSH (199). In contrast, the homodimer of 1-Cys Prx1 from yeast (ScPrx1) can be reduced by Trx (504). Therefore, structural aspects such as protein/protein interactions should probably be considered when contemplating the specificity of the protein sulfenic acid reductions.
Ascorbate is another compound relevant to the reduction of protein sulfenic acids in biological systems but not frequently considered. Indeed, ascorbate is a known reductant for low-molecular-weight sulfenic acids (142). The direct reduction of protein sulfenic acids by ascorbate was first described in GAPDH (742) and later in 1-Cys Prx enzymes and papain (441, 526, 546, 766). Our results indicated that, in most cases, the rate constants for the reactions of diverse protein sulfenic acids lie in the 0.4–2.2 × 103 M −1·s−1 range, indicating that ascorbate is a broad-spectrum reductant (18). Possibly the relevance of ascorbate for the reduction of sulfenic acids might be higher in compartments, where the concentration of this reducing agent is elevated. Notably, the overall reduction of protein sulfenic acids by ascorbate was understood as an underlying mechanism in the noncanonical scurvy phenotype of triple mutant mice, lacking systems to reoxidize PDI (766).
3. Thiol-sulfinic/sulfonic acid switches
In some cases of higher oxidative insult, protein sulfenic acids can be hyperoxidized to sulfinic (R-Cys-SO2H) (Fig. 9) or sulfonic (R-Cys-SO3H) acids that were initially considered irreversible processes and, as a consequence, would provoke the inactivation of redox-active proteins harboring this PTM. In this way, the hyperoxidation of proteins would be toxic to cells due to the loss of function of the corresponding proteins. The finding that motifs that facilitate the hyperoxidation of 2-Cys Prx were positively selected for throughout evolution was surprising, indicating a physiological role for this PTM (720). This observation led to the coining of the so-called flood gate hypothesis (Fig. 9B), in which the inactivation of 2-Cys Prx would increase local H2O2 concentrations, allowing proteins that react slowly with this oxidant to be oxidized (720). Later on, other motifs that facilitate the hyperoxidation of 2-Cys Prx were identified (63, 627). Notably, not only hyperoxidation but also phosphorylation of Prdx1 provides another means to inactivate 2-Cys Prx, and this PTM was also proposed to have a role in increasing local H2O2 concentrations (719). Furthermore, acetylation of mammalian 2-Cys Prx (Prx1 and Prx2) increases their resistance to hyperoxidation (497). Hence, the thiol/sulfinic acid switch (Fig. 9) appears to fine-tune protein control.
Besides the local accumulation of H2O2, another mechanism was proposed to explain the positive selection of hyperoxidation motifs throughout evolution and involves the inactivation of 2-Cys Prx to preserve pools of reduced Trx (Fig. 9B) for other vital cellular processes (446). According to this model, the disulfide form of 2-Cys Prx actively consumes reduced Trx to turn over the peroxidatic cycle, thus reducing the availability of this oxidoreductase to reduce critical enzymes, such as ribonucleotide reductase and methionine sulfoxide reductase (156). When hyperoxidized, 2-Cys Prx enzymes do not react with Trx, increasing the reduced pool of this enzyme in cells and keeping the reducing equivalents required to sustain the DNA synthesis and repair oxidative lesions, among other essential processes.
A breakthrough in the field occurred with the discovery that sulfinic acids in 2-Cys Prx could be reduced back to sulfhydryl forms (717, 718) and the identification of an ATP-dependent system called Srx (58). At that moment, it became clear that the thiol/sulfinic acid pair is a reversible switch (Fig. 9), a convenient feature for a regulatory process. Accordingly, the thiol/sulfinic acid pair is involved in converting 2-Cys Prx enzymes from peroxidases into chaperones (holdase) upon hyperoxidation to sulfinic acids (305). Furthermore, hyperoxidized 2-Cys Prx can recruit chaperones to misfolded proteins in aggregates (255). Other lines of evidence indicate the biological relevance of Srx and sulfinic acids in 2-Cys Prx. For instance, the regulation of adrenal steroidogenesis involves the reduction of sulfinic acids in mitochondrial Prdx by Srx in a circadian pattern (332, 333).
Initially, 2-Cys Prxs were the only known substrates of Srx, an enzyme that reduces sulfinic acid. However, proteomic studies using an electrophilic diazene probe revealed that 55 other proteins have the potential to be also turned over by Srx (8). The reduction of sulfinic acids in protein tyrosine phosphatase (PTP), nonreceptor type 12 (PTPN12/PTP-PEST), and DJ-1 was confirmed by an enzymatic assay using recombinant Srx (8). Despite the notable progress, several aspects of the thiol/sulfinic acid switch likely remain to be elucidated.
Therefore, distinct thiol switches can mediate redox signaling (Fig. 9). Regarding H2O2 signaling, many mechanisms are possible (474), some of which were already mentioned above. The simplest of them is the direct oxidation of signaling proteins by H2O2 (Fig. 9A, arrow I). Perhaps the best example of such a pathway is the activation of the bacterial transcription factor OxyR. OxyR possesses two distinct globular domains, the DNA binding domain (N-terminal) and the regulatory domain (C-terminal domain). A highly reactive Cys in the regulatory domain (Cys-199 in OxyR from E. coli) is rapidly oxidized (∼105 M −1·s−1) to a sulfenic acid intermediate, which then forms an intramolecular disulfide bond with another Cys residue of the same domain (Cys-208 residue in OxyR from E. coli) (22, 310, 359). The formation of disulfide bonds in each of the four subunits of the OxyR tetramer triggers substantial structural changes in this transcription factor, resulting in the induction of the expression of its target genes (116, 310, 359). After oxidative stress subsides, OxyR is deactivated upon the reduction of the intramolecular disulfide bond by the Grx system for OxyR from E. coli (760) or the Trx system for OxyR from Pseudomonas aeruginosa (699).
The formation of sulfenic acids in the EGFR (501) and Src kinase (274) are two other examples consistent with the direct oxidation of protein thiols by H2O2. This proposal is because it is difficult to propose an alternative mechanism for the protein sulfenic acid formation other than the direct oxidation of thiols by H2O2.
However, most thiols react only slowly with H2O2 (10–50 M −1·s−1) (711). In contrast, the peroxidatic Cys (Cp) of Prxs can react with H2O2 1 to 10 million times faster (482, 498, 512, 642, 654). Although most of the protein thiols react slowly with H2O2, some of them are detected in oxidized forms in biological systems (710). One model that proposed to account for this apparent contradiction between biology and chemistry was the so-called relay mechanism (Fig. 9, arrow II), in which a sensor protein (Prx or Gpx) mediates the oxidation of slow reacting thiols by H2O2. These sensor proteins can react very rapidly with H2O2 and, through physical protein/protein interactions, transmit the signal as oxidizing equivalents to a slow-reacting thiol that can be a signaling protein, such as a transcription factor or a phosphatase.
The first experimental support for this model came from studies on the mechanism by which Yap1 is activated. Yap1 is the transcription factor that mediates the responses of the yeast Saccharomyces cerevisiae to H2O2 (235, 301). The finding that Gpx (also called Orp1) mediates the oxidation of Yap1 by H2O2 through thiol/disulfide exchange reactions provided the first experimental evidence for this redox relay mechanism (158).
There are also examples of the redox relay mechanism in mammalian cells. For instance, Prx1 from mammalian cells can physically interact in a redox-dependent pattern with various signaling proteins (94, 307, 661). Interestingly, Prx2-mediated activation of the transcription factor, STAT, displays features similar to the Gpx/Orp1–Yap1 pathway in yeast (596). In all cases, a fast-reacting thiol (Gpx/Orp1 or Prx2) senses H2O2 and then relays the oxidizing equivalents through physical interactions and thiol/disulfide exchange reactions to the transcription factors (Yap1 or STAT). Genetic approaches using deletion or depletion of cytosolic Prxs broadly inhibited protein thiol oxidation by H2O2, which contrasts with what was predicted by a model that assumes direct oxidation of proteins by H2O2 (617). Furthermore, a proteomic approach identified several partners of 2-Cys Prx (617).
The relay mechanism can be even more complicated, with the intermediacy of Trx and Prx (Fig. 9A, arrow III), which we have previously named the Prx-Trx model (474). Accordingly, several signaling proteins are redox-regulated by Trx enzymes, as reviewed (50). For example, only reduced Trx1 binds apoptosis signaling kinase 1 (Ask-1) and, as a consequence, inhibits its enzymatic activity (561). NF-κB is another signaling protein regulated by Trx1, as the binding of this transcription factor to its target DNA sequence requires the reduction of a single Cys residue (268, 416). In this sense, 2-Cys Prxs can participate in redox signaling not only by regulating H2O2 levels but also by modulating the redox status of Trx (474).
4. Relationships between H2O2 signaling and tyrosine phosphorylation
H2O2 signaling is closely associated with phosphorylation-dependent pathways, especially those related to Tyr residues (549). Indeed, the catalytic Cys of PTP enzymes directly involved in removing a phosphate group of its substrate also undergoes reversible oxidation/inactivation by H2O2. This dephosphorylation catalyzed by PTPs is a kind of nucleophilic displacement reaction that proceeds via an essential catalytic Cys residue conserved within the PTP signature motif, Cys(X)5Arg (163, 164, 492). The sulfhydryl group of the catalytic Cys displays an acidic pKa (within the 4.0–6.0 range), indicating that a major fraction of the sulfhydryl group is present as thiolate (RS−) ion at neutral pH, which is a better nucleophile than the corresponding thiol (RSH). This low pKa of the catalytic cysteine is necessary for its dephosphorylation activity; but, at the same time, renders PTPs more susceptible to oxidative inactivation, which appears to be a reversible and regulatory process (485, 644).
As mentioned above, the primary product of the two-electron oxidation of the nucleophilic cysteine of PTPs is sulfenic acid (RSOH). The outcome of sulfenic acids in PTPs varies depending on the enzyme (393). Nevertheless, most of the oxidative intermediates generated in PTPs are reversible, a convenient feature for a regulatory process. For instance, in the cases of Cdc25 (23 –25, 82, 193, 598) and phosphatase and tensin homologue (PTEN) (371), an intramolecular disulfide bond linking the catalytic Cys residue to a proximal Cys (named backdoor Cys) is generated, which can also be an intermolecular disulfide bond in other phosphatases (HePTP) (393). Furthermore, a stable sulfenic acid species is formed as the oxidation-dependent inactive state of other PTPs (164, 309). Finally, cyclic and reversible sulfenilamide is generated in other PTPs, which is described next.
It is important to mention that the PTP superfamily can be divided into “classical PTPs” (38 members) and “dual-specificity phosphatases” (61 members). The classical PTPs are further subdivided into receptor-like transmembrane PTPs (rPTPs) and nontransmembrane PTPs (nPTPs or cytosolic PTPs) (485, 644). While all nPTPs comprise a single catalytic domain linked to diverse regulatory or targeting domains, most rPTPs have two PTP domains arranged in tandem (485, 644).
The dual-specificity phosphatases can often also dephosphorylate phosphoserine/threonine residues, in addition to phosphotyrosine. This group comprises the MAP kinase phosphatases and the tumor suppressor PTEN that dephosphorylates lipids (phosphatidylinositol (3,4,5)-triphosphate [PIP3]). As mentioned above, sulfenilamides, intra- and intermolecular disulfides, and sulfinic/sulfonic acids were all detected as oxidized intermediates in PTPs belonging to these distinct subfamilies using crystallography and MS, among other techniques (485).
In the case of PTP1B, which is perhaps the best-studied among all classical PTPs, oxidation is reversible due to the rapid conversion of the sulfenic acid form of the catalytic Cys to a 5-atom-ring structure, a cyclic sulfenamide. In PTP1B, the juxtaposition of His214 with Cys215 polarizes the amide bond, promoting nucleophilic attack by the amide nitrogen of Ser216 on the sulfur atom of the sulfenic acid in Cys215, leading to the formation of a covalent bond between the sulfur and nitrogen atoms of these neighbor residues. Of note, the cyclic sulfenamide can be readily reduced back to the thiolate form (443, 562). Possibly this sulfenamide might have two roles: (i) prevent irreversible hyperoxidation of catalytic Cys and (ii) facilitate the reduction of the sulfenamide to restore the active form of the PTPs (644).
Sulfenamides were also detected in the crystal structures of the D2 domain of rPTPα, which is structurally very similar to and with an identical chemical composition of sulfenamides generated in PTP1B (730). It has been shown that rPTPs have a tandem arrangement of two PTP domains, with the catalytic residues in the membrane proximal domain (D1). In contrast, the membrane distal domain (D2) predominantly functions as a regulatory domain, displaying little to no catalytic activity (511, 730). Intriguingly, although virtually devoid of phosphatase activity, most rPTP D2 domains share the conserved catalytic Cys and Arg residues of the PTP signature motif, suggesting a redox sensor role (730). As demonstrated by an antibody-based assay for detecting oxidation-inactivated PTPs, the second regulatory domain of rPTPα is more susceptible to oxidation (511).
Remarkably, using this antibody, it was possible to detect platelet-derived growth factor (PDGF)-induced oxidation of endogenous Src homology 2 domain-containing protein tyrosine phosphatase 2 (SHP-2; PTPN11), an nPTP (511), which is a very physiological condition. SHP2 is a ubiquitous multidomain nonreceptor phosphatase implicated in diseases such as cancer, diabetes, and Noonan syndrome. Noonan syndrome is one of the most common genetic disorders associated with congenital heart disease, and approximately half of the patients with Noonan syndrome present mutations in SHP2, such as N308D. Remarkably, SHP2N308D is more prone to oxidation than wild-type SHP2, but an association of this kinetic feature with Noonan syndrome is premature (392).
Methods to detect oxidation of phosphatases in cells are a challenge. Traditionally, most of the methods involved thiol alkylation of the reduced form of PTPs (485). However, there are some drawbacks, as many of these alkylating agents also modify sulfenic acids (523). Therefore, assays based on dimedone, which specifically alkylate and therefore trap Cys sulfenic acids, were developed (473), as described above. Another immunochemical approach to directly profile oxidized PTPs involved the generation of an antibody that recognizes the conserved sequence of the PTP signature (VHCDMDSAG) harboring the catalytic cysteine modified with dimedone (CDMD) that chemoselectively reacts with Cys sulfenic acids to form a stable thioether adduct (217).
Employing these and other methods, PTPs of all subfamilies were found to be oxidized in cells upon physiological stimuli such as growth factor (EGF and PDGF), insulin, and cell receptor (B cell and T cell) stimulation (362, 644). A remarkable feature is the pronounced selectivity for the oxidation of each specific pathway by a different stimulus. For instance, while exogenous H2O2 treatment of fibroblasts caused oxidation of multiple PTPs, stimulation of the same cells with PDGF selectively oxidizes SHP2 phosphatase (421). In contrast, insulin stimulation preferentially oxidized PTP1B and TC-PTP, two PTPs that are relevant for negative regulation of insulin receptors (421).
Although the thiolates of catalytic Cys of PTPs display acidic pKas, their reactivity toward H2O2 is low (kinact = 10–20 M −1·s−1) (164). In contrast, Prx and Gpx react with H2O2 1 to 10 million times faster and are abundant protein thiols. Furthermore, GSH is present in cells in millimolar concentrations. Therefore, it is reasonable to consider that on chemical grounds, Prxs, Gpxs, and GSH outcompete PTPs (710). However, as mentioned above, PTPs are frequently found oxidized in cellular systems, showing an apparent contradiction (710). The relay mechanism (described above) is frequently considered to account for this apparent contradiction, but at least so far, no physical interaction between Prx and PTPs has been described (617).
Although the Prx redox relay model has gained increased appreciation as described above, the other mechanisms of H2O2 signaling cannot be ruled out. Indeed, because the H2O2 reactivity can be increased two to three orders of magnitude by the CO2/bicarbonate pair (656), the mediation of Prx or Gpx enzymes may not be required in some circumstances. Notably, bicarbonate concentrations in mammalian tissues are generally very high (in the millimolar range), as CO2 is a product of energetic metabolism (19). This stimulating effect of the CO2/bicarbonate pair on H2O2 reactivity is likely related to the formation of peroxymonocarbonate (HCO4 −). In equilibrated H2O2 and CO2/bicarbonate solutions and at neutral pH, HCO4 − generation depends on the slow perhydration of CO2 (31). Indeed, at physiological conditions, the CO2/bicarbonate pair can accelerate the oxidation of signaling protein such as PTP1B (139, 761) and other slow reacting thiols, such as bovine serum albumin (652) and AhpE (548). Furthermore, the hyperoxidation of 2-Cys Prx can also be accelerated by the CO2/bicarbonate pair (513, 656), allowing the flood gate process to occur, preserving the reduced Trx pools.
The activity of Prx enzymes can be inhibited not only by hyperoxidation but also by phosphorylation. For instance, Prx isoforms can be phosphorylated at distinct residues, affecting (increase or inhibit) their peroxidase activities (592). Therefore, redox and phosphorylation pathways appear to be a two-way road. Oxidation of critical Cys residues affects the activity of kinases and phosphatases, whereas phosphorylation of each one of the six mammalian Prx changes their ability to remove H2O2.
Of note, the pool of Prx1 located at the plasma membrane can be phosphorylated at Tyr194 by Src kinase in response to stimuli such as EGF and PDGF, leading to its inactivation (719). Consequently, H2O2 locally rises, allowing the oxidation of nearby less reactive thiols, such as the catalytic Cys of PTPs. Therefore, in response to growth factor stimulation, H2O2 generation by NADPH oxidases (NOX1) in lipid rafts is necessary to simultaneously activate Src family kinases and inactivate PTPs sustaining further phosphorylation and inactivation of Prx1 (592). Therefore, NADPH oxidases, Prx1, Src kinases, and PTPs take part in a retroalimentation cycle in lipid rafts that operate in response to growth factor stimulation (592). In contrast, Prx2 is inactivated preferentially by hyperoxidation (719).
In summary, distinct thiol switches (RSH/RSSR; RSH/RSOH; RSH/RS2OH) and various mechanisms of H2O2 signaling can co-occur in cells, possibly in a coordinated manner that can also interact with other pathways, such as those dependent on tyrosine phosphorylation. Understanding how these processes occur from a global perspective may represent a challenge in the redox field that could be circumvented using systems biology (SB) approaches.
B. Protein S-glutathionylation
Protein S-glutathionylation is the process where a mixed disulfide is formed between GSH and protein Cys residues (Fig. 10). GSH is commonly referred to as a redox buffer (e.g., protein S-glutathionylation; reaction with radical and reactive species) although it also plays an essential role as a reductant cofactor of oxidoreductases (e.g., Grxs) and Gpxs and also as a cofactor of GSTs upon a nucleophilic reaction in the detoxification of xenobiotics. A more proper definition of the role that glutathione has in cell metabolism is yet to be established. Protein S-glutathionylation is a reversible process, thereby considered a protein redox cycle (754). Many potential mechanisms have been proposed for protein S-glutathionylation (212), but very few are considered to occur in vivo (754). Indeed, the relatively easy task to demonstrate intracellular protein S-glutathionylation was not followed by attempts to uncover the underlying mechanism. Nevertheless, investigations on protein S-glutathionylation unraveled a powerful mechanism of protein modulation through metabolic redox shifts. The importance of protein S-glutathionylation in redox biology is attested by a search in the PubMed platform that identified 95 reviews on the topic in the last decade.
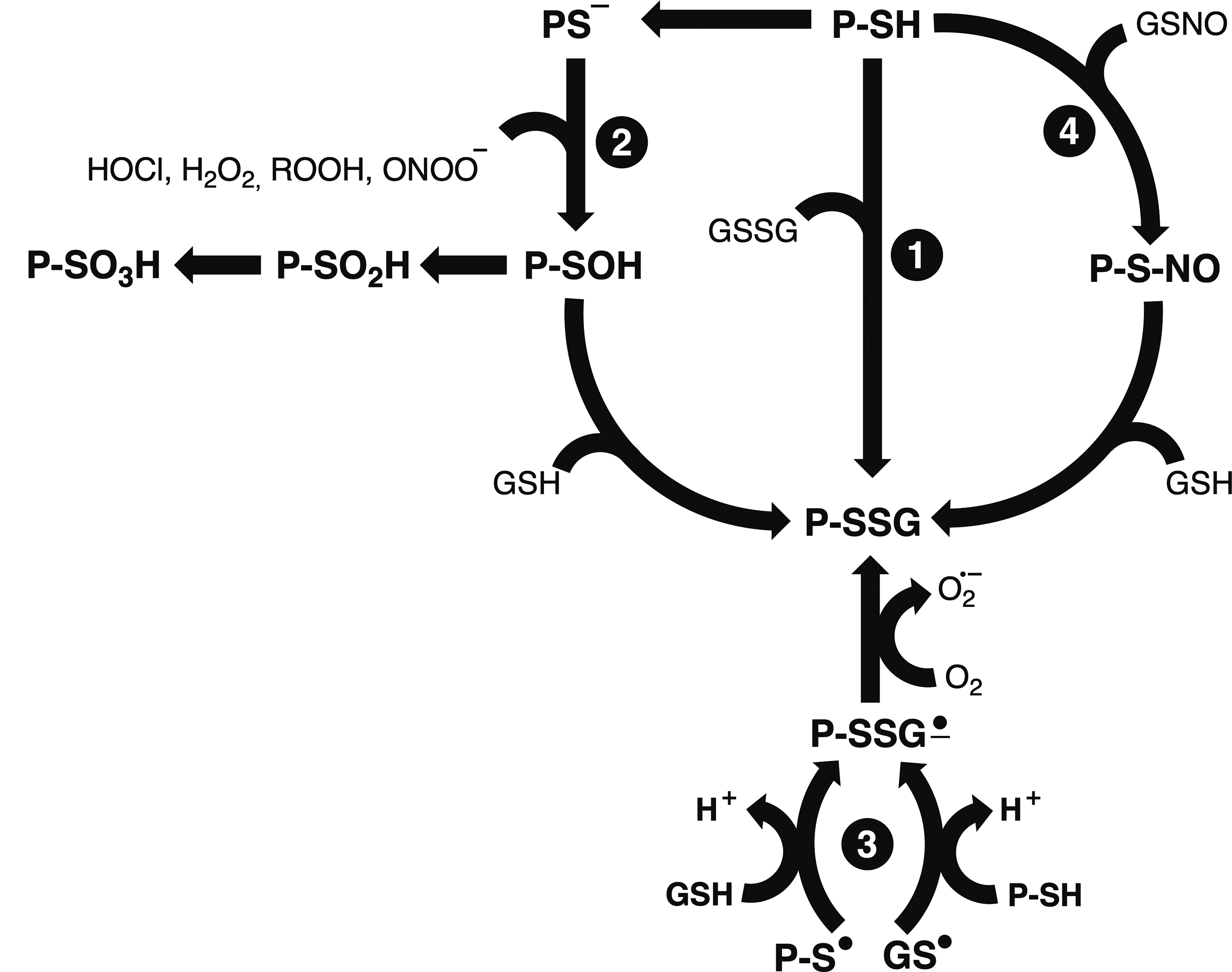
The most studied mechanisms of protein S-glutathionylation claimed to take place inside cells are as follows (summarized in Fig. 10): Oxidation of protein sulfhydryl group (P-SH) by the oxidized form of glutathione (GSSG). Under normal conditions, the GSH/GSSG ratio is very high, around 100:1, but depends on the intracellular compartment and cell type (229). An exception is in the ER, where the GSH/GSSG ratio is around 3:1 (288). For most protein-sulfhydryl groups, the GSH/GSSG ratio should decrease dramatically (around 1:1) to achieve S-glutathionylation conditions. In this sense, such strong oxidative stress conditions are unlikely to be physiologically possible. The deprotonated protein-sulfhydryl group (PS−) can be oxidized by H2O2, organic peroxides, HOCl, or peroxynitrite (H2O2, ROOH, HOCl, and ONOO−, respectively), rendering the oxidized sulfhydryl form, namely sulfenic acid (P-SOH). Protein-sulfenic acids react with GSH rendering the protein-S-glutathionylated adducts (Section III.A). This reaction is considered an essential protective mechanism against overoxidation, which would generate the irreversible sulfinic (P-SO2H) and sulfonic (P-SO3H) forms. However, the physiological condition necessary for the reaction between GSH and P-SOH depends on the intracellular peroxide levels that, in turn, depend on overcoming the rich intracellular peroxidasic activity. Although such a possibility is unlikely, it cannot be discarded based on the Flood gate hypothesis discussed above (Section III.A) Protein (PS•) or GSH (GS•) thiyl radical formation followed by reaction with GSH or PS−, respectively, to form a radical glutathionyl intermediate (PSSG•). The reaction with O2 to form the PSSG and the superoxide anion was suggested to occur in vivo (212). The modification of protein-sulfhydryl (PSH) by the glutathione nitroso-derivative GSNO results in the protein-Cys residue being transnitrosated to PSNO and subsequently forming PSSG through the reaction with excess GSH (233).
It is acceptable to state that protein S-glutathionylation may occur enzymatically or by spontaneous chemical reactions. Glutathione S-transferase π (GSTP) has been proposed to catalyze protein S-glutathionylation (647). Strong evidence supporting the enzymatic process came from the observation that cells lacking GSTP or GSTP-depleted presented attenuated S-glutathionylation of specific proteins. GSTP is found in many cellular compartments (cytosol, nucleus, ER, and mitochondria). The kinetic advantage of the enzymatic reaction is based on the GSH pKa that is lowered from 9.2 to 6.5 by GSTP through proton abstraction by a Tyr residue of GSTP, resulting in a thiolate anion (GS−) that is enzymatically transferred to the protein Cys residue. A well-documented and remarkable example of enzymatic S-glutathionylation is the case of Prx6 that is S-glutathionylated at the Cys in the active site through heterodimerization with GSTP. Glutathione is transferred to the oxidized catalytic Cys residue (Prx6-SOH), generating the also inactive S-glutathionylated form of the enzyme (Prx6-SSG) followed by the release of GSTP and subsequent spontaneous reduction of Prx6 by GSH (402). This sequence of events is an example of a complete redox cycle for restoring enzymatic activity.
1. Reversal mechanism
The most studied enzymes that carry out protein S-deglutathionylation are Grxs (213). As already described, Srx can deglutathionylate specific Prxs at specific Cys-SG sites (92, 496). GSTO1-1 was reported as a deglutathionylating enzyme in epithelial breast cancer cells with specificity toward actin (422). Amazingly, when the same cells were exposed to S-nitrosoglutathione (GSNO), GSTO1-1 was associated with the S-glutathionylation of several proteins because, as mentioned above, some S-nitroso proteins are labile to subsequent S-glutathionylation (233). Importantly, a GSTO1-1 polymorphism has been associated with several diseases (422). Evidence of enzymatic specificity for S-deglutathionylation comes from human Prx1, in which three out of four Cys residues are found to be S-glutathionylated. Two Cys residues are preferentially deglutathionylated by Srx and the last one by Grx (496). Despite these examples, more direct evidence for the specificity of enzymatic deglutathionylation is missing in the literature.
2. Methods to detect protein S-glutathionylation
Several techniques are used to detect S-glutathionylated proteins. The first methods described were based on radio- and immune-labeling (227) and the utilization of biotinylated GSH (77). The most promising approaches are based on trapping the reduced protein Cys residues with alkylating agents, followed by treatment with Grx or reducing agents (e.g., dithiothreitol [DTT]) to remove the –SSG moiety and subsequent exposure of the free protein sulfhydryls to fluorescent probes or biotinylated thiols for detection. MS techniques allow the identification of the protein and the Cys-containing sequences. These methodologies are also able to map linear protein motifs prone to S-glutathionylation (729). However, the few MS studies so far do not permit mapping the motifs, if any, prone to S-glutathionylation.
3. S-glutathionylation as a modulatory mechanism of protein function
The number of proteins found to be S-glutathionylated is not considered significant when considering the total proteome of mammalian cells (240). Among them, there are well-documented examples where the S-glutathionylation is associated with the modulation of protein function coupled to general intracellular or compartmental redox shifts. In these situations, protein S-glutathionylation functions as a regulatory cell response mechanism. As established elsewhere (424), some of the criteria necessary to fulfill this condition would include (i) specificity of S-glutathionylation toward the protein Cys residue; (ii) occurrence of S-glutathionylation at high ratios of GSH/GSSG; (iii) S-glutathionylation occurrence at physiological conditions upon a stimulus, altering a functional or physiological endpoint; (iv) existence of a rapid and efficient mechanism for reversing the reaction thereby re-establishing the initial physiological condition when the stimulus ceases. Among the proteins that fulfill these criteria, examples include Prx, as mentioned above and the yeast catalytic unit of the 20S catalytic unit of the proteasome where 2 out of 32 Cys residues are S-glutathionylated in vivo, promoting the opening of the 20S catalytic chamber and increasing the degradation of oxidized proteins. This process was shown to be in vitro reversed by Grx2 and Trx1 or Trx2 and dependent on the intracellular redox status (160, 587, 588). In addition, many other proteins play a regulatory role when S-glutathionylated. Examples include several mitochondrial proteins that adjust the energy metabolism (398), ER proteins to cope with Ca2+ metabolism (754), and Fas or CD95, members of the TNF family of death receptors in the amplification of cell death pathways (15).
4. Physiopathological processes associated with protein S-glutathionylation
The S-glutathionylation of actin and hemoglobin has been proposed to be biomarkers in patients with Friedreich ataxia, diabetes, hyperlipidemia, and uremia (480, 629). A GSTP polymorphism found in European ancestors (259) alters the substrate-binding site of the enzyme and is considered a risk factor for a response to redox imbalance in at least one example provided by the recycling of Prx6 [(403); see above, the role of GSTP in Prx6 recycling]. The studies reported in cellular models (403) suggest that the GSTP polymorphism implies that individuals will have potential differences in the antioxidant response based on Prx6 activity, impacting the protection of cell membranes against lipid peroxidation, as Prx6 acts toward lipid peroxides. As GSTP interacts with several proteins, there are some speculative interpretations about the consequences the GSTP polymorphism has on protein S-glutathionylation (240). Therefore, more investigation is necessary to clarify that point. The S-glutathionylation of specific substrates is believed to involve the immune response and allergic inflammation (754). The DNA binding of the S-glutathionylated tumor suppressor p53 has been found inhibited in human cancers, a fact that is expected to determine a relationship between redox control of p53 in cancers and healthy tissues (674).
As pointed out, protein S-glutathionylation is promoted by a redox buffer (GSH), interacting with an important redox-sensitive protein residue (Cys). Moreover, as it is reversible, it represents a recycling mechanism. Hence, protein S-glutathionylation might be considered a prominent redox mechanism of protein modulation and signal transduction. However, impaired protein function is also caused by oxidative stress due to ROS and RNS, as is the case with eNOS activity (111, 404). In this example, there is a disruption in the coupling of NADPH oxidase to eNOS, resulting in decreased NO synthesis and superoxide generation, thus suggesting an angiotensin II-induced impairment of endothelium-dependent vasorelaxation (214).
C. Protein persulfidation
Protein persulfidation (P-SSH), also called sulfhydration or sulfuration, is a modification of protein Cys residues formed by the reaction between H2S, in the HS− form, and supposedly oxidized protein thiols (P-SSR or –SOH; Equations 4 and 5, respectively) and also by reaction of inorganic persulfides (HSSnS−) and polysulfanes (HSSnSH) with the protein-Cys thiolate form (P-S−) (198). Toward the end of the 1990s, H2S was recognized as a physiological mediator (231, 500). Cys and its derivatives are substrates for enzymatic H2S formation by the enzymes cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and β-mercaptopyruvate sulfurtransferase. The secondary source of H2S is the gut microbiota. Persulfides and polysulfides are endogenously generated and obtained from dietary intake (231). H2S concentration is estimated to be in the low nanomolar range in most tissues (318). Therefore, the low steady-state concentration of H2S would reflect a high rate of clearance and/or consumption. Besides the direct signaling of H2S, another model of its signal transduction would be via protein persulfidation. Protein persulfidation has been shown to regulate protein function and, consequently, diverse biological processes. Regarding the nomenclature, the term persulfidation is the most accurate one as sulfhydration implies a mechanism based on “hydration,” which is not the case (198).
The most acceptable in vivo mechanism of protein persulfidation is the reaction between H2S and P-SOH (Equation 5). As mentioned above, protein-sulfenic acids readily react with thiols (RSH), forming disulfides. In addition, sulfenic acids react with H2S generating the persulfidated derivatives. This mechanism was shown to occur via the sulfenic acid form of the human albumin Cys4 residue rendering the Cys4-persulfidated protein (137). In agreement, intracellular protein-persulfide levels are increased upon treatment with H2O2, and these levels decrease when H2S-generating enzymes (CBS and CSE) are inhibited. Protein persulfidation resulting from the reaction between H2S and protein-sulfenic acids was shown to overcome the H2S reaction with protein disulfides (Equation 4) (198). As protein sulfenic acids can be further oxidized, generating the irreversible sulfinic and sulfonic acid (P-SO2H and P-SOH, respectively) forms, similarly to protein S-glutathionylation, persulfidation would protect protein-thiols against overoxidation since both mechanisms are reversible.
Crosstalk between NO• and H2S signaling pathways through the formation of HSNO by the reaction of S-nitrosothiols with H2S is expected to contribute to protein modification (318). According to the mechanism proposed, H2S reacts with protein-SNO regenerating the reduced protein thiol (P-SH) with the concomitant production of HSNO. In turn, HSNO, by reaction with H2S, results in the production of HNO and the inorganic persulfide HSSH, which can react with the protein thiolate group (P-S−) and generate the protein persulfide derivative (P-SSH). Accordingly, evidence shows that the same protein Cys residues can be modified by S-nitrosation and S-persulfidation, generating opposite functional effects (456). An example based on both mechanisms is the regulation of GAPDH activity and, consequently, glycolysis and gene transduction. GAPDH is S-nitrosylated or S-persulfidated at residue Cys 150. While the S-nitrosation of this residue inhibits GAPDH glycolytic function and promotes its translocation to the nucleus, the S-persulfidation dramatically increases GAPDH glycolytic activity (456). Another well-documented example of functional protein modulation by the exchange between S-persulfidation and S-nitrosation is the case of NF-κB (576). The Cys 8 residue in subunit p65 of NF-κB is the only Cys residue among the eight S-persulfidated, increasing its DNA binding and the consequent antiapoptotic effect. On the contrary, S-nitrosation of the same residue occurs as it is depersulfidated, consequently reducing NF-κB binding to DNA. These examples provide evidence for the specificity of persulfidation toward specific Cys residues. Both S-nitrosation and S-persulfidation processes are reversed by the Trx system (318, 347). Persulfidation was also reported as a regulatory mechanism linked to the ER stress response (347). In this case, upon inhibition of the catalytic activity of the phosphatase PTP1B through S-persulfidation of the Cys125 residue located in the active site of the enzyme, PERK phosphorylation is maintained and protein translation is suppressed since PERK functions as an ER stress sensor.
Many of the pathways where H2S is involved are supposedly mediated by protein S-persulfidation, for example, inflammation and vasorelaxation (500). Therefore, a well-documented example is the S-persulfidation of KATP and IKCa channels resulting in vasorelaxation (457). Many other notable examples of the modulation of pathways by protein-persulfidation are found in the literature (198).
Protein persulfidation as a modulatory mechanism of protein function relies on its reversibility under physiological conditions. There is convincing evidence for the role of the Trx system in catalyzing depersulfidation (198). Trx was shown to be 200-fold more efficient at reducing Cys-SSH in PTP1B than DTT (347). Also, the treatment of cell lysates with a Trx reductase inhibitor increased the total levels of protein persulfidated (698). The Grx system has been proposed; however, only the in vitro reduction of bovine serum albumin-SSH has been demonstrated (198). Further studies should unravel that potential.
One significant limitation to detect proteins modified by persulfidation is methodological. Recently, a quantitative method termed low-pH quantitative thiol reactivity profiling that improves similar ones was reported (206). By applying this methodology, cell lysates treated with NaHS resulted in identifying 994 proteins modified by persulfidation. Although these data cannot be taken as an actual physiological situation of persulfidation, they reveal potential protein-Cys targets. Very few studies have generated data of persulfidated proteins in the steadystate of cells. In most of them, persulfidated proteins represented 0.15% to 1.2% of the proteome of cells and tissues examined (198).
Protein-persulfidation and S-glutathionylation are firmly associated with functional protein modulation, thereby regulating intracellular redox shifts.
D. Oxidation of protein methionine residues
The oxidation of protein methionine residues generates the sulfoxide moiety (MetO) that is enzymatically reduced back by the Trx-dependent methionine reductases (MsRs) (386). MsRs are highly preserved throughout evolution from bacteria to all kingdoms (386). As MsRs are Trx-dependent proteins, NADPH provides the reducing power to the cycle.
First reports on Met oxidation were on the inhibition of GS of Lacto arabinosus (686), followed by reports on Bacillus subtilis sporulation inhibition due to Met oxidation (346). Later, the reduction of MetO was described (60, 178), and an enzyme with sulfoxide reductase activity was identified (80, 183). During the 1990s, Met residues were proposed to be an antioxidant barrier on the protein surface to protect them from widespread oxidation. The classical work from Stadtman's and Levine's groups showed that 8 out of 16 Met residues of the enzyme GS, all surface exposed, could function as oxidant scavengers (372). Hence, the proposed mechanism of protein protection against widespread oxidation by Met residues and the catalytic recycling provides efficient antioxidant protection to the specific protein and its environment. This early idea of Met residues functioning as oxidant scavengers has been challenged by new data (72, 369). We also direct the reader to the discussion below regarding Met residue oxidation and loss of protein activity.
The oxidation of Met introduces a chiral center at the sulfur atom generating two epimers, namely R-MetO and S-MetO. The two MsRs (A and B) have specificity for the epimers: MsrA for the S-MetO and MsRB for the R-MetO. Mammals have three isomers of class B and one of class A. Studies reported in the literature regarding the knockout and overexpression of MsRA in many species have contributed significantly to the hypothesis of protein Met residues as oxidant scavengers. Knocking out MsRA in several species, including yeast and bacteria, significantly increased their susceptibility to oxidative stress, while overexpression conferred resistance (377). A remarkable study showing the causality of the hypothesis on Met residues as oxidant scavengers replaced 40% of the total Met residues with norleucine in E. coli (390). Although the norleucine-substituted strain grew at the same rates as the control cells, exposing the modified strain to the oxidant HOCl resulted in 100% death. In contrast, the same concentration of the oxidant did not affect the viability of control cells. Another observation that corroborates the hypothesis is the amazing ability of all eukaryotic cells to increase Met content in their proteins upon an oxidative challenge (361).
As Manta and Gladyshev (406) stated, the reaction of protein-Met residues with the majority of oxidants is generally too slow to be considered biologically relevant. Their enzymatic generation by a specific class of monooxidase enzymes is proposed (406). The enzymatic oxidation and reduction of protein-Met residues could function as an essential regulatory or signaling mechanism. Indeed, some reports in the literature are consistent with the notion that MetO is not linked to protein inactivation but a gain of function (184) or interchangeable modulation (291).
Methionine oxidation and recycling provide important mechanisms of on-off switches in cellular regulation. Such a mechanism was demonstrated in the binding of lymphotoxin-α to the tumor necrosis factor receptor 1, where a specific Met residue of the ligand interacts with a specific Tyr residue of the receptor. Met oxidation prevents the interaction, thus providing a regulatory mechanism of lymphotoxin signaling (376).
Another important source of evidence on the role of MetO/Met recycling in the cellular protection against oxidative stress in human pathological conditions is associated with either mutations in genes encoding for MsRs or alterations in activities and concentrations of MsRs. A human polymorphism in msrA is associated with increased cardiovascular diseases (247). Decreased activity of MsRA was reported in the brain of Alzheimer's patients (211). Moreover, the high toxicity of the Aβ(1 –42) peptide is attributed to the Met 5 residue, as its substitution abolishes toxicity (87). It is important to point out that α-synuclein, which plays a crucial role in PD development, contains two Met residues that are highly susceptible to oxidation. When α-synuclein was expressed in Drosophila overexpressing msrA, the locomotor defects were suppressed (696). MsRA was consistently associated with the metastatic potential of hepatocellular carcinoma as msrA expression decreased in patients with metastasis in comparison with tissues of patients without metastasis (366). MsRA levels were also attenuated in cases of advanced grades of breast cancer (157). Loss of MsRB causes human deafness (5).
Despite much evidence on the indirect antioxidant defense and recycling regulatory mechanisms, Met oxidation is associated with protein aggregation (Section IV.A) and loss of protein function that represents a major consequence in most proteins via conformational changes and unfolding (180). Methionine sulfoxide can also be further irreversibly oxidized to the sulfone form (MetO2), but at a significantly lower extent (180). The importance of MetO2 in human pathophysiological conditions remains unclear so far.
Regarding the loss of protein function upon Met oxidation, calmodulin is a well-characterized target (56) together with calmodulin-regulated enzymes such as calcineurin (Ca2+-calmodulin-dependent kinase II) (180). Met oxidation also has a vital role in regulating the immune system by inhibiting the degradation of the transcription factor NF-κB. The oxidation of Met45 of NF-κB inhibitor (IκB) implies on IκB resistance to degradation, and consequently, the inhibition of transcription mediated by NF-κB. However, this mechanism is reversible through MsRs (320, 423). Nonetheless, protein inhibition through Met oxidation should be considered in the level of cellular signal transduction as in many cases, Met oxidation and reduction play a dynamic role as a regulatory mechanism in a variety of cellular cascades.
The most applied methods to detect protein oxidized Met residues are MS analysis that allows detection on a proteomic-wide scale (226) and immune assay (693). Other methods are based on MetO derivatization (180).
IV. Fate of Oxidized Proteins
The primary fate of nonrepairable oxidized proteins is proteolysis or aggregation. Proteolysis would be the best pathway for cellular homeostasis maintenance, whereas aggregation is an important fate of oxidized proteins associated with prevalent human pathologies. The sections below discuss both possibilities.
A. Protein aggregation
In general, oxidative modifications of amino acids alter their charge or steric properties, destabilizing the protein native conformation (334). Surface hydrophobicity is commonly increased (107), facilitating aggregation (453). The mechanism of production and the chemical characteristics of protein aggregates in vivo are not entirely elucidated. However, it is known from in vitro studies that different oxidative modifications of protein residues may participate in the onset and perpetuation of protein aggregates. In the following paragraphs, the term aggregation refers to crosslinked material as well as to noncovalent aggregates.
It is increasingly clear that most organisms bear a group of proteins that aggregate under mild conditions that do not disturb the other constituents of the proteome. Because of that, some research groups refer to those aggregation-prone proteins as metastable proteins (289, 483, 701). These proteins were shown to be large and weakly hydrophobic, with a flexible structure enriched in disordered regions (483). Walker et al. (688) elegantly demonstrated that loose protein folding increases the solvent accessibility to methionine side chains, making them more susceptible to oxidation. Other research groups demonstrated that misfolding predisposes proteins to oxidative damage (181, 348). In support of this idea, a strong inverse correlation between translational fidelity and protein carbonylation was described (348). This relationship is particularly relevant during aging since it is associated with a prooxidant cellular status (36). Indeed, widespread protein aggregation was shown to occur during aging in Caenorhabditis elegans (147), S. cerevisiae (514), and Drosophila (161). However, unfortunately, none of these studies assessed the oxidative modifications present in the age-associated aggregates.
1. Mechanisms involved in protein aggregation induced by protein oxidation
Tanase et al. (632) analyzed aggregates from bone marrow and splenic cells from 3-, 12-, and 22-month-old mice. They found a significant association between carbonylation of the proteome and the accumulation of high-molecular-weight protein aggregates with age. More than that, the authors found that over 90% of the carbonylated proteome was aggregated, suggesting that protein carbonylation is intimately linked to age-induced protein aggregation (632). Aggregates from aged mice had different carbonyl modifications, especially aminoadipic acid and glutamic semialdehyde, generated from the oxidation of lysine and arginine, respectively. The aggregates were also enriched in kynurenine and oxolactone, which are tryptophan oxidation products, and pyrrolidone and pyrrolidinone, which are generated from proline oxidation (632). The cellular turnover of aggregates is slower than of nonaggregated proteins. Thus, it would be interesting to know whether there is a causal relationship between protein oxidation and aggregation or whether increased oxidation is simply due to slower aggregate turnover.
Experimental data clearly show that Met oxidation is intimately linked to human prion protein (PrP) aggregation (540, 714, 744). As shown, sodium periodate-induced Met oxidation increased the aggregation of recombinant human prion protein (rhPrP) proportionally to the number of oxidized Met residues (human PrP has nine of them) (714). Interestingly, when PrP Met was substituted by methoxinine (a hydrophilic chemically stable Met) to mimic oxidized Met residues, a stronger aggregative behavior was observed. The opposite behavior was obtained when norleucine (a hydrophobic and chemically stable analogue that mimics reduced Met) was used (714), indicating that Met oxidation is the main trigger of PrP aggregation. Similar results were obtained in a yeast model of prion disease. Oxidative stress conditions promoted by incubation with hydrogen peroxide, menadione, or silencing of antioxidant enzymes increased the amount of aggregation of the yeast PrP. The presence of aggregates was accompanied by Met oxidation, and both were significantly reduced by overexpression of Met sulfoxide reductase (179).
Met oxidation was also described in amyloid aggregates composed of apoA-I (106, 716) or α-synuclein (282, 521). In the latter case, Met oxidation was shown to enhance the formation of soluble oligomers (757), arguably the main toxic form of aggregate in PD (300). The oxidation of each one of the four Met present in α-synuclein was shown to increase the polarity of the protein, elevating the degree of unfolding and preventing its organization into mature fibrils, locking aggregated α-synuclein in the soluble oligomeric form. Of note, the degree of fibrillation inhibition (i.e., the assembly of α-synuclein into amyloid fibrils) was found to be proportional to the number of oxidized Met residues (282, 521). Interestingly, dopamine-quinone, a product of dopamine oxidation, was shown to form adducts with α-synuclein and was also suggested to inhibit fibrillation (757). Synuclein oligomerization was also shown to be induced by peroxynitrite in the presence of CO2 or MPO in the presence of H2O2 (601), which are possible in vivo aggregation-inducing agents. Finally, the formation of toxic oligomers was also found to occur after the covalent modification of α-synuclein by 4-hydroxy-2-nonenal (HNE) (527).
Oxidation of SOD1 Trp and His residues was shown to occur and be involved in the aggregation of the wild-type human enzyme. While Trp oxidation was the result of the enzyme bicarbonate-dependent peroxidase activity (125, 749), His oxidation to 2-oxohistidine was obtained by MCO (CuCl2 in the presence of ascorbic acid) (536). More recently, Dantas et al. (143) reported that one of the products of cholesterol oxidation, cholesterol 5,6-secosterol aldehyde B, was able to covalently modify SOD1 Lys leading to significant aggregation of the enzyme. Of note, secosterol aldehydes were also shown to promote β-amyloid peptide (670) and α-synuclein (66) aggregation.
Different states of Cys oxidation were described as being involved in protein aggregation. For instance, the folding and stability of the FF domain of the yeast URN1, a model protein, were significantly impacted upon irreversible oxidation of a free Cys residue to sulfonic acid. Indeed, the oxidative modification of this single residue was sufficient to induce the protein aggregation into amyloid structures (407). The establishment of intermolecular adventitious disulfide bonds between two RRM2 domains of the transactive response DNA-binding protein 43 (TDP-43) was described as a determinant for dimerization, unfolding, and aggregation of the TDP-43 protein in vitro (127, 531). Interestingly, it was proposed that those adventitious bonds impair the ability of the RRM2 domain to refold, thus facilitating misfolding and ensuing aggregation (531).
2. Cellular consequences of aggregate accumulation
Highly oxidized and crosslinked proteins were shown to be a significant part of the brownish age pigment lipofuscin. However, the amino acids involved in such crosslinks were not described (280, 487). The mechanisms through which lipofuscin arises are still debated (445), but a failure in the cellular proteostasis system certainly contributes to its accrual (543). Importantly, lipofuscin was shown to incorporate different metals (up to 2%), which afford lipofuscin the ability to produce ROS, further oxidizing proteins and lipids and contributing to accumulation in cells (543). The ability of lipofuscin to inhibit the proteasome (281) exacerbates the accumulation of oxidized proteins and leads to the accumulation of other proteasome substrates such as the proapoptotic protein Bax, consequently facilitating apoptosis (525).
Protein aggregates may disrupt cell homeostasis by promoting the loss of function of the aggregated proteins and/or acquiring a toxic gain of function. The mechanisms through which aggregates exert their toxic effects are far from being completely understood. However, they seem to be dependent on the supramolecular organization of the aggregate (81, 95), the cellular environment, and the cell type. However, some features have been reported in a number of situations in which toxic aggregates are present, such as the failure of the ubiquitin/proteasome system (UPS) (43, 48, 280). Although the mechanisms through which protein aggregates inhibit the UPS are still elusive (281, 381), part of the inhibition may be due to the proteasome sequestration into aggregates, as observed after brain ischemia (223). UPS inhibition is adeterminant for the toxic effects of aggregates. Indeed, aggregate dissolution brought about by HSP 104 overexpression in yeast increased the activity of the proteasome without affecting its levels and normalized the degradation of proteasome substrates in aged yeast cells. Importantly, protein disaggregation allowed an increase in the replicative life span of yeast cells, indicating that the accumulation of aggregates shortens the longevity of this organism (16).
Damage to membranes was also reported as part of the toxicity mechanism of α-synuclein aggregates, although the molecular details are still unclear (300). Importantly, aggregation-prone proteins, which play essential roles in cellular functions, were shown to be sequestered by amyloid-like aggregates in HEK293T cells, which undoubtedly contribute to tissue aging as described in C. elegans (289).
Most of the data gathered so far on protein aggregates used in vitro systems. While valuable to uncover possible formation mechanisms and other crucial molecular details of aggregates, in vitro data may not reflect in vivo situations as aggregate accumulation in living organisms is the result of an interplay between the proaggregative stimuli and the activity of the proteostasis system; thus, it is essential to study protein aggregates in vivo. Those studies will undoubtedly contribute to the understanding of the relative contribution of aggregates to pathologic states and organismal aging, a topic that is actively being investigated.
Despite the vast array of evidence relating oxidation-induced protein aggregation to pathological states, it is important to emphasize that oxidation-induced protein aggregation may have protective or functional purposes. As an example, elegant works by Benjamin Tu's group showed that the yeast ortholog of ataxin-2 (Pbp1) coordinates the target of rapamycin complex 1 (TORC1) signaling and autophagy in response to mitochondrial activity (734). In conditions of intense mitochondrial respiration, Pbp-1 protein organizes into puncta composed of labile β-polymers. Upon addition of small concentrations of hydrogen peroxide, specific Met residues present on a low complexity domain of Pbp1 are oxidized, leading to β-polymer disassembly and subsequent TORC1 activation and autophagy inhibition. The reduction of the oxidized Met residues reverses the process (322). Interestingly, the organization of Pbp1 into labile β-polymers has a functional purpose and depends on the redox state of Met residues located at the low complexity domain, as shown by site-directed mutagenesis (322). For more examples of functional or protective roles of redox-dependent aggregation, the reader is referred to the review by van Dam and Dansen (141).
3. Human pathologies associated with oxidized protein aggregation
Age-associated cataract is a prototypical example of a disease caused by high-molecular-weight protein aggregates, which lead to lens opacity. Oxidative modification of amino acid residues of crystallin proteins was reported to play a significant role in cataract etiology (444). Indeed, ∼90% of protein sulfhydryl groups are lost in lenses with advanced cataracts (655). Moreover, oxidation of Trp 9 and Met 1 are among the major PTMs detected in aggregated α-crystallin, a protein present in all insoluble aggregates in lenses (606). Finally, Trp-Trp and Trp-Tyr crosslinks were recently characterized in human nuclear cataracts (Fig. 6A) (498).
The presence of protein aggregates in brain tissue is a hallmark of many NDs, such as Alzheimer's, Parkinson's, and Huntington's, among others (see for instance Fig. 6B). In the same way, high levels of oxidized biomolecules were reported in postmortem brains of patients with NDs (113). Considering the data obtained in vitro showing that oxidation is a destabilizing factor for protein folding, it is likely that protein oxidation contributes to the abnormal protein deposits found in the brains of patients with NDs. However, the etiology of NDs is still elusive, and no work to date has demonstrated a direct relationship between the oxidation of proteins and their abnormal deposit in the brain or the development of disease in humans. More studies are required to define the participation (if any) of oxidized proteins and their aggregation in the setting of NDs.
B. Proteolysis
The direct repair of oxidized proteins is limited, as herein described. During the 1980s, proteolysis was suggested as part of the intracellular defense against oxidized proteins (148). Initially, in vitro and in vivo evidence accumulated and indicated that the proteasome was the main protease that preferentially recognizes and degrades oxidatively modified proteins (316). Over the last two decades, other proteolytic systems were shown to remove oxidatively damaged proteins and protein aggregates, mainly the mitochondrial LON protease and autophagy. These three protein quality control (PQC) systems are discussed below.
1. Proteasome
The description of the polyubiquitination of proteins as a PTM that directs them to degradation in the early 1980s was a significant achievement to the understanding of proteostasis (121, 122, 275). Afterward, the discovery of the proteolytic complex responsible for the recognition and degradation of polyubiquitinated proteins, namely the 26S proteasome (26SPT), was reported (697). The 26SPT is composed of a central cylindrical-shaped catalytic unit called the 20SPT, coupled on one or both sides to the most abundant regulatory unit, the so-called 19S unit (316, 681). The 19S regulatory unit is responsible for recognizing polyubiquitinated proteins and their subsequent deubiquitination, unfolding, and translocation to the catalytic 20S unit. The polyubiquitination machinery, together with the 26S protease, is presently referred to as the UPS.
Initially, it was assumed that the 20SPT was permanently coupled to the 19S regulatory unit, degrading exclusively polyubiquitinated proteins. Some investigators challenged this proposition about the degradation of oxidized proteins. The 20SPT was mentioned as an independent particle called the “multicatalytic proteinase complex” (707) that was associated with the degradation of oxidized and nonubiquitinated proteins (150, 187, 316). Throughout the last decades, free 20SPT is increasingly accepted as an essential player in the degradation of oxidized proteins through a ubiquitin- and ATP-independent pathway. However, its role in vivo is not fully appreciated (159). A debate is still in place on whether oxidized proteins are polyubiquitinated or not before degradation. Indeed, there is no described biochemical feature that precludes oxidized proteins from being polyubiquitinated. Therefore, the question is whether the prompt degradation of oxidized protein substrates by the 20SPT could bypass their polyubiquitination and subsequent degradation by the 26SPT. In other words, there are no data to consider which mechanism would prevail on kinetic grounds. The mechanism proposed and explored in vitro for the recognition of oxidized proteins by the 20SPT relies on the exposure of hydrophobic patches due to structural rearrangement upon oxidation (149). A recent study with yeast cells exposed to oxidative stress revealed that 50% of highly abundant oxidized proteins were polyubiquitinated (405). However, this study does not discuss the fate of the remaining 50% of the oxidized proteins, suggesting that distinct mechanisms of proteolytic degradation of oxidized proteins can coexist.
Reported alterations of the UPS upon oxidative stress, such as the uncoupling of the 20S catalytic unit from the 19S regulatory unit, the increased expression of the regulatory unit 11S, and decreased polyubiquitination, are interpreted as adaptive responses that UPS uses to cope with oxidative shifts (344) and highlight the role of the 20SPT catalytic unit as the main player in the degradation of oxidized proteins. Nonetheless, all the investigations in the field use oxidative challenges to cells that do not necessarily resemble aging or degenerative processes, in which protein oxidation proceeds at a slow pace. It has been very challenging to design experimental conditions related to these human pathologies. Indeed, a large amount of data are available, showing that proteasomal activation is associated with extended life span (117). On the other hand, proteasomal activity declines with age and in age-related pathological conditions, with consequent accumulation of oxidized proteins that correlate with a decrease in the expression of antioxidant enzymes in aged cells (271, 493, 520). The decline of proteasomal activity in those conditions is widely reported. However, it is unclear whether it is because of its decreased expression or increased PTMs, including oxidative damage. Major oxidative modifications reported are modifications by HNE, the formation of carbonyl moieties, and glycation of the 20SPT catalytic unit, implying inhibition of protein degradation (35). The proteasome likely has a high recycling rate that is decreased during aging and in age-related pathologies. The most significant alteration to compromise protein degradation under these conditions would be the decreased proteasome expression rather than inhibition through oxidative modifications.
In conclusion, regardless of the underlying mechanism, the proteasome is a critical component of the quality control systems involved in the degradation of oxidized and misfolded proteins.
2. Lon protease
Mitochondria are the primary intracellular source of O2 •− and H2O2 (197, 455). As a consequence, the mitochondrial proteome is continuously exposed to oxidative damage. Oxidatively damaged mitochondrial proteins represent a risk to the organelle and need to be repaired or degraded. In human mitochondria, the LONP protease (also known as LomP1 or simply LON) is considered the main component of the mitochondrial PQC system. It plays a central role in the degradation of oxidatively modified proteins (683, 684).
LON is a highly conserved ATP-dependent protease localized in the mitochondrial matrix compartment. While specific membrane-integrated proteases (namely, m-AAA protease and i-AAA protease) cleave damaged mitochondrial proteins of the inner membrane, the LON protease preferentially degrades soluble proteins in the matrix (166). The LON substrates include nonassembled polypeptides that arise from incorrect folding during mitochondrial protein biogenesis and proteins damaged by oxidants (633). The yeast ortholog of the mammalian LON, Pim1, has been widely studied as a model to uncover LON protease activities. Pim1 is a large homo-oligomeric protein complex composed of seven 100 kDa subunits organized in a ring-shaped heptameric structure (611). The proteolytic domain responsible for the hydrolysis of peptide bonds is located inside the internal proteolytic cavity or chamber, shielded from the aqueous environment. Therefore, similar to other proteases such as the proteasome complex, protein degradation by LON requires prior unfolding and translocation of the client protein into the proteolytic cavity. This task is performed by the ATPase domain situated at the edges of the complex (516, 684).
Bender et al. (44) investigated the role of Pim1 in the maintenance of mitochondrial proteostasis under oxidative stress conditions. By using a proteomic approach, the authors identified a set of mitochondrial proteins that are mainly degraded by Pim1 in response to the exposure of isolated mitochondria to three distinct oxidative conditions: H2O2, menadione, and succinate combined with antimycin A (an inhibitor of electron transfer from ubiquinone to complex III). Although some proteins were found to be degraded only under a single type of oxidative treatment, Fe-S cluster-containing enzymes were the primary targets of degradation. It was proposed that after the oxidative modification of their Fe-S clusters, the corresponding polypeptides become destabilized and thus more prone to degradation by Pim1 (44). This observation is consistent with previous studies showing that Fe-S clusters containing enzymes are major targets of oxidative modifications (42, 68, 89, 400).
However, protein oxidative damage is not restricted to Fe-S-containing enzymes. The mechanism proposed is based on the generation of partially unfolded protein regions upon oxidation, which are recognized by LON (41, 306, 400, 484). Hence, the conformational state of a polypeptide chain serves as the primary criterion for its selection as a protease substrate. This idea is further supported by a study that revealed that Pim1 requires an unstructured segment longer than 50–60 amino acids at the N-terminal to initiate protein degradation (306). These unstructured segments can access the interior of the Pim1 proteolytic chamber, allowing degradation to occur. In this scenario, tightly folded substrates representing native enzymes remain mostly resistant to proteolysis. As a result, the proteolysis reaction is selective to unfolded or damaged polypeptide chains.
As mentioned above, LON plays a crucial role in the control of mitochondrial proteostasis. This statement is highlighted by the fact that homozygous Lomp1 knockout mice exhibit embryonic lethality (528). It has been known for a long time that LON activity declines in aged animal models, which is accompanied by the accumulation of oxidatively modified proteins in the mitochondria (70, 360). This decline may contribute to the progress of various age-associated diseases, such as NDs (69, 668).
3. Autophagy
Although autophagy is mainly studied as a catabolic process involved in the degradation of intracellular components during starvation conditions, it is now clear that this process participates in many other pathways necessary for cellular homeostasis (191, 349). In addition to a nonselective process of self-consumption (nonselective autophagy), it has been demonstrated that some cargoes can be degraded with high selectivity in response to diverse conditions (335). Numerous studies have reported the selective autophagic degradation of damaged or superfluous organelles, including mitochondria (321, 468, 469, 743), peroxisomes (165, 192, 653, 753), and ER (330, 430, 467).
Although the degradation of organelles is the best-described type of selective autophagy to date, some evidence indicates that intracellular protein aggregates can also be selectively degraded in a process known as aggrephagy (59, 293, 387, 388, 491, 569). The selective incorporation of protein aggregates into the autophagosome requires selective autophagy receptors (SARs) that bind simultaneously to the protein aggregate cargo and the core components of the autophagic machinery. Examples of these proteins include the p62/SQSTM1 (sequestosome 1), the next to BRCA1 gene 1 (NBR1), and optineurin (OPTN) (222, 311, 618).
The most studied ubiquitin-dependent SAR in the context of aggrephagy is p62. For many years, p62 was considered a common component of ubiquitin-positive protein aggregates, such as Mallory and Lewy bodies, neurofibrillary tangles (β-amyloid), and Huntingtin aggregates (335, 353, 354, 460, 748). Subsequently, it was demonstrated that the recruitment and oligomerization of p62 into ubiquitinated aggregates are required for efficient cargo degradation by the autophagic machinery (59). The trafficking of ubiquitinated protein aggregates into autophagosomes depends on specific motifs present in the p62 structure. This protein simultaneously interacts with the MAP1LC3/GABARAP (ATG8 in yeast cells) family of proteins (a core component of autophagosome membrane) and ubiquitin via the LC interacting region (LIR) motif and ubiquitin-binding (UBA) domain, respectively (491). As a consequence of these interactions, the ubiquitinated protein aggregates are selectively sequestered and delivered to the autophagosome.
In parallel to the description of p62, numerous other SARs that recognize ubiquitinated cellular cargos and LC/GABARAP (Atg8) were described, including the Cue5 from yeast cells, which is required for clearance of aggregation-prone proteins (388). The fact that these SARs recognize ubiquitin molecules attached to cargos led researchers to propose that ubiquitin is the primary signal that cells use for discriminating which cargo is degraded by autophagy (329, 336, 345, 556). Considering that the UPS also uses ubiquitin as a degradation signal for substrates, it was initially thought that both pathways could degrade the same type of substrates. However, it is now acknowledged that they exhibit a high degree of specificity involving selective enzymatic reactions and discriminatory receptors, ensuring delivery of the correct substrate to either the proteasome or lysosome. Substrate size plays an important role in pathway choice. Soluble monomeric proteins are mostly conducted to the UPS. In contrast, larger protein complexes such as protein aggregates, which are unable to enter into the narrow channel of the proteasome and thus resistant to proteasomal degradation, are degraded by the autophagic machinery (518). Moreover, the arrangement of the ubiquitin chains attached to the cargo might play an important role in substrate fate. While Lys 48-linked ubiquitination preferentially targets substrates to proteasomal degradation, Lys 6-linked chains serve as a recognition signal for multiple autophagy receptors (175).
In addition to its function as an SAR, p62 can also escort ubiquitinated substrates to the proteasome by acting as a proteasomal shuttle factor via its Phox and Bem1p (PB1) domain (556). The structural architecture of p62 determines which pathway is used by p62-delivered substrates. While p62 dimerization leads substrates to the proteasome, oligomerization of p62 into ubiquitinated cargos promotes substrate channeling to autophagy (722). The underlying mechanisms that control the switch of this receptor between the dimeric and oligomeric states have been recently unraveled. Under a condition termed ubiquitin stress (a condition characterized by an accumulation of free ubiquitin, as observed upon heat shock or prolonged proteasome inhibition), p62 is ubiquitinated, which disrupts the dimerization of its UBA domain and facilitates its ability to recognize polyubiquitinated cargoes for selective autophagy (506). This model is also supported by a recent work showing that oxidation of p62 redox-sensitive Cys residues in response to oxidative stress induces the formation of disulfide-linked conjugates, which facilitates p62 oligomerization with consequent induction of the autophagy pathway (98). Importantly, oxidized p62-dependent autophagy activation seems to be essential for cell survival in the context of oxidative stress associated with age-related diseases.
In conclusion, the data cited above suggest a functional connection between UPS and autophagy pathways toward ubiquitinated protein degradation. Autophagy would be a compensatory mechanism when the proteasome is overwhelmed or inhibited (175, 415, 518).
Disturbances in PQC pathways are clinical hallmarks of aging and many age-related diseases, including NDs. There is a decline in these pathways during aging with direct consequences, including damaged protein accumulation. On the contrary, accumulating evidence indicates that interventions that stimulate the UPS and autophagy can prolong the maintenance of a healthy proteome and consequently increase longevity (368). Recent findings in the same direction are discussed in Section V.B.2.
Figure 11 illustrates the fate of oxidized proteins, including aggregation and proteolysis.
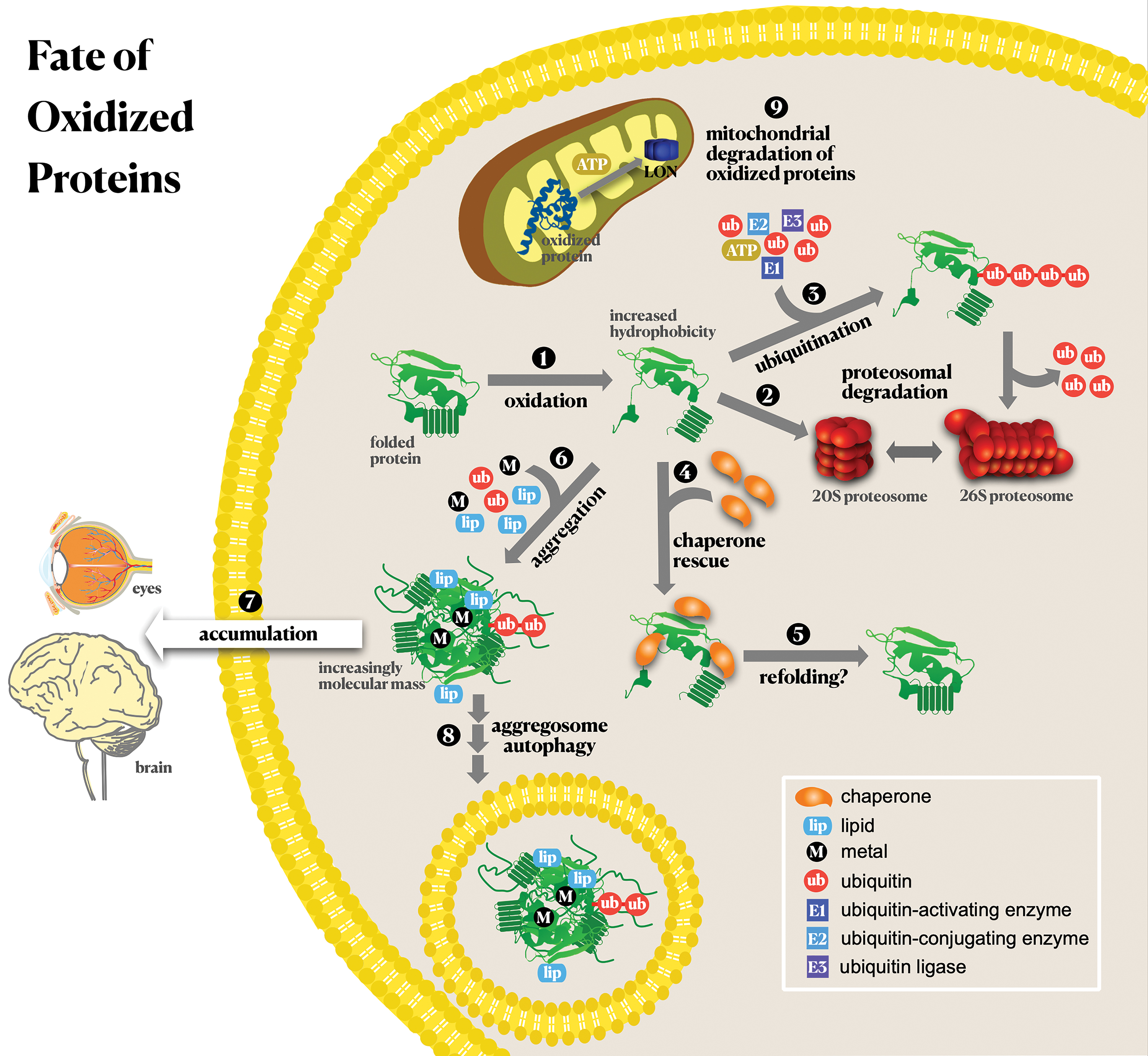
V. Conclusions
A. Oxidatively modified proteins as biomarkers of pathological conditions
The concern in the literature on the existence of redox biomarkers has occupied the attention of many investigators. It is beyond the scope of the present review to cover redox biomarkers. Our interest is toward the search for oxidized proteins as markers of human pathological conditions.
As discussed throughout the present review, oxidized proteins are frequently associated with several diseases. Some clinical investigations focused on the levels of specific oxidative protein modifications in a set of diseases, for example, carbonyl protein levels and AGEs. However, none of those works could conceptually establish any oxidized protein as a biomarker of any of the pathologies investigated. In the present review, we reproduce Frijhoff et al.'s definition of a biomarker, based on the World Health Organization, “as any substance, structure, or process that can be measured in the body or its products and influence or predict the incidence of outcome or disease” (204). Consequently, a clinically useful biomarker must be able to meet the criteria, such as (i) specificity for a particular disease (diagnostic), (ii) prognostic value, and (iii) correlate with disease activity. At the present moment, hemoglobin glycation is the only biomarker utilized in the clinic for diagnostic purposes and to follow the progress of diabetes. However, hemoglobin glycation is mainly related to increased levels of glucose instead of oxidative stress.
Table 3 summarizes the potential association of specific proteins, when oxidized, with human pathologies. The criteria to build up Table 3 were to select only those oxidatively modified proteins found in human samples of pathological conditions. However, the table does not contemplate all of them (e.g., proteins only detected by immunoblotting). Reports of HNE-adducts associated with human pathologies and verified in human samples are reported in Table 1. Many of the proteins shown in Table 3 were also investigated in animal and cellular models corroborating human findings and in vitro, where other oxidative modifications were reported. Nonetheless, one should consider that the identification of protein oxidative modifications is dependent on methodological approaches. While some methodological tools facilitate their detection in human samples, for example, immunological approaches, many other modifications verified in vitro cannot be reliably investigated in human samples. Mass spectrometric approaches have been developed to allow a broader spectrum of modified amino acid side chains, as shown in many sections above. As depicted in Table 3, most of the proteins are related to neurodegenerative and cardiovascular diseases, most probably because these pathologies are widely investigated. The propensity of some proteins to undergo widespread oxidation, generating several oxidative modifications (e.g., albumin, ApoA and ApoB proteins, β-amyloid, LDL, α-synuclein; SODs), is noteworthy. It is also interesting to point out that protein nitration and Tyr-Tyr crosslinks are frequently identified. However, it is unclear if this is because they are investigated more often or because their identification by immunological detection is facilitated. The proteins shown in Table 3 might be considered potential markers of pathological conditions; however, they are neither easily identified for diagnostic purposes nor indicative of disease progression since quantitative and stoichiometric data are still lacking.
Oxidized Proteins Suggested As Potential Biomarkers and Players in Human Pathologies
ALS, amyotrophic lateral sclerosis; apoA-I, apolipoprotein A-I; BB-CK, brain creatine kinase; CA-II, calcium channels II; CYP27A1, stearoyl 27-hydroxylase; GDH. glutamate dehydrogenase; IsoLG, isolevuglandin; MDA, malondialdehyde; MM-CK, myofibrillar creatine kinase; MnSOD, manganese superoxide dismutase; PD, Parkinson's disease; SERCA, sarcoplasmic reticulum Ca2+-ATPase; SOD1, superoxide dismutase-1; TIM, triosephosphate isomerase; VDAC, voltage-dependent anion channel.
B. Prevention of protein oxidation
It is well established that the accumulation of oxidized proteins is harmful to the cells. Indeed, protein oxidation triggers proteotoxicity, which might be the main etiologic contribution to many pathological states. Following this train of thought, prevention and repair (only in the case of sulfur-amino acids) of protein oxidation and the elimination of oxidized proteins would be the most effective defense against pathological processes. On the contrary, functional modulation of some redox-active sulfur-centered proteins influences catalysis, cell signaling, and metabolic regulation (Section IV). Therefore, a better understanding of these mechanisms may lead to means of controlling redox homeostasis, perhaps using drugs that interfere with specific metabolic points of redox homeostasis maintenance.
Another consideration is biological plasticity in the onset of antioxidative response, where hormesis would play an important role. Hormesis is a dose/response phenomenon characterized by low-dose stimulation and high-dose inhibition (91) of many biological processes. In redox metabolism, hormesis is related to low doses of oxidants promoting stimulatory effects of the antioxidative and repair responses (91, 552). In contrast, high doses are related to macromolecule damage and consequent loss of cellular homeostasis. In agreement with this concept, supplementation with antioxidants was shown to preclude significant consequences of oxidant production, such as those promoted by physical exercise (553). While hormesis can have undesirable effects, for example, anticancer drugs, it has been considered useful in pathologies related to the oxidative spectrum.
Below, some points on the prevention of protein oxidation are explored.
1. Antioxidants
Prevention of macromolecule oxidation relies on the maintenance of the redox homeostasis, whose physiological regulation is not yet completely understood. One of the most explored tools to counteract redox imbalance is the utilization of antioxidants, particularly natural sources. A search in the PubMed platform utilizing the term antioxidant therapy returned more than 11,800 reviews among 84,000 publications in the last decade. By taking only publications based on Human Randomized Controlled Trials presenting antioxidant therapy in the title in the last decade, about 4500 results emerged. In these publications, a set of nutritional proposals and specific compounds or natural products were tested and proposed as therapies against several pathologies. There is no consensus established by the scientific community on the real benefits of such therapies. The idea of the importance of healthier nutritional intake is gaining more and more traction, including advisements by worldwide public health services to the general and specific populations (e.g., scholars) to increase the intake of natural products (fruits and vegetables), even though the main goal is the control of obesity, diabetes, hypertension, and others. It is important to emphasize that such recommendations are not yet based on systematic scrutiny on their benefits. It is out of the scope of this review to explore the subject.
Noteworthy is the rereading of natural “antioxidants” based on the hormesis phenomenon. Instead of antioxidant protection, many phytochemicals induce cellular stress response (454). This is based on the fact that humans consume many toxic phytochemicals from natural sources at very small doses within the hormetic range. For many years it was assumed that the free radical scavenging property of phytochemicals would be responsible for their benefit as antioxidants. However, many epidemiological studies have failed to demonstrate the benefits of dietary supplementation. Instead, much evidence suggests that hormesis underlies the health benefits of phytochemicals (91). A general hypothesis is that these phytochemicals would induce adaptive cellular stress response pathways that induce the expression of genes encoding antioxidant enzymes, chaperones, detoxifying enzymes, and other protective proteins. Nevertheless, more experimental data are needed for a better understanding of the role natural products play in redox biology.
2. Elimination of oxidized proteins and aggregation prevention
Besides the loss of function, one of the most critical outcomes of protein oxidation is the proteotoxic event of aggregation, as discussed above (Section IV.A). Aggregation takes place if oxidized proteins are not promptly eliminated. Three intracellular systems accomplish the degradation of oxidized proteins: proteasome based or not on the labeling by polyubiquitin chains, autophagy, and the mitochondrial LON protease. However, those systems are usually compromised under the same pathological conditions; thus, the pool of oxidized proteins increases. There is an emerging field of investigation for proteasome activators as pharmacological tools (128, 313, 676, 677), but no drug has been clinically tested so far.
In the last years, a powerful technique has been developed based on the induction of protein degradation by proteolysis-targeting chimeras, namely PROTAC (84, 120, 464). The methodology is based on the utilization of small bifunctional molecules capable of interacting with the protein substrate and ligases to induce the polyubiquitination of the protein substrate, directing it for proteasomal degradation. Polyubiquitination is a three-step process where the final one is the PTM of the protein substrate by ubiquitin through E3 ligases that transfer ubiquitin to an Lys residue of the substrate either directly or indirectly. The PROTAC-mediated degradation is based on targeting the E3 ligase-specific substrate by degraders that combine affinity for both substrate and E3 ligase. This approach promotes the degradation of selected proteins as E3 ligases are substrate specific. Now, it has become an important tool in both experimental biology, for the understanding of cell regulation and signaling, and in drug discovery. Several PROTAC studies on the degradation of aggregate-prone proteins have been conducted (118, 215, 294, 589). Moreover, the same technique has also been used to direct proteins to the lysosome (628). This approach seems robust in the way it targets proteins considered undruggable so far and introduces a new perspective on pharmacological interventions (120). In principle, this approach might be extended to the degradation of any protein. In the case of oxidized proteins, as important as the identification of the oxidative process these proteins undergo is identifying the proteins prone to oxidation in all pathologies where they accumulate. That might be a possibility to intensify their degradation and establish either their pathological role or avoid proteotoxicity.
Chaperone-like molecules might be an important tool for avoiding the aggregation of oxidized proteins. Although numerous proteins were described to have moonlighting chaperone-like activity (315, 593, 624), some were described to prevent protein aggregation in an oxidative scenario. One example is the 19-amino acid sequence derived from α-crystallin, which prevents the aggregation of UV-irradiated or H2O2-treated γ-crystallin proteins in vitro (351). The same group of researchers modified the original 19-amino acid sequence giving rise to a set of short peptides with varying degrees of chaperone-like activity, some of which have improved activity compared with the original sequence (535). Peptides derived from α-crystallin are taken up by primary human fetal retinal pigment epithelium cells in culture and were able to prevent apoptosis induced by H2O2 (605). In vivo assays also demonstrated that the peptides prevented selenite-induced cataracts when administered intraperitoneally to rats (463), evidencing the therapeutic potential of these molecules.
Another compound with interesting chaperone-like properties is polyphosphate. Polyphosphate is made up of a thousand phosphate monomers linked through phosphoanhydride bonds. Numerous functions were reported for polyphosphate in a wide range of organisms, including humans (723), and some authors argue that polyphosphate is a primordial chaperone (238). Both in vitro and in E. coli exposed to conditions that lead to oxidative protein unfolding, polyphosphate was able to stabilize those proteins, significantly decreasing the amount of protein aggregation (238). Moreover, polyphosphate significantly accelerates the formation of mature fibrils from amyloid proteins in vitro, effectively inhibiting amyloid cytotoxicity (135). While polyphosphate has significant effects in vitro, its therapeutic potential in vivo remains unexplored.
C. Emerging approaches to integrate current knowledge on oxidative processes, protein oxidation, and human pathologies
Systematic studies on protein oxidation started 40 years ago (see Section I). Over the decades, the knowledge on the nature of protein oxidation and the mechanisms involved has made significant advances. Despite the presence of oxidized proteins in many human pathologies, the literature poorly describes either the participation of specific oxidized proteins in the time course of each pathology or their role as etiological agents. This observation is not surprising because the quantification of oxidative PTMs under pathological conditions and in human samples remains limited. Also limited is the knowledge of how specific oxidative protein modifications affect protein structure and function. In addition, it is likely that some protein modifications, such as crosslinks, remain undiscovered.
After the birth of functional genomics in the 1990s and the explosion in computing power, computational SB has become a useful and reliable tool for modeling and connecting components of biological pathways. Through quantitative measures and data integration with mathematical models, SB establishes networks of multiple components simultaneously (634). Currently, the massive increase in data from omics, together with the increased performance of bioinformatics, culminated with the intensification of SB applications in cell biology. In the case of redox biology, the goal of identifying interactions among redox processes, oxidized biomolecules, and pathological conditions depends on the integration of redox metabolomic, lipidomic, and proteomic (oxidized lipids and proteins, respectively) databases compiled with data from tissues or cellular models of a specific condition, for example, the redox omics (metabolome; oxidized lipids and proteins) of the brain during aging. Despite considerable data already available in the field for many models, the use of SB in redox biology is limited. Another notable approach gaining relevance in biology and medicine is artificial intelligence (AI) (338). Here freely defined, AI is an algorithm-based analysis of a set of data from different sources culminating with conclusions on the subject under analysis otherwise hardly achieved with conventional analytical approaches. Of note, a powerful, successful application of AI has been in medical diagnostics, including guidelines for treatment protocols. Indeed, AI is bringing new perspectives for investigations in every scientific field.
In conclusion, to establish more reliable connections between redox biology (signaling, regulation, damage) and a specific pathological condition at the functional and causal level, future studies should deepen the basic knowledge, emphasize quantification, create a network of information, and use emerging approaches.
Footnotes
Acknowledgment
Special acknowledgment to Prof. Ohara Augusto (OA) for the critical reading of the review.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
M.D., O.A., F.M.C., F.G., S.M., and L.E.S.N. are members of the CEPID Redoxoma supported by São Paulo Research Foundation (FAPESP) grant 2013/07937-8; L.M.R., L.T., and A.D. acknowledge the Comisión Sectorial de Investigación Científica, CSIC, Universidad de la República, Programa de Desarrollo de las Ciencias Básicas, PEDECIBA, and Agencia Nacional de Investigación e Innovación, ANII, Uruguay; SM is also supported by CNPq grant 424094/2016-9; E.J.H.B. and C.S. are supported by FAPESP grant 2017/22501-2; E.J.H.B., L.E.S.N., S.M., and O.A. acknowledge CNPq, grants: 306460/2016-5, 310224/2017-9, 309083/2017-6, and 301597/2019-9, respectively. F.G. and R.N.B. are also supported by FAPESP, grants 2017/09443-3 and 2016/11724-8, respectively. M.D. is also supported by Butantan Foundation.