
Abstract
Aims:
Oxidative stress and mitochondrial dysfunction play a role in the process of skin photoaging via activation of matrix metalloproteases (MMPs) and the subsequent degradation of collagen. The activation of nuclear factor E2-related factor 2 (Nrf2), a transcription factor controlling antioxidant and cytoprotective defense systems, might offer a pharmacological approach to prevent skin photoaging. We therefore investigated a pharmacological approach to prevent skin photoaging, and also investigated a protective effect of the novel mitochondria-targeted hydrogen sulfide (H2S) delivery molecules AP39 and AP123, and nontargeted control molecules, on ultraviolet A light (UVA)-induced photoaging in normal human dermal fibroblasts (NHDFs) in vitro and the skin of BALB/c mice in vivo.
Results:
In NHDFs, AP39 and AP123 (50–200 nM) but not nontargeted controls suppressed UVA (8 J/cm2)-mediated cytotoxicity and induction of MMP-1 activity, preserved cellular bioenergetics, and increased the expression of collagen and nuclear levels of Nrf2. In in vivo experiments, topical application of AP39 or AP123 (0.3–1 μM/cm2; but not nontargeted control molecules) to mouse skin before UVA (60 J/cm2) irradiation prevented skin thickening, MMP induction, collagen loss of oxidative stress markers 8-hydroxy-2′-deoxyguanosine (8-OHdG), increased Nrf2-dependent signaling, as well as increased manganese superoxide dismutase levels and levels of the mitochondrial biogenesis marker peroxisome proliferator-activated receptor-gamma coactivator (PGC-1α).
Innovation and Conclusion:
Targeting H2S delivery to mitochondria may represent a novel approach for the prevention and treatment of skin photoaging, as well as being useful tools for determining the role of mitochondrial H2S in skin disorders and aging. Antioxid. Redox Signal. 36, 1268–1288.
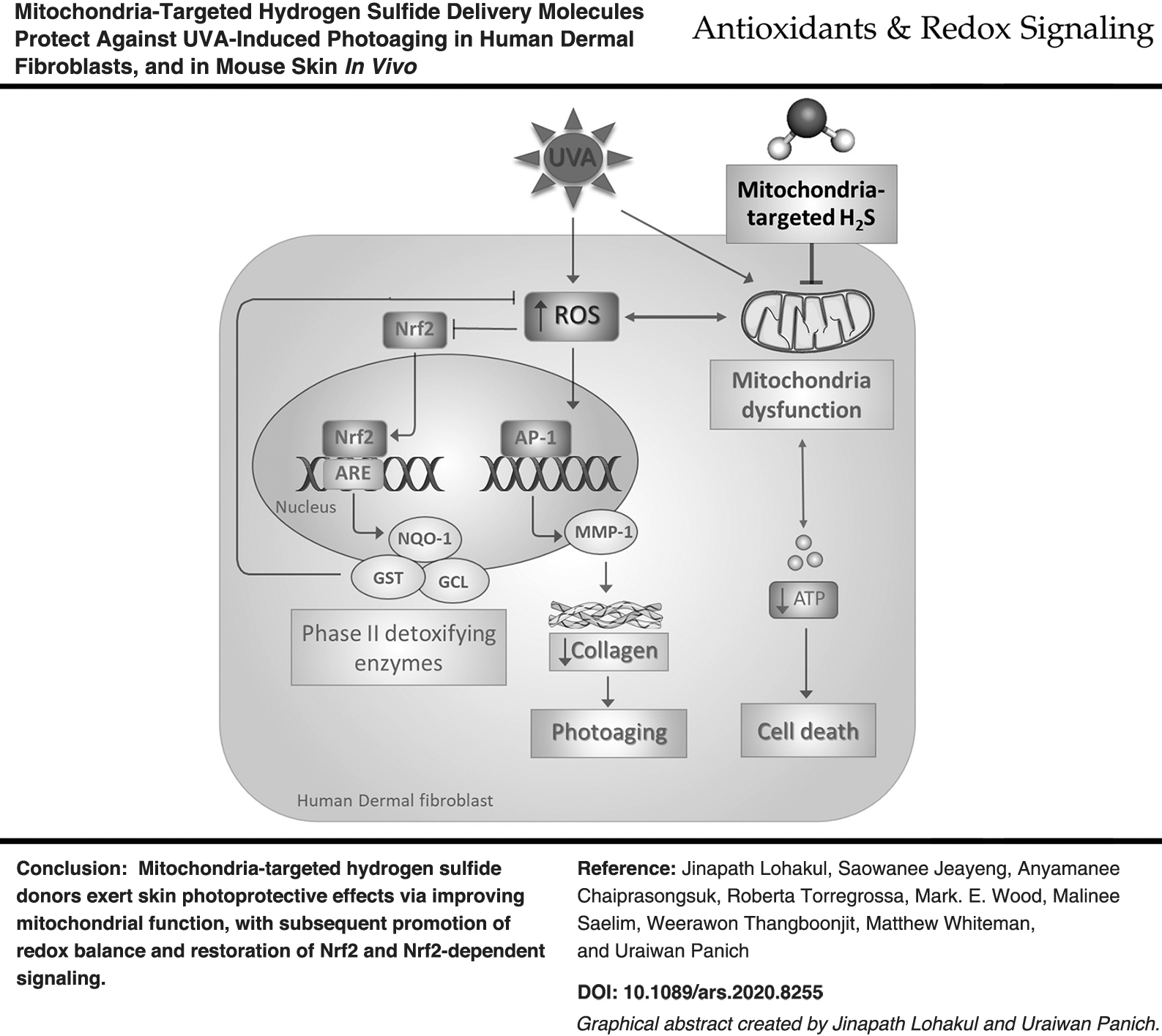
Introduction
Exposure to ultraviolet radiation (UVR/ultraviolet A light [UVA]) is the primary factor of extrinsic skin photoaging, or photodamage responsible for the majority of dermatological problems associated with age, predominantly through mitochondrial dysfunction, oxidative stress, and excessive extracellular matrix degradation (13, 56, 62). Mitochondrial dysfunction and the resulting oxidative stress can precipitate the aging process in skin by upregulation of matrix metalloprotease (MMP)-1 activity, and dampening nuclear factor E2-related factor 2 (Nrf2)-mediated cellular defenses (13, 14, 33, 42). As such, mitigating or reversing mitochondrial damage may offer an approach to protect against (or at least partially delay) aging, and compounds capable of promoting mitochondrial integrity and Nrf2 activity would potentially exert photoprotective effects against UVA-induced skin damage (4, 9).
Innovation
Prolonged skin exposure to ultraviolet A light (UVA) radiation is a risk factor for premature skin aging, actinic keratosis, and solar elastosis, as well as basal and squamous cell carcinomas and melanoma and nonmelanoma skin cancers. As such, there is a need to protect skin from the detrimental effects of UVA exposure and the resulting skin lesions. Molecules such as AP39 and AP123, and future related compounds, may offer a novel approach to investigate how mitochondrial sulfide protects skin from UVA radiation. This could lead to novel therapeutic strategies to prevent and/or mitigate skin disorders and premature aging.
Mitochondria are a primary source of cellular oxidants in skin exposed to UVR/UVA and specifically targeting/protecting mitochondria may represent a novel approach to prevent, limit, and/or reverse UVR/UVA-induced damage and help rejuvenate skin (5, 64) [reviewed in Brand et al. (9)]. We therefore postulated that one novel approach to do this would be through modulation of mitochondrial hydrogen sulfide (H2S). H2S is a “third gasotransmitter” [comprehensively reviewed in Wang (74, 75)] alongside carbon monoxide (CO) and nitrogen monoxide (•NO). It is endogenously synthesized in a variety of human and mammalian cells from cystathionine-γ-lyase (CGL) and cystathionine-β-synthase (CBS) from sulfur containing amino acids such as cysteine and homocysteine, and by 3-mercaptopyruvate sulfurtransferase (3-MST)/cysteine aminotransferase or 3-MST/D-amino acid oxidase from 3-mercaptopyruvate [reviewed in Coavoy-Sanchez et al. (17)]. CGL and (25) CBS (23, 66) are cytoplasmic enzymes but under conditions such as oxidative stress, translocate to mitochondria and generate H2S, whereas 3-MST is predominantly mitochondrial (17, 75), suggesting that mitochondria are a key target for cellularly generated H2S as a mechanism to protect against cellular injury. Human and dorsal mouse skin can generate H2S through one or more of these enzymes (17, 58) although the cellular characterization of each enzyme is not well understood. Punch biopsies show CGL and 3-MST messenger ribonucleic acid (mRNA) and protein expression and activity (28, 39). H2S production has been observed in isolated human keratinocytes containing CGL and CBS (39, 41), normal human dermal fibroblasts (NHDFs) containing predominantly CBS, as well as normal human epidermal melanocytes containing detectable levels of CGL, CBS, and 3-MST (52, 60). Pharmacological inhibition or genetic silencing of H2S generating enzymes exacerbates mitochondrial dysfunction, cellular oxidant generation, and tissue injury (10, 24, 38). In addition, induction of H2S synthesis has been shown to be cytoprotective and anti-inflammatory in several animal models, at least in part, through preventing oxidative stress (31, 71) and mitochondrial dysfunction (7 –9) induced by various stress stimuli including UVR/UVA exposure (51, 63). Therefore, given the detrimental effects of UVR/UVA on mitochondria and the predominantly mitochondrial effects of H2S on cell protection (53, 67), we investigated novel mitochondria-targeted slow release H2S delivery molecules we have developed [AP39 (40) and AP123 (26)] on UVA-induced cellular injury using NHDF in vitro, and a rodent model of UVA-induced photoaging in vivo.
Results
AP39 and AP123 prevented UVA-induced cytotoxicity in vitro
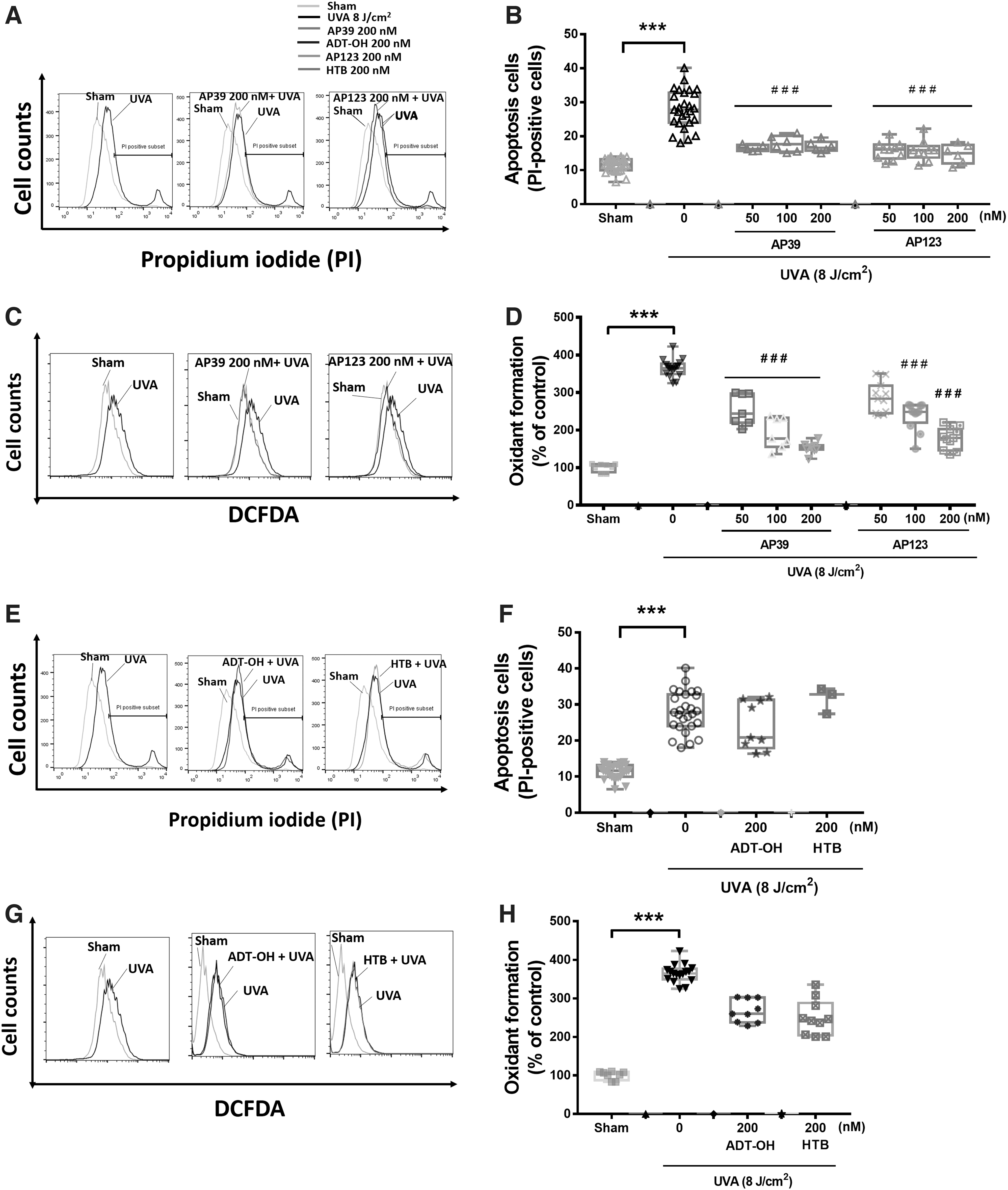
Control experiments using AP39 and AP123 or their respective control compounds (ADT-OH and 4-hydroxythiobenzamide [HTB]) showed that at the highest concentration used in the in vitro part of our study (200 nM), each compound alone did not exhibit any significant cytotoxicity or oxidative stress alone (Supplementary Fig. S1). Preliminary investigations showed that UVA treatment of NHDFs caused a concentration-dependent increase in oxidative stress and cellular damage and MMP-1 activation, and from these experiments (data not shown) an ultraviolet (UV) exposure of 8 J/cm2 was used for further studies. Figure 1 shows UVA treatment induced significant (p < 0.001) apoptotic cell death (Fig. 1A, B) and cellular oxidant production (Fig. 1C, D). AP39 and AP123 inhibited UVA-induced apoptosis (Fig. 1A, B) and cellular oxidant generation (Fig. 1C, D) in a concentration-dependent manner. In contrast, nontargeted H2S donor control molecules (ADT-OH and HTB), respectively, had no effect on these parameters (Fig. 1E–H).
AP39 and AP123 attenuated MMP-1 activity and collagen degradation in NHDFs in vitro
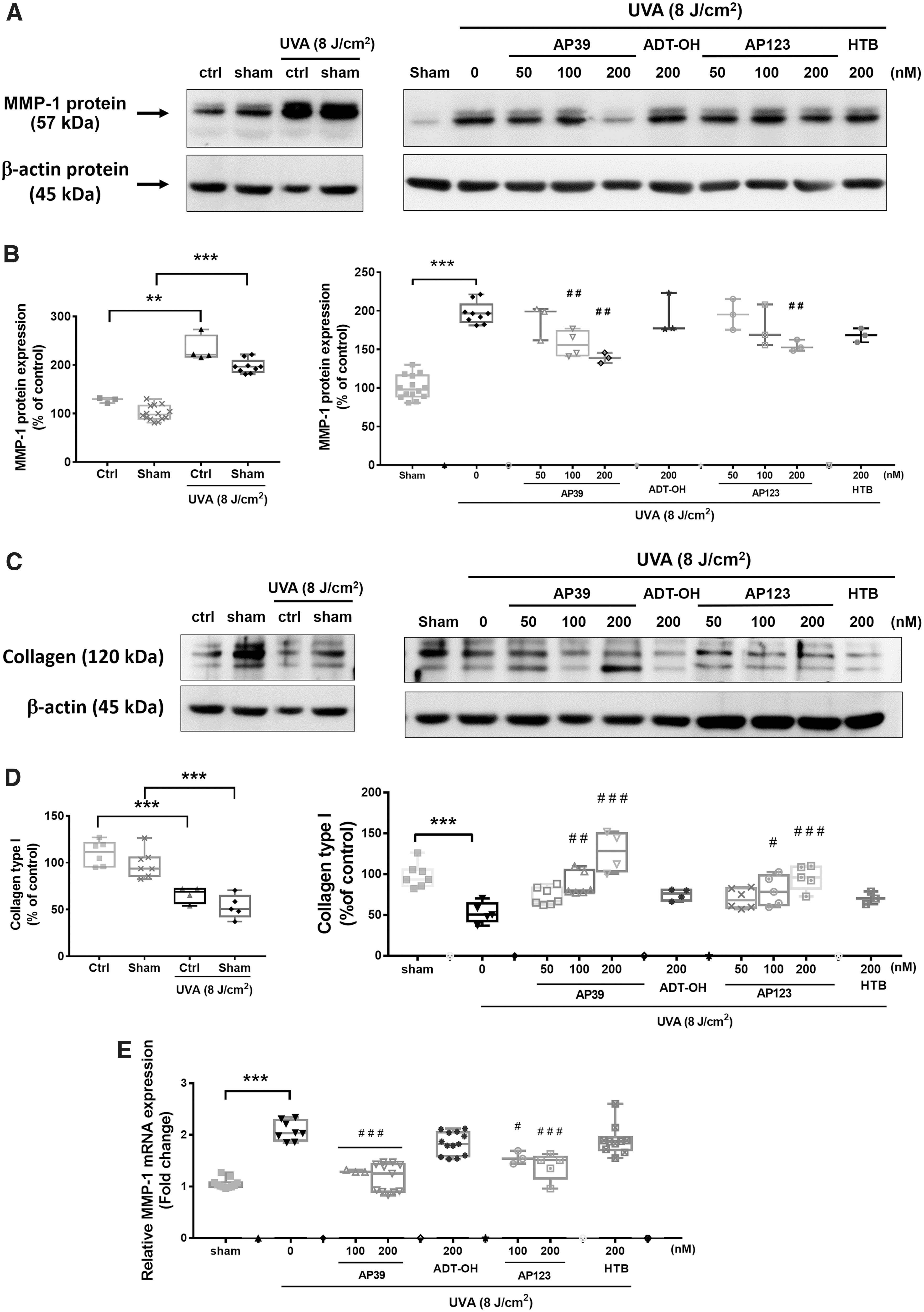
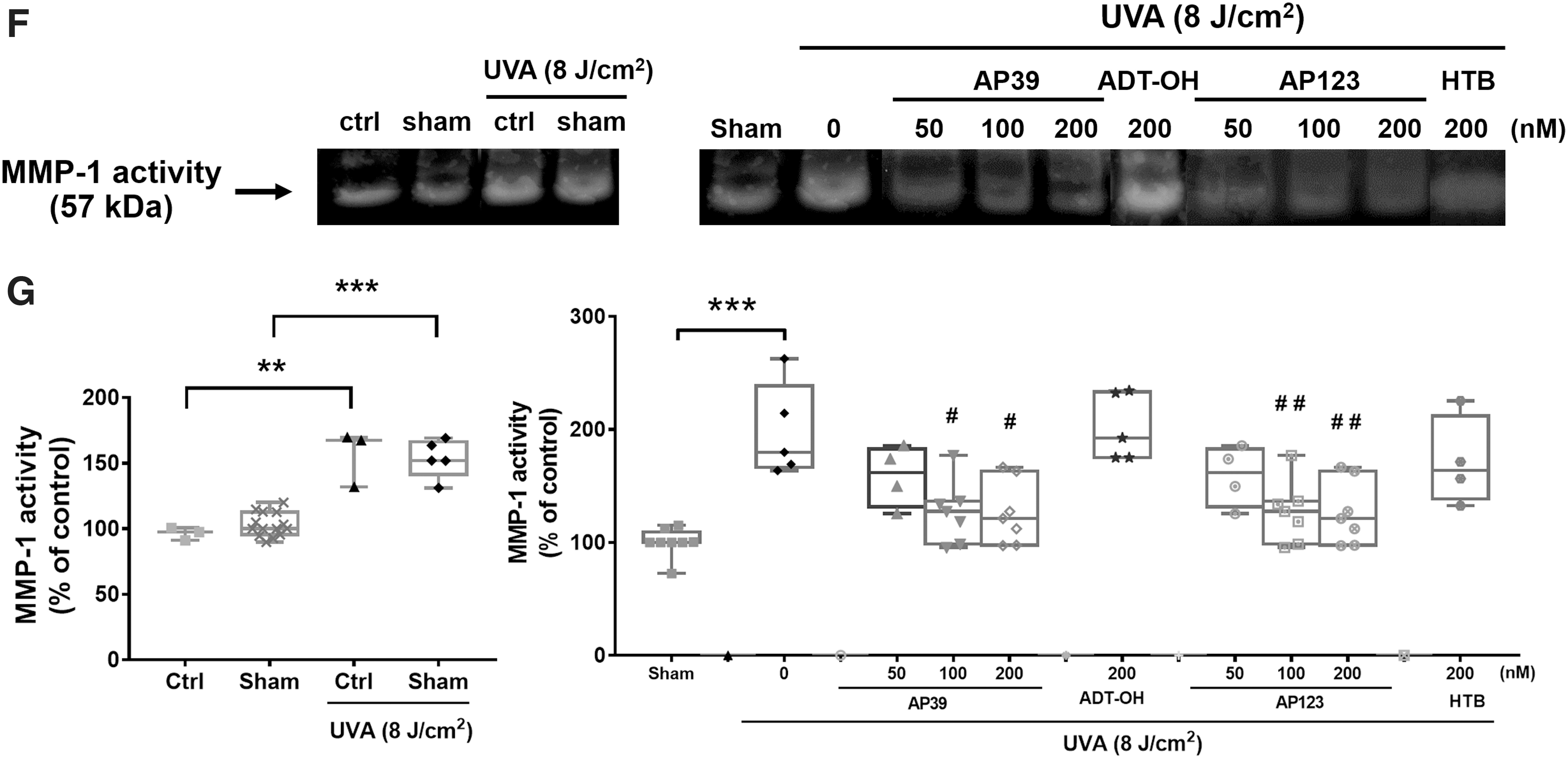
NHDFs were exposed to UVA (8 J/cm2) and after 24 h, clear induction of MMP-1 protein (Fig. 2A, B) and collagen-1 degradation (Fig. 2C, D) were observed. MMP-1 mRNA levels (Fig. 2E) and MMP-1 catalytic activity (zymography; Fig. 2F, G) were also elevated in response to UVA exposure. Each of these parameters was significantly inhibited by AP39 (100 and 200 nM) and AP123 (200 nM). In contrast, nontargeted H2S donor control molecules (ADT-OH and HTB), respectively, had no effect on MMP-1 induction, activity, or collagen degradation.
AP39 and AP123 attenuated UVA-induced mitochondrial dysfunction in vitro
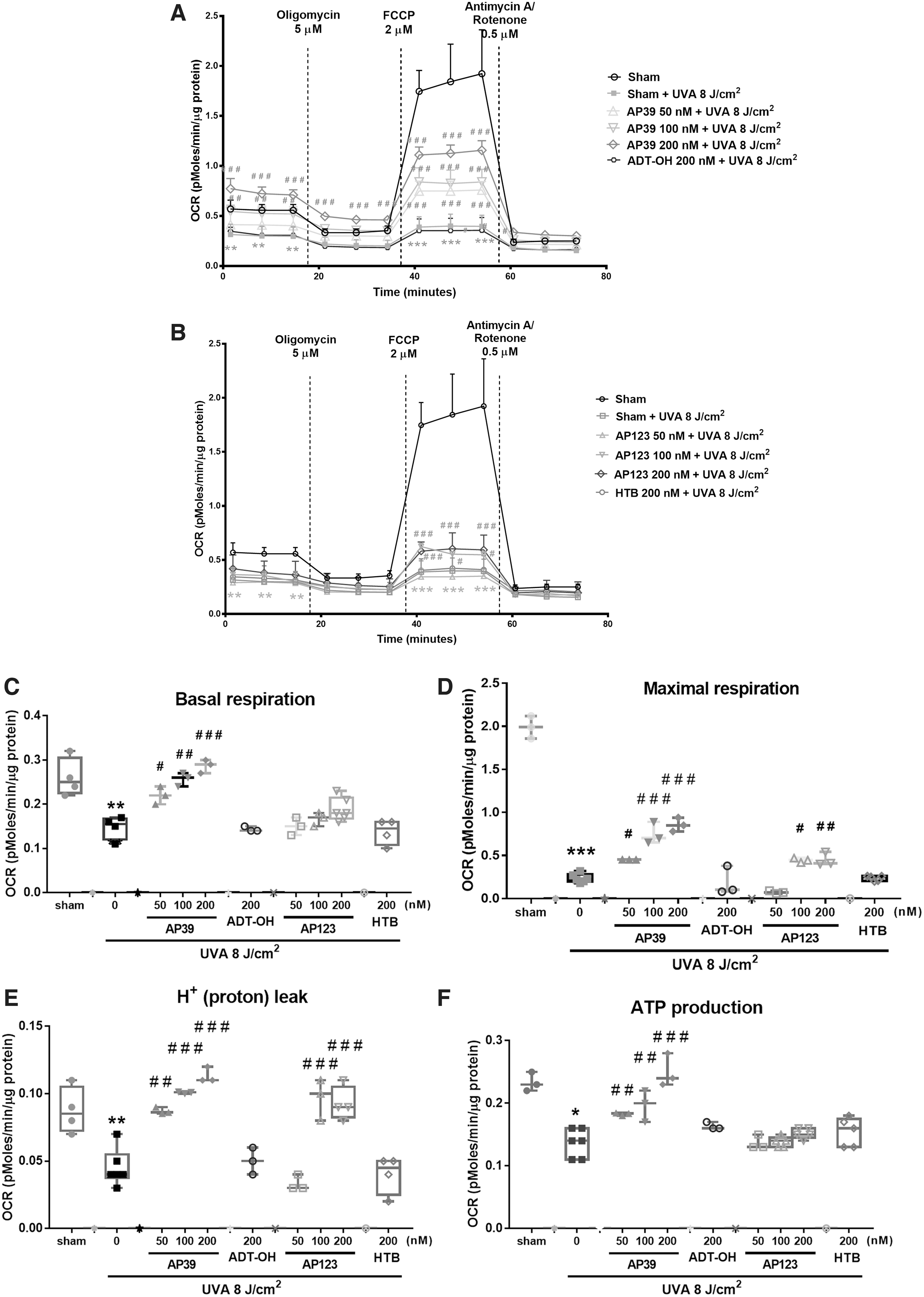
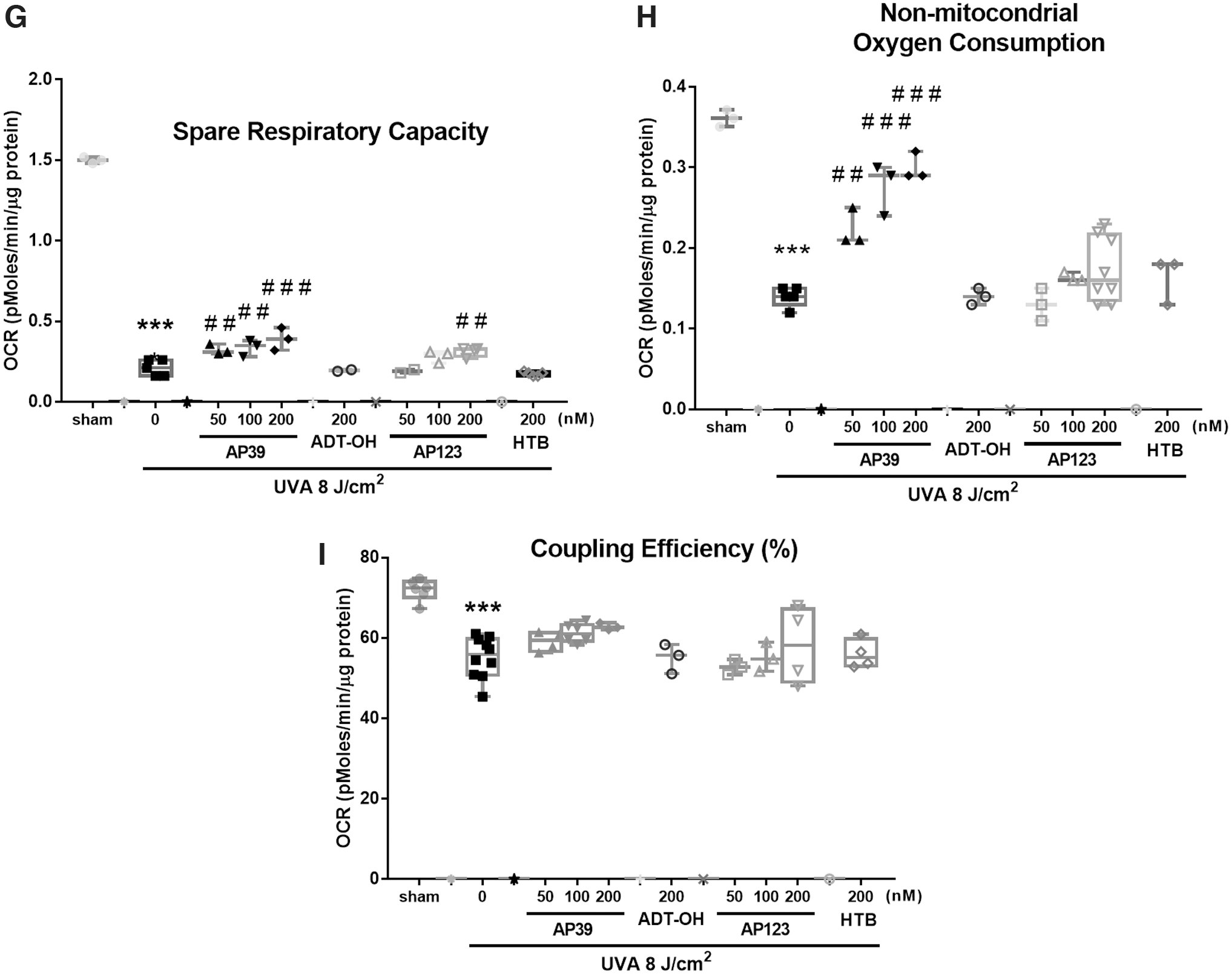
We assessed cellular bioenergetics 1 h after UVA treatment (8 J/cm2) using a Seahorse XFp extracellular flux analyzer (Fig. 3A–I). UVA treatment significantly impaired each bioenergetic component: lowered O2 consumption rate (Fig. 3A, B), lowered basal respiration (Fig. 3C), maximal respiration (Fig. 3D), proton leak (Fig. 3E), ATP production (Fig. 3F), spare respiratory capacity (Fig. 3G), nonmitochondrial oxygen consumption (Fig. 3H), and respiratory coupling efficiency (Fig. 3I). However, AP39, and to a lesser extent AP123, significantly prevented UVA-induced decline of each bioenergetic component in a concentration-dependent manner (50–200 nM). This protective effect was not observed with the control compounds ADT-OH or HTB indicating that mitochondria-targeted H2S rather than just H2S generation alone was responsible for mitochondrial protection. Further control experiments showed that vehicle treatment alone had no effect on cellular bioenergetics (Supplementary Fig. S2) and neither AP39 nor AP123 without UVA irradiation substantially altered any metabolic parameters (Supplementary Fig. S3).
AP39 and AP123 inhibited UVA-induced downregulation of Nrf2 in vitro
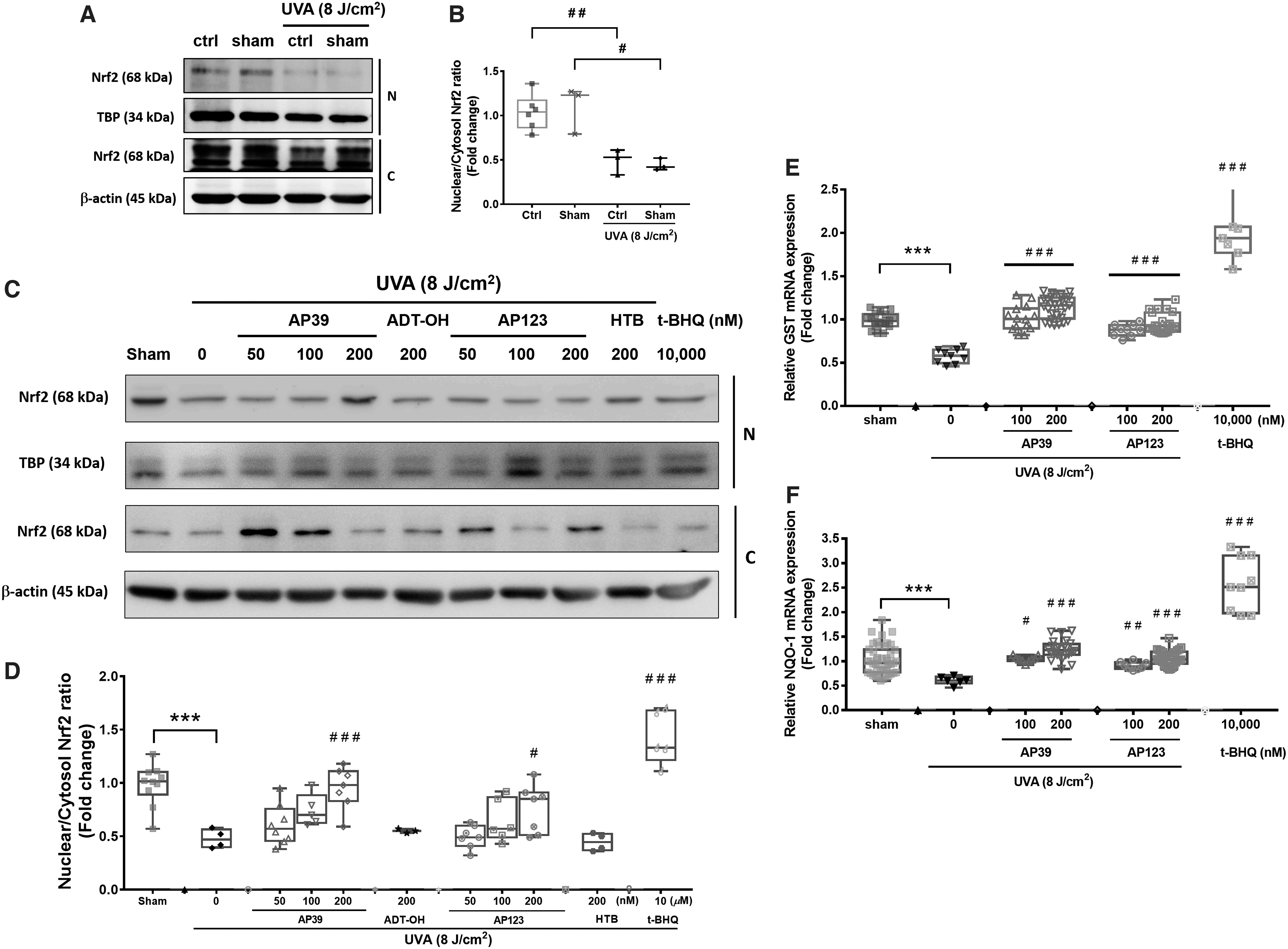
UVA exposure significantly decreased nuclear and cytoplasmic Nrf2 levels (Fig. 4A) and the nuclear:cytoplasmic Nrf2 ratio (Fig. 4B). In contrast, treatment of NHDFs with AP39 and AP123 (200 nM) significantly increased cytoplasmic and nuclear Nrf2 levels and nuclear:cytoplasmic Nrf2 ratios, but this was not observed with the control compounds ADT-OH and HTB (Fig. 4C, D). Further control experiments showed that a 30-min treatment of NHDFs with AP39, AP123, ADT-OH, or HTB did not significantly change the nuclear:cytoplasmic Nrf2 ratio compared with naive cells without UVA radiation (Supplementary Fig. S4). We further investigated the effects of UVA exposure on downstream Nrf2-mediated gene expression. Figure 4E and F shows UVAcexposure significantly decreased mRNA expression of the Nrf2-target genes GSTP1 and NAD(P)H quinone dehydrogenase 1 (NQO-1), respectively, whereas AP39 and AP123 (100 and 200 nM) significantly prevented this decline. As expected, tert-butyl hydroperoxide (t-BOOH) (10 μM; positive control) induced GSTP and NQO-1 (Fig. 4D–F). Supplementary Figure S5A–D shows the Nrf2 inhibitor brusatol partially attenuated AP39- and AP123-induced Nrf2 nuclear translocation (Supplementary Fig. S5A), mRNA expression of Nrf2 target genes (Supplementary Fig. S5B), and MMP-1 protein (Supplementary Fig. S5C) and mRNA (Supplementary Fig. S5D) expression after UVA exposure.
Effects of AP39 and AP123 against UVA-induced skin damage in BALB/c mice in vivo
All animal experiments were reviewed and approved by the Siriraj Animal Care and Use Committee (SiACUC), Si-ACUP 012/2559 (COA#014/2559). We previously showed that UVA irradiation of mouse skin caused a dose-dependent upregulation of MMP-1 expression, collagen I degradation, and increased epidermal thickness at 24 h after the final irradiation (10 J/cm2, cumulative dose, 60 J/cm2) (13, 14). We therefore used this model for our studies with AP39 and AP123, and controls. Compounds (0.3 and 0.1 μM/cm2) were applied topically to dorsal mouse skin for 1 h and then irradiated with UVA (total dose 60 J/cm2). UVA treatment markedly reduced epidermal thickness (Fig. 5A, D), increased MMP-1 (Fig. 5B, E), and decreased collagen levels (Fig. 5C, F), and this effect was not attenuated by nontargeted H2S (1 μM/cm2, ADT-OH or HTB). However, AP39 and AP123 (0.3 and 1 μM/cm2) decreased epidermal thickness (Fig. 5A, D), inhibited MMP-1 activation (Fig. 5B, E), and increased collagen-1 levels (Fig. 5C, F). As expected (13), these protective effects were also observed with the positive control, Nrf2 activator
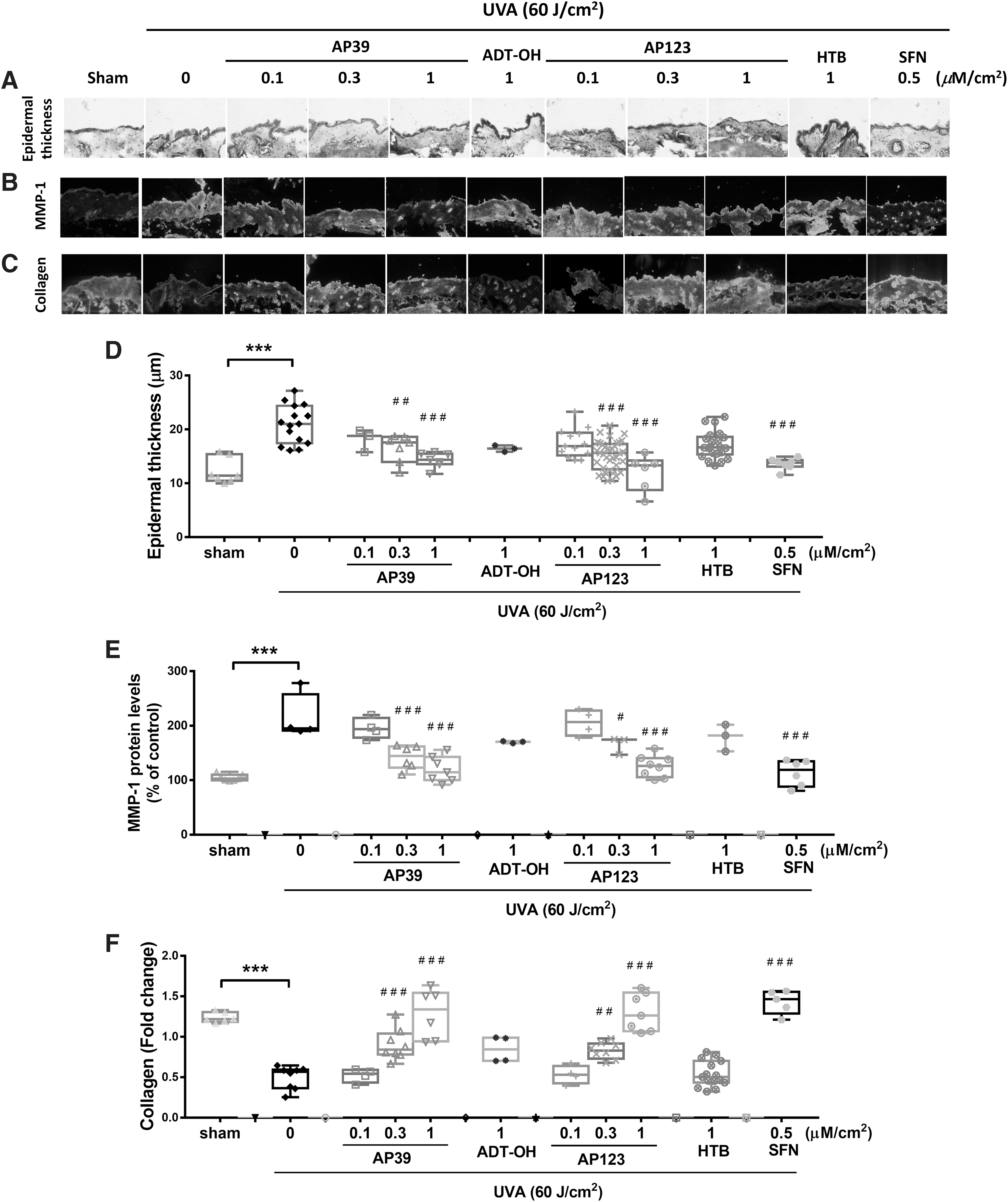
1-isothiocyanato-4-methylsulfinylbutane (sulforaphane [SFN]; Fig. 5A–F). Further control experiments showed that neither AP39, AP123, control compounds ADT-OH and HTB, or SFN (3 μM/cm2) had any effect on epidermal thickness on nonirradiated mouse dorsal skin (Supplementary Fig. S6) confirming that protection by AP39 and AP123 was by mitochondria targeting of sulfide rather than an effect of sulfide generation in general.
AP39 and AP123 activated Nrf2-regulated redox signaling in mouse skin in vivo
Figure 6A and F shows UVA exposure (60 J/cm2) lowered nuclear Nrf2 levels in mouse skin, and this decline was dose dependently inhibited by AP39 and AP123 (0.1–0.3 μM/cm2), but not by nontargeted controls (ADT-OH and HTB, respectively). In addition, AP39 and AP123 (but not controls) significantly and dose dependently inhibited UVA-induced decline in glutamate/cysteine ligase glutathione-S-transferase (GST) and NQO-1 levels (Fig. 6G–I, respectively) as well as prevented oxidative DNA damage (8-hydroxy-2′-deoxyguanosine [8-OHdG]; Fig. 6E, J). AP39 and AP123 also prevented UVA-induced decline in skin expression of the major mitochondrial antioxidant defense enzyme manganese superoxide dismutase (MnSOD) (Supplementary Fig. S7A, C) and regulator of mitochondrial biogenesis (proliferator-activated receptor-gamma coactivator [PGC-1α]) (Supplementary Fig. S7B, D). SFN used as a positive control also attenuated these parameters, as expected (Fig. 6 and Supplementary Fig. S6).
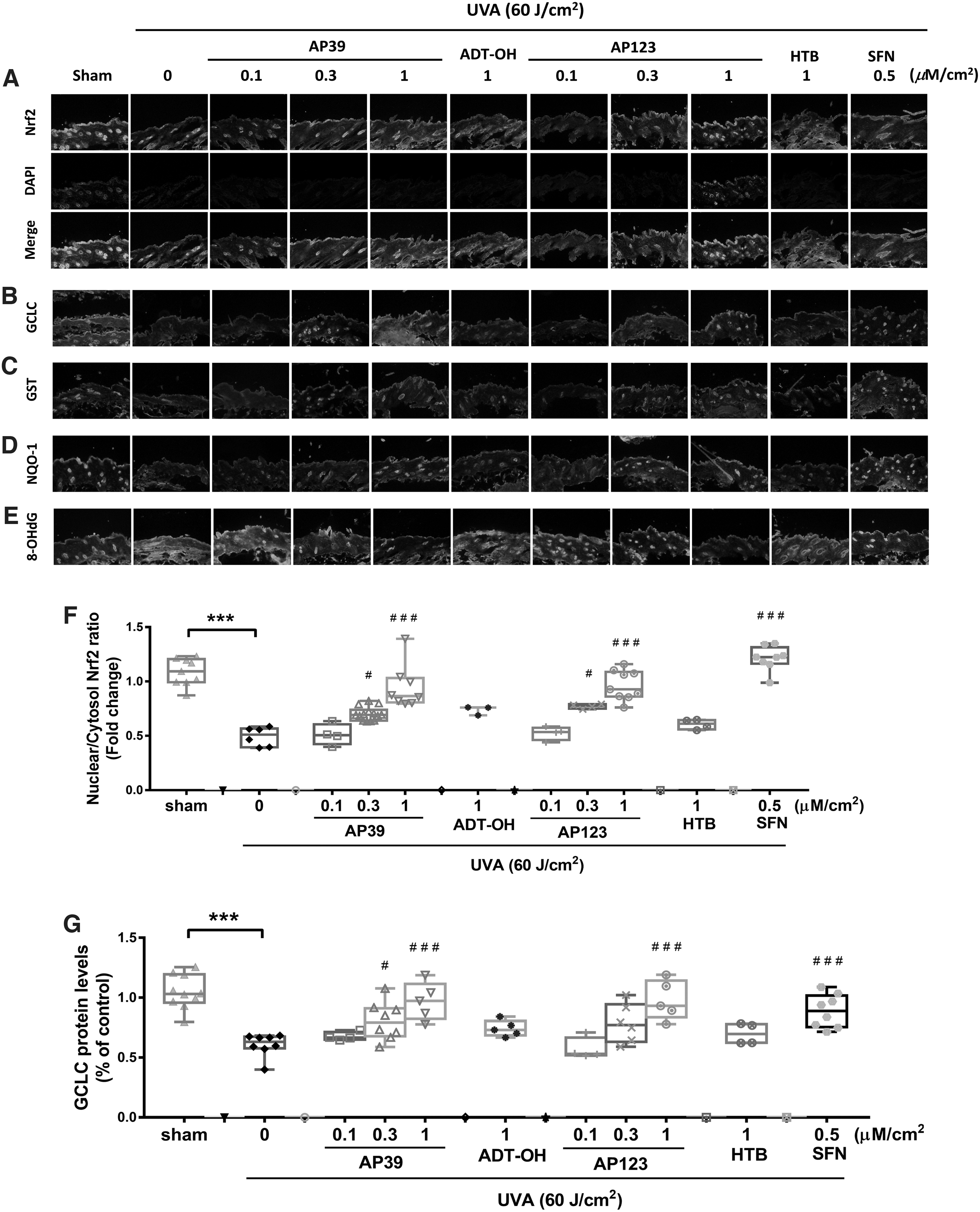
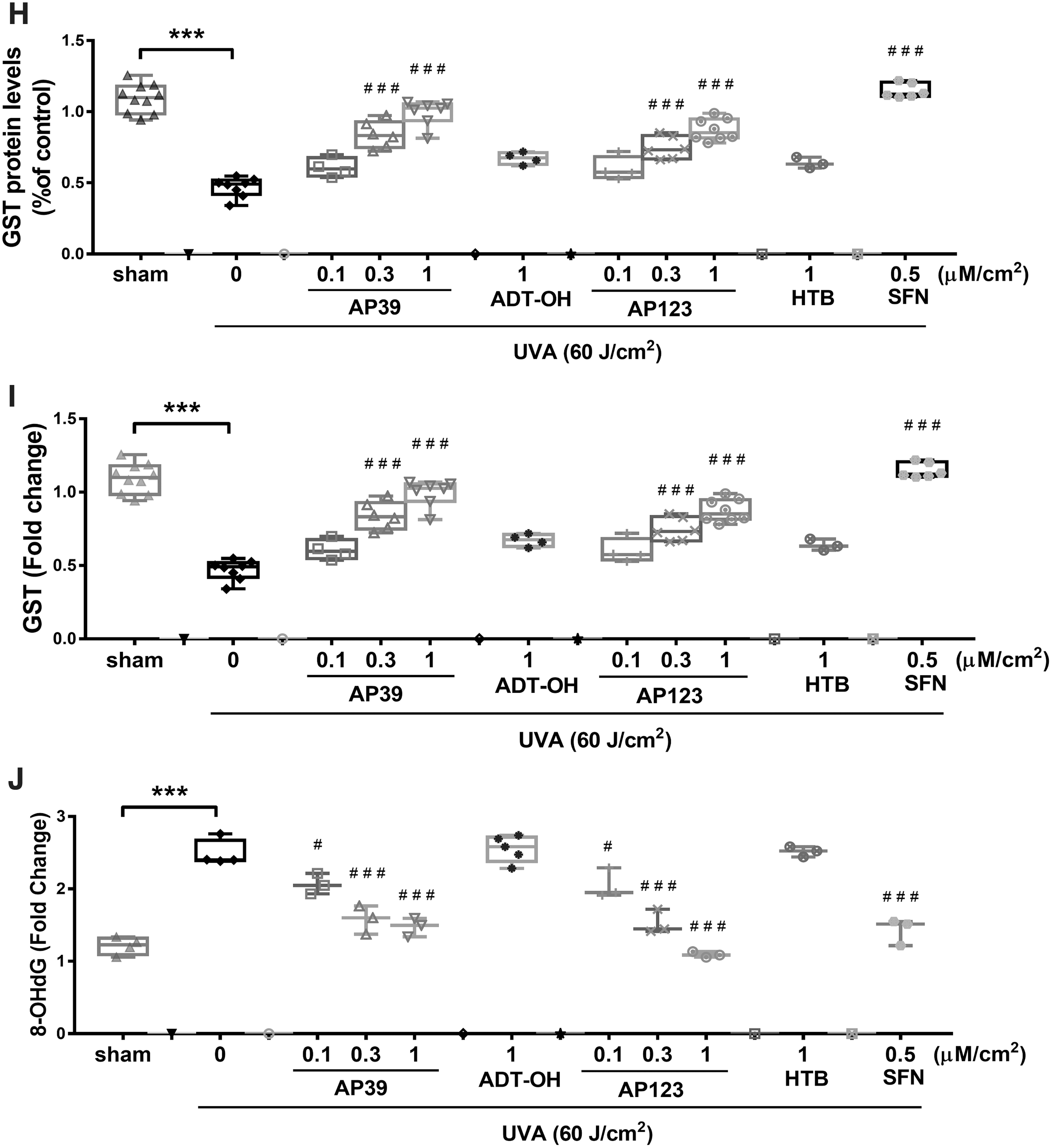
Discussion
UVR/UVA exposure is the principal cause of photoaging, as well as a primary risk factor for melanoma and nonmelanoma skin cancer development. Exposure of UVA in mammalian skin cells in culture, or skin in vivo results in substantial tissue damage through MMP induction and collagen degradation through oxidative stress, mitochondrial damage, and suppression and/or depletion of endogenous antioxidants such as glutathione (GSH) and MnSOD. Mitochondria are the major source of oxidant production as well as key sources of cellular oxidants during UVA exposure, and pharmacological approaches to selectively target this organelle have been proposed to prevent/limit UVA-induced photoaging (9). In the skin, endogenous H2S plays a crucial role in regulating skin physiology, and H2S delivery molecules have a therapeutic potential for treatment of dermatological diseases (e.g., psoriasis, atopic dermatitis, pruritus, and acute injuries including burns; reviewed in Coavoy-Sanchez et al. (17). H2S-generating compounds (e.g., sodium hydrosulfide [NaSH] and GYY4137) exert protective effects against oxidative damage, cytotoxicity, and inflammatory responses in skin keratinocytes (80), as well as promote skin wound healing (79). With these observations in mind, we evaluated the effects of two potent novel mitochondria-targeted H2S delivery molecules, AP39 (40) and AP123 (26), in UVA-induced photoaging.
H2S has recently been considered a “central player” in the aging process, and tissue levels decline with age across species (19, 57, 85). Genetic silencing or pharmacological inhibition of endogenous H2S generating enzymes, particularly mitochondrial 3-MST, shortens the life span in Caenorhabditis elegans and Drosophila melanogaster and increases susceptibility to cellular oxidative damage (85). In mammals, CGL knockout exacerbates inflammation and induces vascular damage (83), highlighting the critical need to maintain cellular H2S synthesis for health. Modulating cellular H2S levels through H2S generating molecules such as NaSH and the slow-release H2S donor GYY4137 has been shown to extend the life span in nematodes and other small animals (21, 57), although the effects of H2S on skin and skin photoaging have not been fully investigated.
Lower systemic levels of H2S (i.e., “H2S deficiency”) have been seen in skin diseases such as psoriasis where systemic H2S negatively correlated with blood levels of proinflammatory mediators (3). Overcoming “H2S deficiency” using exogenously applied H2S generating molecules (e.g., 50–800 μM NaSH) inhibited tumor necrosis factor-α-induced interleukin (IL)-6 and IL-8 synthesis in HaCaT keratinocytes in vitro (3, 80) and against proinflammatory signaling in psoriasis in vivo using the “slow-release donor” GYY4137 (17, 58), suggesting H2S modulators/donors as a useful therapeutic tool for skin treatment. However, due to the “untargeted approach,” very high concentrations/doses of these molecules were required [e.g., NaSH 50 μM–4 mM; (3, 46, 80); GYY4137, 400–500 μM (27, 46)], presumably because they do not deliver H2S to where it is needed, that is, to the mitochondria. We therefore used mitochondria-specific H2S delivery molecules AP39 and AP123 (26, 40) and compared their effects against UVA-induced skin damage with their respective control compounds, which generate H2S but do not target mitochondria (ADT-OH and HTB, respectively). AP39 and AP123 have previously been shown to stimulate mitochondrial respiration and preserve cellular bioenergetics after oxidative insult at concentrations/doses orders of magnitude lower than nontargeted H2S (e.g., <100 nM), through increased mitochondrial respiration, notably complex II and III activities (26, 68), and cytochrome c reduction (73), and shown considerable potency (e.g., <1 mg/kg) and therapeutic efficacy in other animal models of mitochondrial dysfunction, for example, myocardial infarction (16, 36), organ transplantation (35, 84), shock (78), cardiac arrest (34), acute kidney injury (1), and skin burn injury (2). Although our current study is the first time these compounds have been applied to skin and photoaging, our findings are generally consistent with those reported elsewhere with AP39/AP123 (i.e., mitochondrial protection). Indeed, in NHDFs, both compounds (but not their respective nontargeted controls) decreased UVA-induced oxidative stress, MMP-1 induction, and collagen degradation (Figs. 1 and 2). Treatment with AP39/AP123 also prevented UVA-induced loss of cellular bioenergetics, mitochondrial electron transport chain (ETC) activity, and ATP synthesis (Fig. 3) with concomitant increases in Nrf2 nuclear accumulation and its target gene levels (Fig. 4), and Nrf2 activation and signaling were prevented with the Nrf2 inhibitor brusatol (Supplementary Fig. S7D–F). These findings were generally reproduced in vivo and both compounds (but not their respective controls) dose dependently decreased MMP-1 induction, collagen loss (Fig. 6), and oxidative DNA damage (Fig. 7). AP39 and AP123 also induced a significant Nrf2 activation in mouse skin, concomitant with the upregulation of cytoprotective defense enzymes (GST and NQO-1) and mitochondrial MnSOD and increased mitochondrial biogenesis (Supplementary Fig. S6). The preserved mitochondrial bioenergetics (Fig. 3) was observed concomitantly with suppression of cellular oxidant production (Fig. 1) and the subsequent restoration of downstream Nrf2 signaling (Figs. 2, 4, 6, and 7).
The precise “antiaging” and cytoprotective mechanism(s) for cellular H2S is/are not completely understood. Exogenous H2S has been reported to activate cellular defense mechanisms such as increasing GSH levels through increasing cysteine uptake, and induction of GST (77) (Fig. 6), as well as Nrf2 activation (61, 82). It is unlikely that the effects we observed were due to “oxidant scavenging” or direct “antioxidant” effects as the rate constants of reaction of even bolus sulfide (e.g., 5 mM NaSH) in vitro with biological oxidants O2 •−, hydrogen peroxide (H2O2), and ONOO− are far too slow to account for this (12), and AP39 and AP123 were used at nM levels and hydrolyze very slowly (26) generating insufficient H2S to compensate for the unfavorable reaction kinetics. In addition, no cell protection was observed (in vitro or in vivo) with equal or higher concentrations/doses of untargeted H2S generating control compounds (ADT-OH or HTB). As such, it is likely that the decrease in cellular oxidant production in vitro (Fig. 1) and the subsequent oxidative skin damage in vivo (Fig. 6) were due to the suppression of mitochondrial oxidant formation resulting from the increased ETC efficiency or preserved cellular bioenergetics (Fig. 3), and decreased electron leakage and O2 •− and H2O2 formation, rather than by direct oxidant scavenging. One possible mechanism for the photoprotection we observed could have been UV absorption and a simple “sunscreen”-like effect. However, control experiments (Supplementary Fig. S8) and previous literature on related triphenylphosphonium-containing compounds (29, 37, 50) showed that this is very unlikely as neither AP39 nor AP123 absorbed UVA. For example, although ADT-OH showed some UVA absorption characteristics (although at 50 μM in Hammers et al. (29) and Supplementary Figure S8A and B c.f. 1 μM used in our current study), it was not able to prevent either UVA-induced damage to isolated NHDF in vitro (Figs. 1–4) or UVA-induced damage to rat skin in vivo (Figs. 5 and 6).
H2S-mediated protein S-persulfidation may be one mechanism accounting for the protective effects we observed. AP39-induced protein persulfidation appears solely mitochondrial (76) and catalyzed by specific reduction of mitochondrial cytochrome c (73, 85) resulting in S-persulfidation of the mitochondrial antioxidant enzyme, MnSOD (85). This H2S-derived post-translational modification of MnSOD leads to a “gain of function” and increased enzyme catalytic activity with resistance to oxidative inactivation by H2O2 or ONOO− allowing it to function for longer and at greater rates than “nonpersulfidated” MnSOD (85). In addition, mitochondrial H2S-mediated cytochrome c reduction also catalyzed caspase-9 persulfidation and the subsequent inactivation, reducing oxidant-induced cytotoxicity (76). ATP synthase is also susceptible to H2S-mediated persulfidation, which enhances catalytic activity and ATP synthesis (48). Key transcription factors regulating cell fate and inflammation are also reported to be persulfidated by H2S, including Nrf2 (resulting in activation (82); as we have observed in vitro, Fig. 3, and in vivo, Fig. 6) and nuclear factor kappa beta (NF-κB) (resulting in suppression) (61). SFN, a well-known Nrf2 inducer, has been suggested to function as a source of cellular H2S, exhibiting several pharmacological effects, including the promotion of Nrf2-regulated antioxidant defenses and downregulation of inflammatory cytokines, and was observed to induce H2S generation of prostate cancer cells in vitro (54) and in vivo (45), and although the mechanisms responsible for this have not been fully characterized, it is possible that Nrf2 activation induced by SFN in our study had a sulfide-dependent component. Nrf2 activators have also been suggested to improve mitochondrial function and integrity (20, 32), at least in part, through upregulation of PGC-1α and resulting in normalization of redox homeostasis and mitochondrial biogenesis, and further work is needed to examine the cross talk between mitochondrial H2S, protein S-persulfidation, and Nrf2 signaling (Fig. 7), and identify and then fully characterize “persulfidated” proteins to critically dissect their role in the photoaging of skin in vivo. However, it is likely that the primary effects of AP39 and AP123 were on mitochondrial preservation, with the subsequent promotion of redox balance and restoration of Nrf2 and Nrf2-dependent signaling rather than direct Nrf2 activation. In this respect, AP39 and AP123 may have preserved endogenous cytoprotective signaling, for example, upregulation of Nrf2-dependent gene products (Figs. 4 and 6). Although the Nrf2 inhibitor brusatol prevented AP39- or AP123-mediated suppression of UVA-activated MMP-1, future studies utilizing Nrf2 knockout animals would be useful to further dissect and distinguish mitochondrial- and Nrf2-driven signaling processes initiated and/or preserved by targeted delivery of H2S to mitochondria.
In our study, we have focused on the pharmacological effects of AP39 and AP123 rather than re-exploring the established basic pathological mechanisms by which UVA induces mitochondrial dysfunction, mitochondrial and cellular oxidant production, and the subsequent detrimental downstream signaling leading to skin damage and tissue remodeling. At present it is not known whether the effects of UVA depleted or increased cellular H2S levels and this, as well as the molecular mechanisms mediating this pathological event, will need to be determined in the future. However, our demonstration of efficacy in the in vitro and in vivo models of photoaging, and supplementing cells and tissues with very low concentrations (nM) of compounds, is unlikely to be dependent on cell or tissue changes of endogenous H2S. For example, AP39 has been shown to be efficacious in shock (78), which exhibits elevated systemic H2S levels (43), as well as in Duchenne muscular dystrophy (21) and hypertension (70), which exhibit lower H2S levels (21, 81), respectively. Indeed, current clinical trials using H2S delivery molecules have been successfully evaluated in conditions showing either increased or decreased H2S bioavailability, for example, ATB-346 (NCT03978208) in osteoarthritis [impaired H2S synthesis c.f. controls (11)] and NCT03291418 in colitis (higher colonic H2S levels c.f. controls (23). Similarly, SG-1002 in cardiovascular diseases (NCT01989208 and NCT02278276) either showing elevated endogenous H2S levels (55) or markedly lower endogenous H2S levels (72).
An additional limitation of our current study is that we have treated cells and animals with compounds before UVA exposure, in line with other similar studies (13 –15, 22), and shown significant cellular and tissue protection. Further detailed studies are needed to determine the extent to which AP39 and/or AP123 can rescue tissue previously damaged by UVA exposure. However, since skin aging (and aging in general) is so far irreversible and not preventable, there is still merit in compounds, which could delay this inevitability and possibly prevent age-related morbidity and the associated health care burden.
To conclude, we have shown for the first time that selective mitochondria-targeting of H2S prevented UVA-induced photoaging in mouse skin in vivo. Our study provides insight into the novel pharmacological role of the mitochondria-targeted H2S to prevent and/or delay photoaging in skin. Mitochondria-targeted H2S from AP39, AP123, and future related compounds may represent a novel approach for the prevention and treatment of skin photoaging as well as being useful novel tools for determining the role of mitochondrial H2S in skin disorders.
Materials and Methods
In vitro characterization
Compound synthesis, cell culture, and UVA irradiation in vitro
Full experimental details are supplied in Supplementary Data. Synthesis of AP39 (40) and AP123 (26) was carried out in-house as previously described. These novel molecules contain the mitochondria-targeting moiety (triphenylphosphonium ion [TPP+]), an aliphatic linker molecule, with dithiolethione (ADT-OH) and HTB as the sulfide source for AP39 and AP123, respectively. To determine whether any observed effects of these novel compounds were due to mitochondria-generated H2S, we also used the nonmitochondrial H2S generating moieties, ADT-OH (for AP39) and HTB (for AP123), without the mitochondria-targeted motif as negative controls throughout the study, at 200 nM. Concentrations were chosen based on preliminary studies and those previously stated in the literature (1, 26, 40, 68).
NHDF cells (CC-2511: Lonza, Basel, Switzerland) were grown in high-glucose (4.5 g/L) Dulbecco's modified Eagle's medium (DMEM, Cat # 12800017; Life Technologies, USA) supplemented with 10% fetal bovine serum and 1% penicillin (100 units/mL)/streptomycin (100 μg/mL) at 37°C in a humidified air of 5% CO2 (PCO2 = 40 Torr) (14). The 80%–90% confluent NHDFs were then assigned to experimental treatments after washing by phosphate-buffered saline (PBS) and seeded overnight on six-well plates (105 cells/well). The stock solutions of AP39, AP123, and the control compounds were prepared in DMSO. In all in vitro and in vivo experiments, the stock solutions were then diluted in the mixture of serum-free DMEM and 80% ethanol (the final concentration not exceeding 0.1%), used as a vehicle for this treatment. Control experiments were performed using SFN and t-BOOH (to activate) and brusatol (to inhibit) Nrf2. After a 30-min treatment of NHDFs with test compounds, cells were irradiated with UVA under a thin layer of Dulbecco's phosphate-buffered saline (DPBS).
After irradiation, NHDFs were immediately incubated with serum-free DMEM and harvested as required: 6 h postirradiation for apoptosis (6), 1 h postirradiation for cellular oxidants, Nrf2 nuclear translocation (14), and mitochondrial function (69), 24 h postirradiation for enzyme activity, MMP-1, and collagen type I expression (49), 12 h postirradiation for MMP-1 gene expression (65), and 2 h postirradiation for Nrf2 downstream gene expression (13) (as mentioned in the method of western blot analysis, zymography, and realtime RT-PCR).
Determination of UVA-induced cell damage in vitro
Cell death was assessed using a propidium iodide (PI) commercial kit (BD Biosciences, USA) (14, 18), and intracellular oxidant generation was determined using dichlorofluorescein diacetate by flow cytometric analysis (BD FACSCalibur™) (44). Data were analyzed by FlowJo Software v10 and expressed as a percentage of control (100%, sham treatment in nonirradiated cells or UVA-irradiated cells; vehicle only).
UVA exposure depletes endogenous antioxidants such as Nrf2 activation in response to phase 2 detoxifying enzymes, upregulates MMP activity, as well as downregulates collagen protein expression (13). After a 30-min treatment of NHDFs with test compounds, the cells were irradiated with UVA under a thin layer of DPBS. The UVA intensity at a distance of 21 cm from the UVA lamp was 9–10 mW/cm2 determined using a UVA meter (Hand-held UV-meter; Honle UV technology, Germany) equipped with a UVA sensor (320–400 nm) (13, 14). The UVA source was a xenon arc lamp (Dermalight ultra 1; Hönle, Martinsried, Germany) with emission maximum at 360 nm. Cells cultured in six-well culture plates were exposed to UVA irradiation for 5–6 min to achieve a single dose of 8 J/cm2 depending on electric power of the lamp.
Collagen zymography for the determination of MMP-1 activity in vitro
Collagen expression and MMP-1 activity were determined in primary NHDFs by standard zymography. Briefly, primary NHDFs were cultured for 48 h or until the cells reached 70%–80% confluence. After UVA irradiation, the cells were incubated in the serum-free DMEM for 24 h and the activity of MMP-1 released from the cells into the culture media was then detected by collagen zymography. Sample supernatants of each sample were mixed with 1:1 (v/v) with nonreducing SDS sample buffer (125 mM Tris-HCl [pH 6.8], 20% glycerol, 2% SDS, 0.002% bromophenol blue) and were subjected to electrophoresis on 10% polyacrylamide gels containing 1 mg/mL collagen solution. After electrophoresis, the gels were then rinsed twice with 2.5% Triton X-100 for 30 min at RT to remove SDS and renature the MMP-1 activity in the collagen gel. Renatured gel was rinsed with fresh developing buffer and incubated in the same buffer for 36 h at 37°C to induce collagen lysis. After incubation, the gels were briefly rinsed in distilled H2O and later stained with 0.006% Coomassie brilliant blue G-250 (50% methanol and 10% acetic acid) for 2 h with agitation and destained using 50% methanol and 10% acetic acid for 5 min at RT. The digested bands become clear (colorless) on a blue background, indicating the expression of MMP-1 activity or collagenase. The enzymatic activity was interpreted by a computer analysis of the intensity of each collagen lysis band. Determination of MMP-1 activity was performed by scanning the gels using a CAMAG TLC scanner (Muttenz, Switzerland), and the integrated density for each band was calculated using the ImageMaster software (Hoefer Pharmacia Biotech). The 25,000 cells per group were used for densitometric units of MMP-1 activity. Data shown are expressed as a percentage of control (100%, sham treatment in nonirradiated cells or UVA-irradiated cells).
Determination of cellular bioenergetics: extracellular flux analysis in vitro
Mitochondrial respiration in NHDF cells was characterized using a Seahorse XFp Extracellular Flux Analyzer (Agilent Technologies) (68). Briefly, NHDFs were performed with the Cell Mito Stress test kit according to the manufacturer's protocol. One day before analysis, 20,000 cells per well of NHDF cells were seeded overnight on an eight-well Seahorse XFp Fluxpak at 37°C with a CO2 incubator and the sensor cartridge was hydrated overnight with the XF Calibrant solution (Agilent Technologies) at 37°C with a non-CO2 incubator. On the day of analysis, the XFp miniplate was treated with H2S donors and UVA irradiation (8 J/cm2) and was allowed in SF-DMEM for 1 h. XF Assay media were supplemented with 25 mM glucose, 2 mM sodium pyruvate, and 2 mM L-glutamine. The XFp miniplate was washed twice with XF assay media and was pre-equilibrated in the non-CO2 incubator at 37°C for 1 h before assay initiation. The oxygen consumption rate (OCR) was analyzed by a sequential injection of modulators; oligomycin (5 mM) was used to measured ATP production rate and proton (H+) leak and the measurement of OCR after carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP; 2 μM) was used to assess the maximal respiration and the spare respiratory capacity. Antimycin A and rotenone combination (0.5 μM) was used together to detect the residual nonmitochondrial respiration rate. Basal respiration was calculated by subtracting the nonmitochondrial respiration from the last rate measurement before oligomycin injection.
The coupling efficiency (%) was determined by the percentage of ATP production rate divided by the basal respiration rate. Three compounds were suspended in prewarmed XF assay medium and loaded into the injection ports of hydrated sensor cartridge according to the order of injection. The loaded XFp sensor cartridge and XFp utility plate were placed into the Seahorse XFp analyzer for calibration. After the calibration, the XFp utility plate was replaced by a miniplate containing cells. Measurement cycle consisted of 12 measurements within 80 min. First, three basal OCR measurements were performed before adding oligomycin, followed by three measurements by the oligomycin injection, three measurements after the FCCP injection, and three measurements of the rotenone/antimycin A. Assay parameters were determined as following: basal respiration, ATP production, proton leak, maximal respiratory, spare respiratory capacity, coupling efficiency, and nonmitochondrial respiration.
Determination of Nrf2 nuclear translocation in vitro
Total cytoplasmic and nuclear fractions were extracted using the NXTRACT CelLytic NuCLEAR Extraction kit (Sigma-Aldrich) according to the manufacturer's protocol. Both protein concentrations were quantified using the Bradford method (Bio-Rad). Samples (40 μg per lane of the cytosolic part and 70 μg per lane of the nuclear part) were separated using SDS-PAGE (10% Tris-HCl polyacrylamide gel) and electrotransferred to PVDF membranes. The membrane was blocked with a blocking buffer and a primary antibody against Nrf2 (1:2500; ab31163; Abcam) overnight and then with the HRP-conjugated secondary antibody for 2 h at RT. Protein expression was quantified and normalized to loading control: β-actin (1:5000; ab8227; Abcam) for cytosolic extracts and antibody against TBP for nuclear extracts (1:2500; ab63766; Abcam). Signals were detected using the Bio-Rad Clarity Western ECL chemiluminescence (Bio-Rad), and band densities imaged using an ImageQuant LAS 4000 digital imaging system (GE Healthcare). The integrated optical density of the bands was analyzed by ImageJ software v1.45 (National Institutes of Health). The nuclear-to-cytosolic Nrf2 ratio (fold change) was calculated by dividing the cytosol Nrf2 fraction to the nuclear Nrf2 fraction, which was normalized by each loading control protein, the β-actin for cytosol fraction, and the TBP for nuclear fraction, respectively. Nrf2 nuclear translocation was expressed as a fold change (sham treatment in nonirradiated cells or UVA-irradiated cells).
Quantitative real-time reverse transcriptase-polymerase chain reaction
NHDFs were treated with AP39, AP123, and controls, and exposed to UVA irradiation (8 J/cm2), as described above. Total RNA of Nrf2-targeted genes, including GST and NQO-1, was collected 2 h postirradiation, and total RNA of MMP-1 gene was collected 12 h postirradiation. The procedure, amplification reaction, polymerase chain reaction (PCR) primer sequence, and analysis are listed in Supplementary Table S1. All RNA products were extracted using the illustra RNAspin Mini RNA Isolation Kit (GE Healthcare) and then reverse transcribed by the ImProm-II Reverse Transcriptase (Promega, Madison, WI) following the manufacturer's protocol. Real-time reverse transcriptase-PCR analysis was determined in triplicate for each sample and each primer. The amplification reaction was performed under the following conditions: 95°C for 10 min, followed by 40 cycles of 15 s at 95°C, 40 s at 59°C, and 40 s at 72°C. A 10 μL total reaction volume contains 2 μL of cDNA with SYBR Green Master (ROX) and 8 μL of PCR primers. Single-product amplification was analyzed by the melt curve analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was used to normalize mRNA content. The mean Ct from mRNA expression in cDNA from each sample was compared with the mean Ct from GAPDH determinations from the same cDNA samples. The results are shown as fold change in gene expression calculated using the 2−ΔΔCt calculation. For the control (nonirradiated cells), the -ΔΔCt vehicle group was set to 0 and 2° was set to 1, and therefore, the fold change in gene expression relative to the vehicle is equal to 1. For the UVA-irradiated and compound-treated cells, 2−ΔΔCt indicates the fold change in gene expression compared with the vehicle group.
In vivo characterization
Determination of UVA-induced cell damage in vivo
This study was approved by the SiACUC, Siriraj Hospital, Mahidol University, Thailand; Certificate of Approval # 016/2559, SiACUP # SU-ACUP 012/2559. Three-week-old female BALB/c wild-type mice were purchased from the National Laboratory Animal Center, Mahidol University, and maintained under controlled conditions (25°C ± 2°C with 55% ± 5% relative humidity in a 12-h light:12-h dark cycle) using an isolator caging system. Water and food were available ad libitum during the experimental period. Mice were randomized into 13 groups of 4 mice each (47, 59) and AP39, AP123, respective controls, or SFN (positive control) was applied to shaved dorsal skin (1 cm2 area, 50 μL in 80% ethanol) for 1 h before UVA irradiation (13, 14): Group I (control), without UVA exposure or topical treatment. Group II (sham), vehicle (topical), without UVA irradiation. Group III (control with UVA). Group IV (sham with UVA), vehicle treatment and irradiation. Group V-VII (AP39 with UVA) with 0.1, 0.3, and 1 μM/cm2 of AP39. Group VIII (ADT-OH with UVA) received 1 μM/cm2 of ADT-OH. Groups IX-XI (AP123 with UVA) received 0.1, 0.3, and 1 μM/cm2 of AP123. Group XII (HTB) received 1 μM/cm2 of HTB. Group XIII (SFN with UVA) received 0.5 μM/cm2 SFN as a positive control. All UVA exposures were UVA at 10 J/cm2/session three times a week for 2 weeks (a cumulative total dose of 60 J/cm2). Mice were anesthetized by an intraperitoneal injection (i.p.) of 100:10 mg/kg of ketamine/xylazine cocktail. Dorsal skin was removed at different time points after the last exposure of UVA, embedded in Tissue-Tek® OCT compound, directly snap-frozen (liquid nitrogen [N2]), and stored at −80°C until microtome sectioning. Skin thickness was assessed by hematoxylin and eosin (H&E) and immunofluorescence (IF) staining. MMP-1 and collagen-1, oxidative stress (8-OHdG, GCL, GST) and mitochondrial function (MnSOD, PGC-1α), and Nrf2 activation were assessed by IF, and DAPI was used to counterstain nuclei. Image analysis was performed using ImageJ. Full details of these procedures and antibodies used are supplied in the Supplementary Data.
IF analysis for determination of skin aging, oxidative stress signaling, and mitochondrial function
The frozen skin tissue samples were collected at different time points following the last exposure of UVA irradiation as follows: 24 h postirradiation for MMP-1 and collagen; 1 and 6 h postirradiation for Nrf2 and its target proteins, respectively; 6 h postirradiation for 8-OHdG; and 24 h postirradiation for MnSOD and PGC-1α. These tissue sections were blocked with PBS containing 2% bovine serum albumin for 30 min. After removing the excess blocking buffer, the slides were incubated with Nrf2 Ab (1:50, ab31163; Abcam), GCL (1:50, ab53179; Abcam), GST Ab (1:50, sc-459; Santa Cruz Biotechnology, Santa Cruz, CA), NQO-1 Ab (1:50, ab34173; Abcam), 8-hydroxy-2′-deoxyguanosine (8-OHdG) [N45.1] Ab (1:50, ab48508; Abcam), MMP-1 Ab (1:50, ab137332; Abcam), SOD2/MnSOD Ab (1:50, ab13533; Abcam), PGC-1α Ab (1:50, ab54481; Abcam), and collagen I (C-18) Ab (1:50, sc-8784; Santa Cruz Biotechnology) for 1 h. The slides were then rinsed three times with PBS and incubated for 1 h at RT with FITC-conjugated the secondary antibodies (green) and with DAPI (blue) to counterstain the nuclei. For detection of nuclear Nrf2, the secondary Ab Alexa Fluor 488 goat anti-rabbit (1:200; Abcam) was used.
H&E staining and analysis of skin thickness
Eight-micrometer frozen tissue sections were collected at 24 h after irradiation of UVA and then were fixed in ice-cold acetone and air-dried for 30 min at RT. H&E staining was performed for the histological evaluation of epidermal thickness. Tissue sections were rinsed twice in distilled H2O for 2 min for rehydration, incubated for 4 min in hematoxylin solution, and then washed in distilled H2O for 10 min for nuclear staining. The slides were then incubated with eosin for 1 min, followed by rinsing in 95% ethanol for dehydration in two steps (15 s each), two-time rinse of absolute alcohol (15 s each), two-time rinse of acetone (15 s each), and three-time rinse of xylene (15 s each). Stained slides were mounted in a permanent aqueous mounting medium with a coverslip for examining under the brightfield microscope.
Immunohistological analysis
ImageJ software (NIH, Rockville, MD) was used to quantify the thickness and fluorescence intensity of each protein. Predefined scale bars of known distance (50 μM) were used as a distance calibration. A straight line was manually drawn perpendicularly to the epidermal layer and the length was read off directly from the software. For fluorescence intensity analysis, an outline was manually drawn around the area of fluorescence emission to define regions of interest (ROI). For all analyses of protein expression data, the corrected total cryosection fluorescence (CTCF) was calculated using the following equation: CTCF = integrated density − (area of each ROI × mean fluorescence of background readings) and the data were presented as percentage of control. Quantitative fluorescence data from ImageJ were then imported into Microsoft Excel for generating histograms for further analysis. For analysis of Nrf2 nuclear localization, we determined Nrf2 subcellular localization based on the ratio of nuclear-to-cytoplasmic intensity of Nrf2. The nuclear and cytoplasmic compartments were manually drawn. Intensity from each compartment was corrected by the background intensity.
Data and statistical analysis
ImageJ software (NIH) was used to quantify the thickness and fluorescence intensity of each protein. Predefined scale bars of known distance (50 μM) were used as a distance calibration. A straight line was manually drawn perpendicularly to the epidermal layer and the length was read off directly from the software. For fluorescence intensity analysis, an outline was manually drawn around the area of fluorescence emission to define ROI. For all analyses of protein expression data, the CTCF was calculated using the following equation: CTCF = integrated density − (area of each ROI × mean fluorescence of background readings) and the data were presented as percentage of control. Quantitative fluorescence data from ImageJ were then imported into Microsoft Excel for generating histograms for further analysis. For analysis of Nrf2 nuclear localization, we determined Nrf2 subcellular localization based on the ratio of nuclear-to-cytoplasmic intensity of Nrf2. The nuclear and cytoplasmic compartments were manually drawn. Intensity from each compartment was corrected by the background intensity.
All data are reported as mean ± SD from at least three biological replicates (n ≥ 3) performed on different days in duplicate, and statistical significance evaluated by independent t-test (Student's; two populations) or one-way analysis of variance followed by Dunnett tests, using Prism (GraphPad Software, Inc.).
Footnotes
Authors' Contributions
Conceptualization: U.P. and M.W. Methodology: J.L., A.C., M.S., and S.J. Software: J.L., A.C., M.S., and S.J. Validation: J.L., A.C., M.S., and S.J. Formal analysis: J.L., A.C., M.S., and S.J. Investigation: J.L., A.C., M.S., S.J., and U.P. Resources: U.P. Data curation: J.L., A.C., M.S., and S.J. Writing—original draft preparation: J.L., A.C., U.P., and M.W. Writing, review, and editing: J.L., A.C., S.J., W.T., M.W., and U.P. Visualization: U.P. Day to day supervision: U.P. Project administration: U.P. Funding acquisition: U.P. and M.W. All authors have read and agreed to the published version of the article.
Author Disclosure Statement
M.W., R.T., and the University of Exeter have intellectual property (patent filings) related to hydrogen sulfide delivery molecules and their therapeutic use. M.W. is a consultant to MitoRx Therapeutics (Oxford). The other authors declare no conflicts of interest in relationship to this study.
Funding Information
This work was supported by the Royal Golden Jubilee (RGJ) [grant number PHD0142/2558]; Young Researcher Development Program from National Research of Thailand (NRCT) [grant number 21/2561]; Siriraj Research and Development Fund Type I [grant number R016031033]; Thailand Research Fund and “Mahidol University” grant [grant number RSA6280101]; and the “Chalermphrakiat” Grant and “Siriraj Graduate Scholarships” Faculty of Medicine Siriraj Hospital, Mahidol University, Thailand. We are grateful to the Department of Biochemistry, Faculty of Medicine, Siriraj Hospital, Mahidol University, for the Seahorse instrument and technical assistances. We thank Miss Oranit Boonphang, Department of Pharmacology, Faculty of Medicine, Siriraj Hospital, Mahidol University, for the technical support. We would also like to acknowledge the Medical Research Council UK (R/M022706/1; M.W.) and the Brian Ridge Scholarship (R.T.) for their generous research support.
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Table S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.