
Abstract
Significance:
Most hepatopathies are primarily or secondarily cholestatic in nature. Oxidative stress (OS) is a frequent trait among them, and impairs the machinery to generate bile by triggering endocytic internalization of hepatocellular transporters, thus causing cholestasis. This is critical, since it leads to accelerated transporter degradation, which could explain the common post-transcriptional downregulation of transporter expression in human cholestatic diseases.
Recent Advances:
The mechanisms involved in OS-induced hepatocellular transporter internalization are being revealed. Filamentous actin (F-actin) cytoskeleton disorganization and/or detachment of crosslinking actin proteins that afford transporter stability have been characterized as causal factors. Activation of redox-sensitive signaling pathways leading to changes in phosphorylation status of these structures is involved, including Ca2+-mediated activation of “classical” and “novel” protein kinase C (PKC) isoforms or redox-signaling cascades downstream of NADPH oxidase.
Critical Issues:
Despite the well-known occurrence of hepatocellular transporter internalization in human hepatopathies, the cholestatic implications of this phenomenon have been overlooked. Accordingly, no specific treatment has been established in the clinical practice for its prevention/reversion.
Future Directions:
We need to improve our knowledge on the pro-oxidant triggering factors and the multiple signaling pathways that mediate this oxidative injury in each cholestatic hepatopathy, so as to envisage tailor-made therapeutic strategies for each case. Meanwhile, administration of antioxidants or heme oxygenase-1 induction to elevate the hepatocellular levels of the endogenous scavenger bilirubin are promising alternatives that need to be re-evaluated and implemented. They may complement current treatments in cholestasis aimed to enhance transcriptional carrier expression, by providing membrane stability to the newly synthesized carriers. Antioxid. Redox Signal. 35, 808–831.
Introduction
Bile formation is a well-controlled process, which is regulated by several bile constituents, hormones, and nerves (52). These regulatory mechanisms are aimed to cope with the flexible physiological requirement for biliary depuration of endo- and xenobiotics, as part of the normal liver function (96). These functional modulations are accomplished by changes in the intrinsic activity, localization, and constitutive expression of hepatocellular carriers localized either at the basolateral (sinusoidal) or at the apical (canalicular) plasma membrane domains (73).
In cholestatic liver diseases, dysregulation of those physiological mechanisms that downregulate hepatocellular transporter function occurs, leading to impairment of bile formation (164). In these alterations, oxidative stress (OS) has a major role (32).
Recently, evidence has emerged that OS impairs bile secretion by altering hepatocellular transporter localization via endocytic internalization, a phenomenon mediated by signaling pathways activated by subtle alterations in the redox state (170). Such an internalization not only impairs transporter capacity (V max) in minutes/hours by reducing the transporter density in the plasma membrane, but also leads, eventually, to downregulation of the protein transporter expression due to exacerbated degradation (169). This post-transcriptional pathomechanism can well act in concert with other deleterious effects of OS on bile secretion, such as changes in the transcriptional expression of these transporters, impairment of intrinsic transporter activity via phosphorylation by redox-sensitive protein kinases, or plasma membrane lipid microenvironment changes (170).
This article focuses on the mechanisms involved in OS-induced changes in transporter internalization to endosomal compartments, and their role in several OS-related pathological conditions where these mechanisms contribute to the cholestatic manifestations.
To this end, we will review the current information on (i) the dynamic localization of hepatocyte transporters in normal and cholestatic conditions, and the molecular mechanisms underlying this process; (ii) the signaling cascades implicated in its dysregulation under oxidative conditions; and (iii) the role of endogenous antioxidant systems, particularly bilirubin, in counteracting these alterations.
Hepatocellular Transporters and Their Role in Bile Formation
Bile generation involves osmotic mechanisms driven by the polarized biliary transfer of solutes into bile. For these biliary compounds to drive osmotic water flows from blood into bile, they must be concentrated and retained into the canalicular space, whose hermeticity is conferred by paracellular tight-junctional complexes. Once excreted, these solutes promote water movement through the water channels aquaporin (AQP)-9 (sinusoidal) and AQP-8 (canalicular) (129).
Solutes that fulfill these requirements are bile salts (BSs), which account for the so called “bile salt-dependent fraction of bile flow” (BSDBF), and both bicarbonate (HCO3 −) and glutathione in its reduced (GSH) and oxidized (GSSG) states, which drive the “bile salt-independent fraction of bile flow” (BSIBF) (20) (Fig. 1).
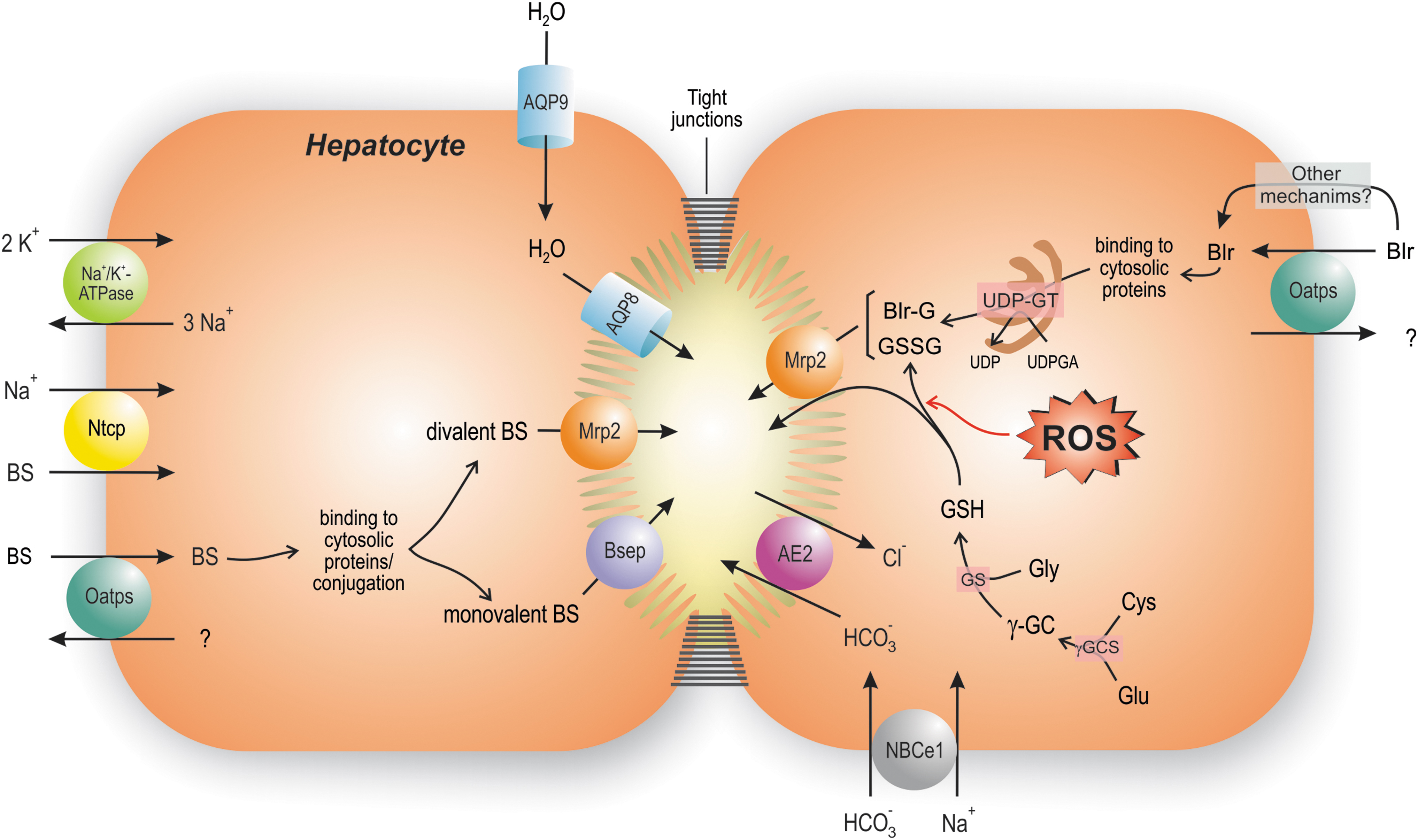
BSs are the main organic biliary solutes. Their chief sinusoidal transporter is Na+-taurocholate cotransporting polypeptide (Ntcp/Slc10a1 and NTCP/SLC10A1 for human and murine isoforms, respectively) (38). Ntcp/NTCP uses the Na+ electrochemical gradient generated by the sinusoidal Na+/K+-ATPase to drive transport of BSs into the cell (17). Ntcp/NTCP is responsible for the uptake of >80% of glycine- and taurine-conjugated BSs, and only 40% of unconjugated BSs (102). The uptake of the remaining ones is carried out in a Na+-independent manner by the carrier family organic anion-transporting polypeptides (Oatps/OATPs), which also transports unconjugated and glucuronidated bilirubin, “type II” organic cations, estrogens, leukotrienes, and numerous cholephilic dyes (69).
After binding to cytosolic proteins, conjugated BSs traverse the cytosol by Fick's diffusion, and are excreted into bile by the canalicular ATP-binding cassette (ABC) transporter, bile salt export pump (BSEP/Bsep; ABCB11/Abcb11) (199). On the contrary, bipolar, divalent glucuronidated or sulfated BSs are excreted by the multidrug resistance-associated protein 2 (MRP2/Mrp2; ABCC2/Abcc2). This export pump also transfers endogenous and exogenous non-BS organic anions conjugated with glucuronic acid, sulfate, or glutathione, such as bilirubin glucuronides, sulfated steroids, and leukotriene C4 (144).
As for the generation of BSIBF, the liver has the highest glutathione concentration of all tissues (126), and excretes this peptide into bile (61). MRP2/Mrp2 is considered the main canalicular transporter of both GSH and GSSG (49). HCO3 − is, in turn, mainly taken up by electrogenic sodium/bicarbonate cotransporter-1 (NBCe1/SLC4A4), and excreted into bile by the Cl−/HCO3 − antiporter anion exchanger 2 (AE2/SLC4A2) (62).
General Mechanisms of OS-Induced Cholestasis
OS impairs bile flow by affecting the hepatocyte secretory machinery (hepatocellular cholestasis). This occurs very rapidly after exposure to oxidizing compounds, including tert-butylhydroperoxide (t-BuOOH) (10, 14, 130, 181, 203), menadione (202), chlorodinitrobenzene (181), allyl alcohol (202), hydrogen peroxide (5, 10), ethylhexanol (202), lindane (13), ethacrynic acid (88), and CCl4 (47), either in the whole rat (13, 47, 88), in isolated perfused rat livers (IPRLs) (5, 10, 14, 202, 203), or in isolated rat hepatocyte couplets (14, 203); this last model consists of pairs of hepatocytes that remain attached after isolation, which reorganize their canalicular structures as a central vacuole where transport of fluorescent analogs of Bsep and Mrp2 substrates can be monitored (135).
Some therapeutic compounds, including cyclosporine A (21), dapsone (206), and nitrofuran derivatives (78), also induced cholestasis due to their pro-oxidant effects. Finally, experimental procedures generating hepatocellular OS, such as aluminum intoxication in rats (65), hepatic ischemia–reperfusion (98), or exposure of cultured hepatocyte couplets to tumor necrosis factor-α (TNF-α) in the extracellular medium (31), also impaired biliary secretory function.
These cholestatic effects more often involve BSDBF failure. In some cases, the impairment in the total bile flow can be masked by the superposed BSIBF elevations due to the concomitant increase in the output of the intracellularly formed GSSG (10, 116), or the osmotic choleretic effect driven by the output of metabolites of the oxidant compounds (e.g., ethacrynic acid) (88). However, all these situations should be considered cholestatic as well, since cholestasis is nowadays defined as an exacerbated BS retention in the hepatic parenchyma secondary to the excretory failure (219).
The classical interpretation of the cholestatic effects associated with OS is centered on the occurrence of the following pathomechanisms: OS-induced hepatocellular apoptosis or necrosis, depending on the degree of the oxidant damage; this reduces the amount of living and hence functional parenchymal cells (128) (see Obstructive Cholestasis-Induced Hepatocellular Transporter Internalization: Role of BS-Induced NOX Activation section for details). i) BSDBF failure due to competitive cis-inhibition of Bsep by the excess of GSSG formed by the oxidant insult (5, 10). GSSG cis-inhibited Bsep in liver canalicular membrane vesicles (68), and an inverse relationship between GSSG and the output of BSs was shown when different oxidant agents were administered to IPRLs, such as menadione (4), t-BuOOH (5, 10), and hydrogen peroxide (5). ii) BSIBF impairment due to a drop in the biliary output of glutathione, due, in turn, to depletion of glutathione hepatic levels after sustained efflux as GSSG, to keep the intracellular GSH/GSSG ratio constant (98).
The previous mechanisms are more likely to occur under high oxidant conditions. However, under low or even very low OS levels, impairment of bile secretion is also apparent, thus suggesting that more subtle cholestatic mechanisms operate under low OS condition.
After exposure to different oxidant compounds to IPRLs, cholestasis occurred before the release of cytosolic hepatic enzymes or the rise in intracellular GSSG (10, 88). Similarly, in studies in hepatocyte couplets, the efflux of fluorescent analogs of BSs to the apical vacuole was reduced by low t-BuOOH concentrations, without affecting cellular viability or GSSG content (2). t-BuOOH exerts its oxidizing effects by generating tert-butyl alkoxyl radical (t-buO•) in reaction with Fe2+, and the further production of tert-butyl peroxyl radical (t-buOO•) when reacting with another t-BuOOH molecule, or with the Fe3+ formed previously (80). This renders alkoxyl and peroxyl radicals that may react with lipids and proteins to produce lipid peroxidation and protein thiol modifications, respectively. This eventually opens Ca2+-dependent mitochondrial permeability transition pores (MPTPs) and promotes the further release of the electron carrier cytochrome c, which affects the electron flow through the electron transport chain (54), thus promoting superoxide anion mitochondrial formation and leakage (117). By using two GSH molecules, glutathione peroxidase reduces t-BuOOH to tert-butyl alcohol (t-buOH) to form GSSG, thus preventing t-BuOOH oxidizing effects.
Endocytosis of hepatocellular carriers engaged in bile generation is a key causal factor of t-BuOOH- and other oxidizing compounds-induced cholestasis, and a common trait in both experimental and clinical cholestatic hepatopathies involving oxidative damage (169) (Table 1).
Structural Alterations and Signaling Mechanisms Involved in the Endocytic Internalization of Hepatocellular Transporters in Experimental and Clinical Cholestatic Conditions Associated with Oxidative Stress
AP2, adaptor protein 2; BDL, bile duct ligation; Bsep/BSEP, rodent/human bile salt export pump; cPKC, “classical” protein kinase C; ERK1/2, extracellular signal-regulated protein kinases 1/2; F-actin, filamentous actin; HuH-NTCP, HuH-7 cells transfected with NTCP; IL, interleukin; JNK1/2, c-Jun N-terminal kinase 1/2; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MARCKS, myristoylated alanine-rich C kinase substrate; Mrp2/MRP2, rodent/human multidrug resistance-associated protein 2; NAFLD, nonalcoholic fatty liver disease; NO, nitric oxide; NOX, NADPH oxidase; nPKC, “novel” protein kinase C; Ntcp/NTCP, rodent/human Na+-taurocholate cotransporting polypeptide; PI3K, phosphoinositide 3-kinase; PKA, protein kinase A; PKC, protein kinase C; PP-1, protein phosphatase-1; ROS, radical oxygen species; SUMO, small ubiquitin-like modifier; TNF-α, tumor necrosis factor-α.
Rapid internalization of Bsep has been demonstrated by us and other groups in several experimental cholestatic contexts, including those related to obstructive cholestasis, hyperosmotic shock, and administration of different cholestatic agents, such as cyclosporine A, taurolithocholate, and estradiol-17β-
A similar phenomenon was reported for Mrp2 after exposure of IPRLs to t-BuOOH (14, 181, 203), chlorodinitrobenzene (181), and ethacrynic acid (88, 184), and in hepatic ischemia–reperfusion as well (108). Since Mrp2 is the main canalicular GSH transporter (152), and GSH is a major hepatocellular antioxidant (11), Mrp2 endocytosis in the OS context may aid to preserve intracellular GSH content (208). In keeping with this, hypertonicity-induced Mrp2 endocytosis delayed radical oxygen species (ROS) generation and the consequent oxidative damage induced by the GSH-depleting agent, ethacrynic acid (88).
GSH acts as a scavenger of electrophilic and oxidant species either directly, by acting as a free radical scavenger, or indirectly via enzymatic mechanisms. For example, GSH is the cosubstrate of glutathione peroxidase, an enzyme that reduces hydrogen and lipid peroxides to render GSSG (63). GSH also counteracts electrophilic stress by helping to depurate electrophilic compounds (e.g., drugs/pollutants and their phase I metabolites), which are conjugated with GSH by glutathione-S-transferases. Apart from being substrates of MRP/Mrp2, these glutathione conjugates are substrates of γ-glutamyl transpeptidase, the first step in the mercapturic acid pathway that forms mercapturic acids to be secreted into blood and delivered to the kidney for urinary excretion (76).
All the information above points to OS as a major determinant of the localization status of hepatocyte transporters involved in bile formation, and we will discuss next the structural and signaling events underlying these effects.
Cellular Structural Basis of Hepatocellular Transporter Localization
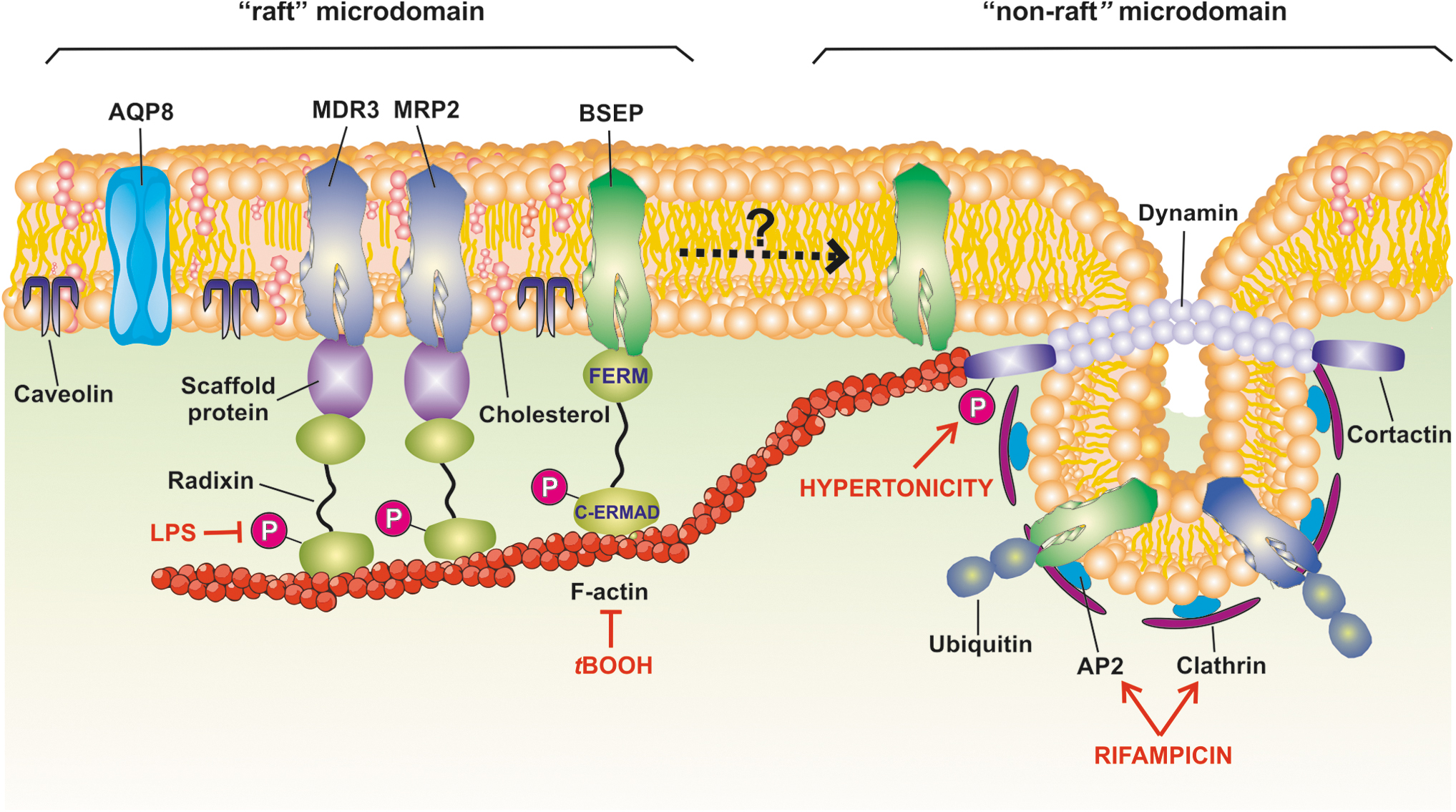
The actin cytoskeleton is a dynamic network consisting of both filamentous actin (F-actin) and associated actin-binding proteins (30). Both integrity and proper dynamics of this complex are crucial for the distribution and stability of integral membrane proteins in their membrane domains, including transporters (104). The proper localization in cholesterol-rich membrane microdomains (“lipid rafts”) of most hepatocellular transporters involved in bile formation is essential for their optimal transport activity. Therefore, the transporter stability in such lipid structures is required for a normal biliary secretory function (8, 97).
Stability in lipid rafts is achieved through a complex network of interactions with neighbor structures from the plasma membrane itself and with cortical F-actin (28), which is tethered to transporters through different actin-membrane linkers (30). It is therefore expected that F-actin-cytoskeleton disruption, a common event under OS conditions (110), plays a major role in ROS-induced endocytosis of hepatocyte carriers (155). Figure 2 schematizes the main findings concerning the membrane localization of canalicular transporters and the cellular components involved in membrane transporter stability, as well as the structures that are altered by OS, thus leading to increased transporter internalization.
At the basolateral pole, vesicular transport of Ntcp to the basolateral membrane involves both an initial microtubule-dependent delivery to the nearest of the plasma membrane and a final microfilament-dependent insertion (44). This occurs from a subapical, early recycling endosomal compartment (178), which functions as a reservoir of carriers for rapid insertion under physiological demand.
Both the normal trafficking of Ntcp to its membrane domain and its stability in membrane require the occurrence of, at least, one glycan molecule bound to the N5 or N11 asparagine residues (109); otherwise, Ntcp is rapidly degraded in lysosomes (109), at difference of the glycosylated one, which is degraded by proteasomes (7). Ntcp activity is also affected by the basolateral membrane lipid composition. In mouse, Ntcp is largely localized in “raft” microdomains, with its plasma membrane distribution and transport activity being dependent on membrane cholesterol content (139).
Regarding the basolateral transporters OATPs, their interaction with the scaffold protein PDZK1 (PDZ domain containing 1, aka NHERF3) is required for a correct sinusoidal membrane localization (213). PDZK1 regulates the intracellular trafficking of the Oatp family member Oatp1a1 by recruiting kinesin-1, a plus-end-directed microtubule-based motor of vesicles containing Oatp1a1; without PDZK1, these vesicles lack the capacity to recruit kinesin-1 and associate with the minus-end directed motor, dynein, to move toward the microtubule minus-end, thus resulting in internalization of these vesicles (215). In addition, phosphorylation of Oatp1a1 at Ser634 and Ser635 (64), which does not affect the transporter capability to associate with PDZK1 (29), induces inhibition of Oatp1a1 transport activity (62) by promoting the rapid endocytosis of the transporter (29).
At the canalicular pole, de novo-synthesized ABC carriers are trafficked, via the Golgi complex, to the canalicular membrane domain (94); they can alternatively be inserted at the canalicular membrane via recycling from apical recycling endosomes (ARE).
The latter compartment also functions as a reservoir for pre-existing canalicular carriers, from which they can be rapidly reinserted on physiological demand; this is followed by endocytosis and the return of the transporters back to ARE, once demand is satisfied (212). This endocytosis is microtubule independent but actin dependent in nature, and involves the participation of apical early endosomes (AEEs), as the first compartment where endocytosed proteins are delivered (77). Then, transporters are trancytosed to ARE via microtubules (6), and can be reinserted by a microtubule-dependent mechanism, when on demand (95). Alternatively, after endocytosis, the carriers stored in AEE can be trafficked, through late endosomes, toward lysosomes for final disposition (159).
This exocytic/endocytic process is unbalanced in many experimental and clinical cholestatic conditions, either by an exacerbation of the endocytic process and/or by impairment of the reinsertion of the endocytosed transporters, thus leading to intracellular vesicular accumulation (169).
Plasma retention of carriers once inserted into the canalicular membrane domain requires tethering to the actin cytoskeleton via different F-actin crosslinking structures, as for example proteins belonging to the ezrin-radixin-moesin (ERM) family (22).
Functional activation of ERM proteins highly depends on protein kinase-mediated phosphorylation (55). All ERM proteins contain two ERM association domains (ERMADs): N-ERMAD, the membrane-binding domain localized at the N-terminal end, and C-ERMAD, the F-actin binding domain localized at the C-terminal end (55). Small GTPase Rho-mediated phosphorylation of C-ERMAD at a threonine residue is essential for the detachment of the binding of N- and C-ERMAD to each other, thus generating the “open,” active forms of the ERM proteins (132). The main hepatocellular ERM protein is radixin (59), which is preferentially localized at the canalicular domain (59), where it links plasma membrane proteins to the actin cytoskeleton, either directly or through adaptor proteins.
The latter indirect association between ERM and transmembrane proteins occurs via PDZ domain-containing scaffolding proteins, including PDZK1 and ERM-binding phosphoprotein of 50 kDa (EBP50, aka Na+-H+ exchanger regulatory factor 1, NHERF1) (81).
Both Mrp2 (120) and the human canalicular phospholipid transporter MDR3 (207) associate with radixin through the binding of their PDZ domain to EBP50. In addition, inhibition of the binding of the MRP2 C-terminal domain to the fourth PDZ domain of PDZK1 induces loss of canalicular MRP2 localization associated with intracellular buildup, as shown in the human hepatoblastoma cell line, HepG2 (51). Instead, binding of BSEP to radixin does not require EBP50, but rather occurs via direct binding of BSEP to the amino-terminal FERM domain of radixin (157). A crucial role of radixin in Mrp2 membrane retention has been confirmed in collagen sandwich-cultured rat hepatocytes with downregulation of radixin (214) and in radixin knockout (KO) mice (92).
Other plasma membrane transporters, such as the MDR family of ABC transporters Mdr1/MDR1, Mdr2/MDR3, and Bsep/BSEP, lack an evident PDZ-interacting motif, and can alternatively bind to actin via both HS1-associated protein X-1 (HAX-1) (60) and the receptor for activated C-kinase 1 (RACK1) (82). HAX-1 also associates with cortactin (146), an F-actin binding protein that induces reorganization of the F-actin cytoskeleton (37), and that is implicated in clathrin-dependent endocytosis (26); of note, MDCK cells expressing dominant-negative cortactin have significantly higher steady-state levels of BSEP in their plasma membrane, thus suggesting a role of cortactin in BSEP endocytic internalization (146).
Regarding RACK1, its siRNA knockdown in HepG2 cells reduced the membrane localization of MDR3 (the human homolog of rat Mdr2) and increased its localization in intracellular compartments, thus impairing MDR3-mediated transport activity of phosphatidylcholine (82).
An additional actin-binding protein that anchors plasma membrane carriers is myristoylated alanine-rich C kinase substrate (MARCKS), a widely distributed protein kinase C (PKC) substrate that, when in an unphosphorylated state, can crosslink F-actin (9). When basic serine residues of its effector domain, which are necessary to interact with membrane acidic lipids, are phosphorylated by PKC, its interaction with acidic lipids is abolished, thus causing its detachment from the plasma membrane (148); even though phosphorylated MARCKS continues attached to actin, its release from plasma membrane leads to local disturbances of the cytoskeleton with increased plasticity, which can affect carrier localization as well.
From the above mentioned, it is expected that situations impairing actin integrity and/or expression or localization of radixin and other scaffolding proteins trigger canalicular transporter endocytosis. Such an event was described after the disruption of actin-cytoskeleton integrity induced by phalloidin (174) or administration of pro-oxidants, such as t-BuOOH (155) or taurochenodeoxycholate, a hydrophobic BS (175). This BS also altered the normal localization of radixin, which is relocalized in intracellular structures (175).
Nevertheless, in other cholestatic situations, carrier endocytosis was also observed without any evidence of actin disarrangement, for example, in estradiol-17β-
The same applies to a number of cholestatic liver diseases in humans, including obstructive cholestasis, autoimmune hepatitis, drug-induced liver injury, primary sclerosing cholangitis (PSC), and stage III primary biliary cholangitis (PBC) (99, 100). Ezrin rather than radixin was later reported to be preferentially linked to MRP2, but not BSEP, in human livers, with hyperphosphorylation of ezrin leading to MRP2 internalization in obstructive cholestasis (27).
It has also been shown in HuH-7 cells transfected with NTCP (HuH-NTCP) cells that taurolithocholate induces MRP2 endocytosis by PKCɛ activation, with further MARCKS phosphorylation and MARCKS plasma membrane detachment (183). On the contrary, changes in cholestatic diseases of the localization or function of other scaffolding proteins, such as HAX-1 (for Mdr2, Mrp1, and Bsep), PDZK1 (for Mrp2), or RACK1 (for MDR3), remain to be corroborated.
Another putative factor that determinates membrane stability of a transport protein is its relative localization in caveolin-enriched, “raft” microdomains versus clathrin-enriched, “nonraft” microdomains. “Rafts” are cholesterol- and sphingomyelin-enriched membrane microdomains, organized in an ordered and tightly packed manner. Several canalicular transporters and channels involved in bile generation, such as Bsep, Mrp2, AE2, and AQP8, reside in “rafts” (83). It has been proposed that “raft” microdomains, which are resistant to dissolution by detergent BSs (83), provide an adequate environment for canalicular transporter stability in membrane, by preventing them from being removed by detergent BSs. Both Mrp2 (83) and Bsep (150) are also more active in “raft” domains, since cholesterol positively modulates their transport activities.
Preferential association of canalicular transporters to “raft” microdomains does not necessarily imply that their endocytosis should occur via the caveolae-dependent pathway. Indeed, Bsep is mainly localized in “raft” domains, but it undergoes physiological, constitutive endocytosis via clathrin-dependent mechanisms, with its C-terminus tyrosine motif being determinant for endocytosis (111). Clathrin-mediated endocytosis takes place exclusively in “nonraft” regions, through the recruitment of clathrin from cytosol to the inner leaflet of this microdomain to form a clathrin-coated pit and further stabilize the formed vesicle (134).
Through the C-terminal tyrosine motif, Bsep associates with adaptor protein 2 (AP2), a key mediator of Bsep endocytosis (74). Inhibition of either ubiquitination (3) or clathrin-dependent endocytosis (3, 146) blocked BSEP internalization. Therefore, it is plausible to hypothesize that, at the steady state, BSEP/Bsep can suffer a constitutive recycling involving membrane transfer from raft microdomains, where it is mostly enriched and shows highest transport activity, to nonraft microdomains, from which it becomes available for clathrin-dependent endocytosis, being later recycled to its membrane domain. The first two proendocytic processes seem to be exacerbated in cholestasis, as we recently demonstrated by using the model cholestatic compound estradiol-17β-
Little information exists about constitutive Mrp2/MRP2 endocytosis. Unlike BSEP, MRP2 seems to undergo endocytic internalization via a clathrin-independent but dynamin-dependent process, as has been shown in Flp-In T-Rex 293 cells (3). Like BSEP, MRP2 is modified by short-chain ubiquitination, and blockage of this process with 4-phenylbutyrate hindered endocytosis and the subsequent MRP2 degradation. Furthermore, an enhancement of plasma membrane MRP2 content and a reduction of plasma bilirubin levels occurred in patients on this treatment (75).
Similar to Bsep, Mrp2 endocytosis occurring in cholestasis induced by estradiol-17β-
Role of Redox-Sensitive Signal Transduction Pathways in Internalization of Hepatocellular Transporters
The role of signaling pathways in rapid cholestatic phenomena, such as those activated by Ca2+ and PKC agonists (23), was already a well-recognized fact by the time hepatocellular transporter internalization was first identified. Therefore, activation of these cholestatic signaling pathways was soon causally linked to the endocytosis of hepatocellular transporters by us and other groups.
Nowadays, an incremental number of cellular signaling pathways are documented to participate in cholestasis, including those mediated by “classical” (Ca2+/diacylglycerol-dependent) and “novel” (diacylglycerol-dependent) PKC isoforms (cPKC and nPKC, respectively) and mitogen-activated protein kinase (MAPK) (169). Most of them seem to be dependent on ROS generation, either from extracellular sources (e.g., neutrophils, pro-oxidant toxics) or from intracellular sources, such as BS-induced mitochondrial dysfunction and NADPH oxidase (NOX) activation.
We will describe next the key role of these signaling pathways in endocytosis of hepatocellular carriers in several types of cholestasis where OS plays a key pathogenic role.
The Ca2+-cPKC signaling pathway: effects on the actin cytoskeleton and actin-binding proteins
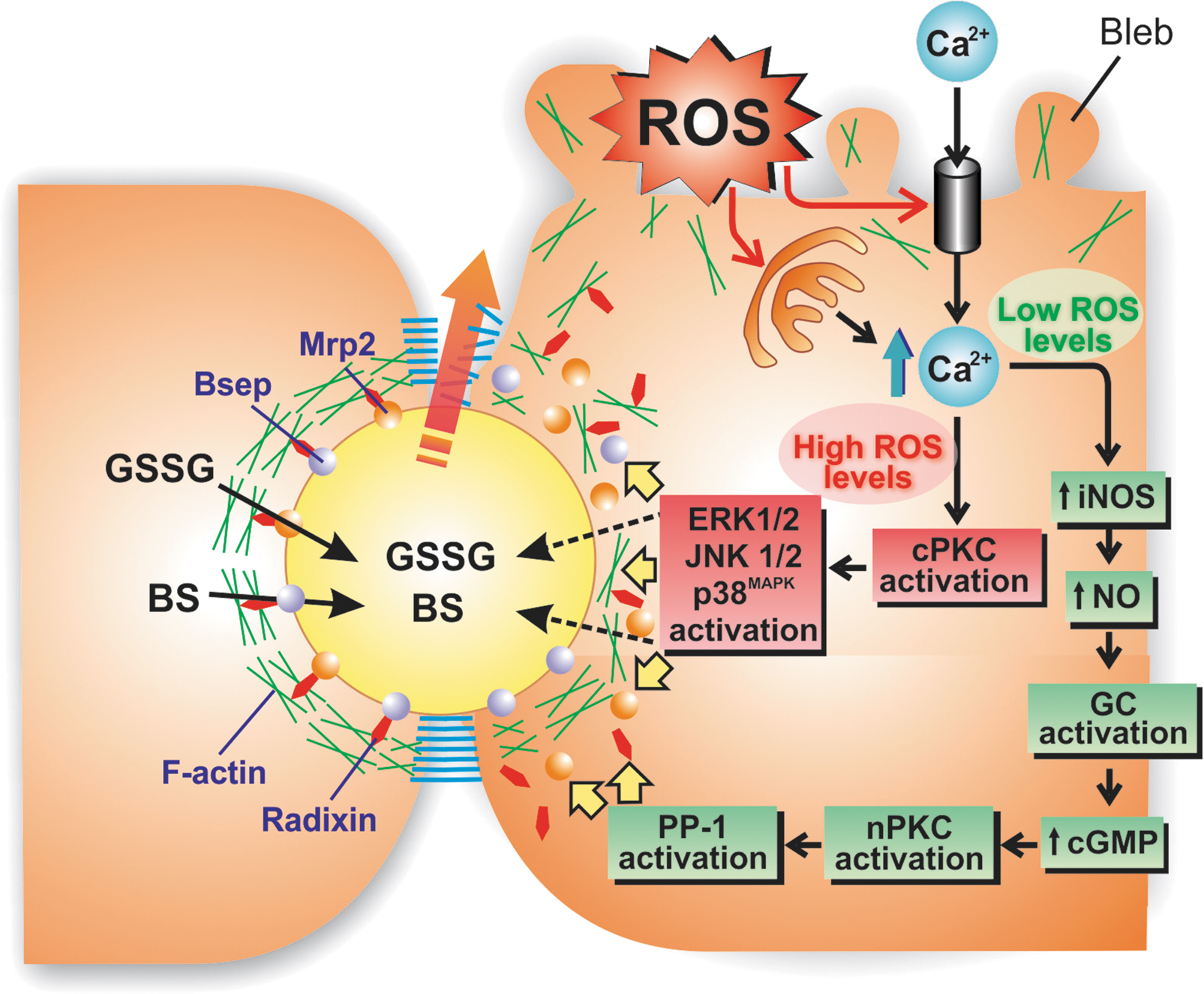
In the hepatic OS context, cytosolic Ca2+ elevation is a central pathogenic event. It involves both the Ca2+ release from intracellular stores (calciosome) and the influx of extracellular Ca2+ via receptor-operated Ca2+ channels at the sinusoidal membrane (163).
Subtle alterations in biliary secretion occur at low (non-necrotic) levels of OS, and Ca2+ elevations are crucially involved. We showed that BAPTA/AM, a cytosolic Ca2+ chelator, fully blocked the quick (minutes) alterations induced by low t-BuOOH concentrations in the capability of hepatocyte couplets to accumulate and retain in their canalicular vacuoles fluorescent BS analogs (197). Suggestively, the actin-cytoskeleton disorganization was fully counteracted by Ca2+ sequestration as well, further suggesting a causal link between these two events. Moreover, A23187, a Ca2+ ionophore, mimicked the damaging effects of t-BuOOH on both actin-cytoskeleton organization and canalicular transport function (197). Collectively, our findings indicate that Ca2+ signaling, rather than the direct F-actin oxidation, is involved in the hepatocanalicular dysfunction induced by low OS levels.
What are the downstream signaling mediators of these Ca2+ effects? Among the multiple events regulated by Ca2+, cPKC activation, a common feature under OS conditions (66), seems to play a key role. cPKC isoforms have a Ca2+-binding domain, and after cytosolic Ca2+ elevations, plasma membrane recruitment of cPKC occurs, thus inducing its activation (40). A role of cPKC in hepatocellular transporter endocytosis is somewhat anticipated from studies on selective cPKC activation under nonoxidative conditions by thymeleatoxin exposure. This compound induced both bile flow drop and endocytosis of Bsep in IPRLs (106), and the same holds true for Ntcp (198).
Our group demonstrated that t-BuOOH induces cytosolic Ca2+ elevations and cPKC activation in isolated hepatocytes (154), and that this was associated with actin-cytoskeleton disarrangement (154) and canalicular secretory dysfunction (155) in hepatocyte couplets, two events that were fully prevented by cPKC inhibitors; alkoxyl and peroxyl radicals generated by t-BuOOH potentiate the lipid peroxidation-dependent mechanisms on Ca2+ signaling, which would lead to greater release of Ca2+ (50).
What is even more important from the curative point of view, both alterations were reversed within 1 h by cPKC inhibition, even having maintained t-BuOOH in the culture medium. The BS secretory failure was due to Bsep internalization, which was completely prevented by cPKC inhibitors as well (155). The same happened with the alterations in tight-junctional permeability, another outcome of actin disorganization (154). The involvement in these effects of other protein kinases acting downstream of cPKC should be considered.
We recently demonstrated in rats that t-BuOOH is a pan-activator of hepatic MAPKs (extracellular signal-regulated protein kinases 1/2 [ERK1/2], p38MAPK, and c-Jun N-terminal kinase 1/2 [JNK1/2]), with cPKC being the mediator for the latter two activations, and that these three MAPKs cooperated to promote actin disarrangement and the further endocytosis of the canalicular transporters Bsep and Mrp2 (203). The mechanism by which this cPKC/MAPK-mediated F-actin reorganization occurs is currently unknown. t-BuOOH, like others Ca2+-elevating, cPKC-activating agents such as vasopressin (171), induces F-actin disruption, thus leading to membrane bleb formation. This phenomenon first involves F-actin cortex disruption, which makes the plasma membrane to become locally detached from the actin cortex. This is followed by the rapid formation of plasma membrane protrusions promoted by the contraction of the cortical actomyosin, which, in turn, generates the increase in cytoplasmic hydrostatic pressure that drives this process (53); this explains the protection conferred by “migrastatics” that, like blebbistatin, inhibit myosin II-ATPase activity.
Actomyosin contractility requires not only Ca2+ but also the PKC-dependent activation of Rho-kinases (ROCKs), thus explaining the role of the Ca2+/PKC-dependent pathway in ROS-induced bleb formation (84). In addition, p38MAPK has been pointed as a downstream target of ROCK to promote cellular blebbing, thus perhaps explaining our finding that this MAPK is involved downstream of cPKC in ROS-induced blebbing (203).
The nature of the canalicular transporter endocytosed under OS conditions and the signaling molecules implicated depends on the amount of ROS generated. Low levels of the oxidant agent ethacrynic acid, a glutathione-depleting agent, did not trigger cPKC but nPKC activation in rats. In this context, ethacrynic acid selectively triggered Mrp2 endocytosis without affecting Bsep. This occurred via the Ca2+-mediated activation of inducible nitric oxide synthase (iNOS), followed by nitric oxide (NO)-mediated cyclic guanosine monophosphate (cGMP) elevations, and the subsequent cGMP-mediated nPKC activation (184). On the contrary, higher ethacrynic acid doses capable to activate both nPKC and cPKC induced both Bsep and Mrp2 endocytosis (184).
OS-induced endocytosis of Mrp2 is associated with a reduction in the proportion of the phosphorylated, active form of radixin (p-radixin) via nPKC-dependent protein phosphatase-1 (PP-1) activation, which disrupts Mrp2 plasma membrane stability (186). This endocytosis was reverted by GSH refilling with GSH-ethyl ester, a membrane-permeable GSH vehicle that releases the antioxidant after intracellular esterase-catalyzed hydrolysis (185).
Interestingly, the association of p-radixin with Mrp2 is enhanced by the protein kinase A (PKA) activation that occurs during the recovery phase induced by GSH replenishment (186). This is in line with our finding that t-BuOOH-induced F-actin disarrangement was also reverted by cAMP-induced PKA activation (154), so that not only the binding of p-radixin with Mrp2, but also the actin cytoskeletal integrity as a whole, is protected by PKA. Hypoxia-induced OS also led to Mrp2 internalization in the rat hepatoma hybrid cell WIF-B cells, with radixin being redistributed from the pericanalicular region to the remaining cytoplasmic region (200).
Contrary to situations where OS is rapidly reversed, long-lasting OS conditions gradually promote Mrp2 degradation, as shown in rats exposed for 8 h to a GSH-depleting agent (187). A proteasomal degradation pathway has been proposed, since a short-chain Mrp2 ubiquitination occurred concomitantly (187). However, short-chain ubiquitination targets Mrp2 for lysosomal rather than for proteasomal degradation (75). Since post-translational Mrp2 conjugation with small ubiquitin-like modifier (SUMO) proteins, an event that boosts Mrp2 expression (136), is inhibited as ubiquitination is enhanced in long-lasting OS situations (187), a balance between SUMOylation and ubiquitination was proposed as a determining factor of the fate of Mrp2 after endocytosis (i.e., reinsertion into the canalicular membrane vs. degradation) (187).
As depicted in Figure 3, a picture is emerging of how acutely induced OS can impair canalicular transporter function. Under OS conditions not impairing hepatocyte viability, the elevations of cytosolic Ca2+ induce both cPKC and nPKC activation or, selectively, nPKC activation, depending on the magnitude of the OS levels. These signaling pathways mediate several functional and morphological alterations in several cellular constituents crucial for bile generation, such as the F-actin network, canalicular carriers, and tight-junctional structures. This, in turn, impairs the biliary excretion and the further retention in the canalicular space of compounds that generate the osmotic driving force for canalicular bile production. Sustained oxidative conditions lead to degradation of the internalized transporters, through mechanisms that need to be better characterized.
NOX-derived ROS as signaling molecules that induce hepatocellular transporter internalization in cholestasis
The membrane-associated NOX family of superoxide and hydrogen peroxide producing proteins is an important source of ROS involved in signal transduction cascades that control numerous physiological processes (physiologically generated ROS), and also deleterious signaling pathways that impair cellular function and integrity (pathologically generated ROS) (113).
The NOX family is composed of five NOX proteins (NOX1-5) and two dual oxidases (Duox1 and Duox2) (113). Each NOX forms a multisubunit enzyme complex that catalyzes the transfer of electrons from an electron donor (NADPH) to an electron acceptor (oxygen [O2]). This leads to the formation of superoxide, the main ROS molecule of biological systems.
The NOX2 complex, taken here as an illustrative example because it is the most complete in terms of the subunits assembled, consists of two subunits, NOX2 itself (gp91phox) and p22phox, thus forming the plasma membrane-bound cytochrome b558 (Fig. 4). The activation of this complex takes place when three cytosolic subunits, p40phox, p47phox, and p67phox, and the small G protein Rac traffic to the plasma membrane and associate with cytochrome b558 (112).
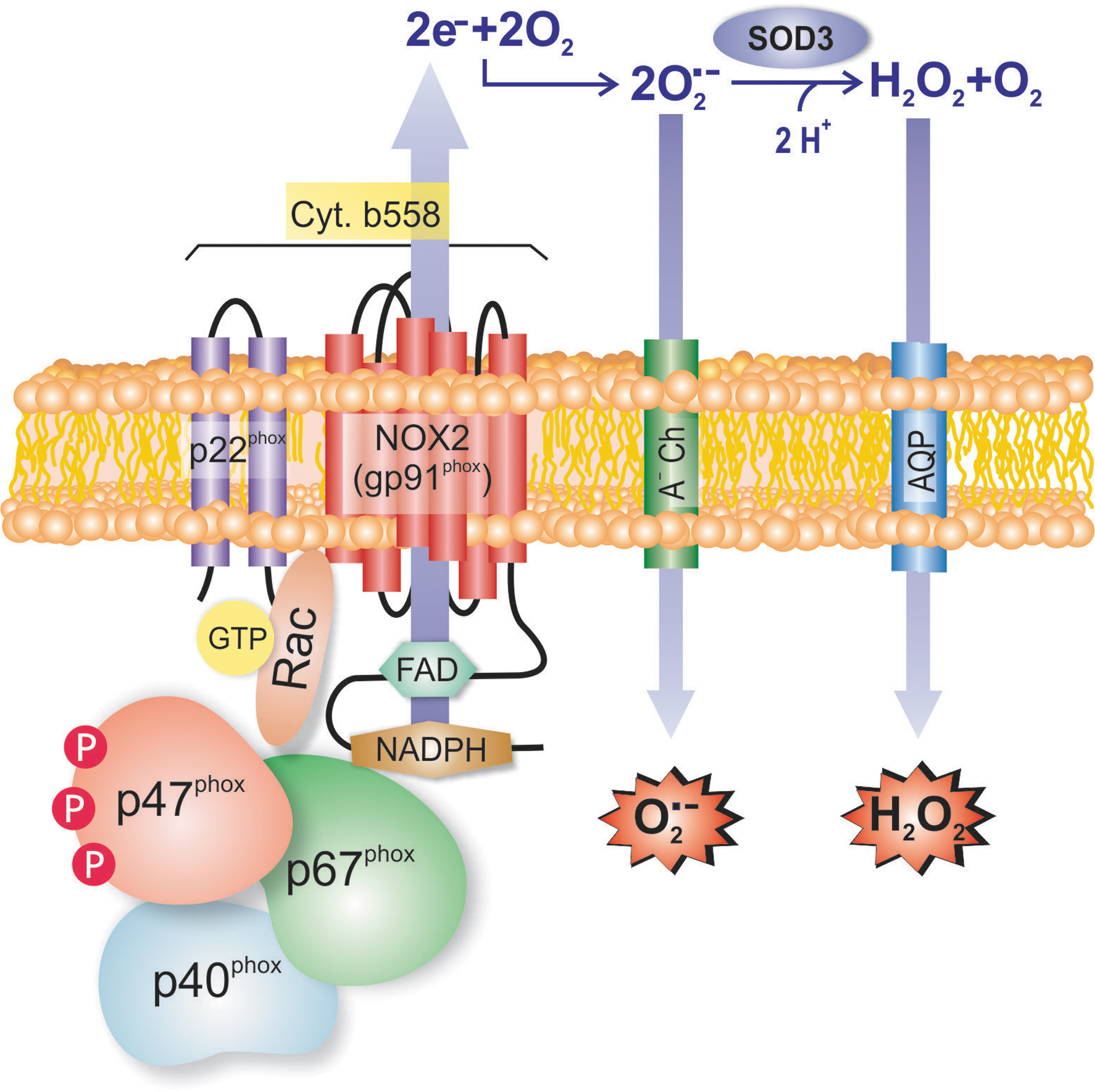
The remaining NOXs form similar complexes with other proteins, with p22phox being shared by NOX1, NOX3, and NOX4 complexes. For example, NOX1 associates with the same cytosolic subunits as does NOX2, except that p40phox is absent, and p47phox and p67phox are replaced by their homologs, NOXA1 and NOXO1, respectively (112). NOX4 is a constitutively active membrane-bound isoform, and it associates only with p22phox (112). Human (140) and murine (165) hepatocytes express NOX 1, 2, and 4, as well as Duox-1 and 2, and the same holds true for hepatic stellate cells (HSCs) (140). On the contrary, Kupffer cells and other immune cells express only NOX2, whereas sinusoidal endothelial cells express NOX1 and NOX4 (140). The production of ROS in HSCs and Kupffer cells is, actually, predominantly dependent on NOXs (15).
We will discuss next three cholestatic situations where NOX activation seems to play a cholestatic role by promoting endocytosis of hepatocellular carriers, namely hypertonicity, inflammation, and obstructive cholestasis.
Role of NOX in hypertonicity-induced hepatocellular transporter internalization
Among the physiological roles of NOX, the modulation of the localization status of basolateral and canalicular carriers as part of the hepatocyte adaptation to perturbations of cell hydration by changes in ambient osmolarity is a good illustrative example (72). Modulations of the hepatocyte hydration status can be induced by extracellular ambient aniso-osmolarity or, alternatively, by hormonal mediators (e.g., glucagon, insulin), substrates (e.g., BSs), and OS (179). Hepatocyte swelling increases bile flow by enhancing BS transport, while hepatocyte shrinkage induces rapid bile flow decrease through reducing BS output (71). The latter event is associated with a rapid NOX2-dependent increase in hepatocellular ROS, mainly superoxide anion (166).
To ascertain the mechanisms behind this cholestatic effect, the acute influence of anisotonicity on the localization status of the canalicular export pumps Bsep and Mrp2 (180) and the basolateral transporter Ntcp (195) has been studied in the IPRLs.
Extracellular hypertonicity induces internalization of these canalicular (25) and basolateral (195) transporters by a mechanism that involves Fyn, a constituent of the Src protein-thyrosine kinase family (Fig. 5). Regarding canalicular transporters, a Fyn-dependent phosphorylation of cortactin, a pericanalicular F-actin/dynamin-crosslinking protein that binds, via its Src homology-3 (SH3) domain, to the C-terminal, proline-rich moiety of dynamin, has been shown. Fyn facilitate clathrin-dependent transporter endocytosis by regulating actin dynamics, which produces forces that deform the plasma membrane, thus triggering vesicle invagination and fission; this occurs when the F-actin crosslinking activity of cortactin is impaired after Fyn-dependent phosphorylation (79). On the contrary, Ntcp is endocytosed after direct Fyn phosphorylation at Ser-226 (195).
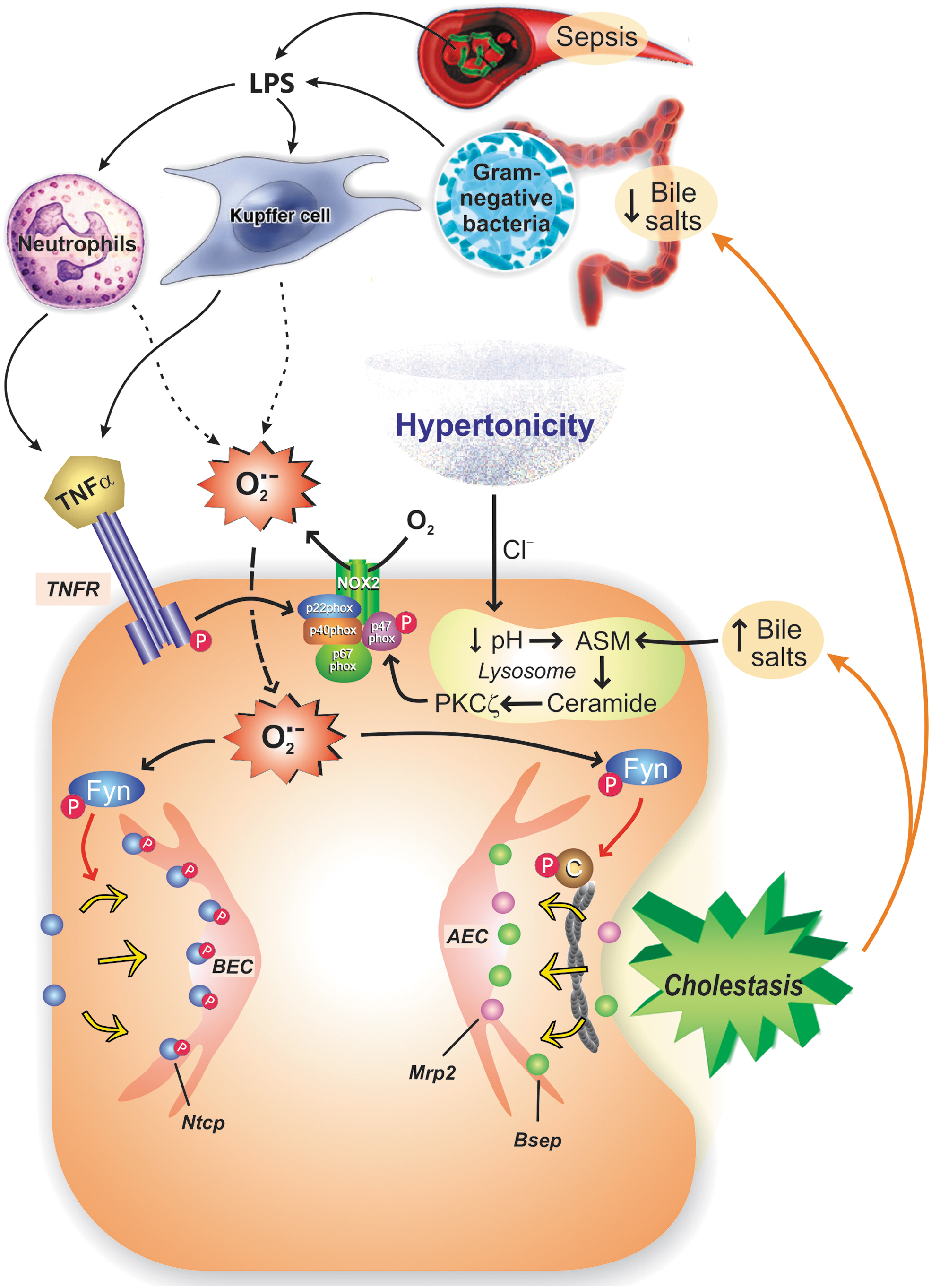
The earliest known event in this pathway is hypertonicity-induced endosomal acidification via a Cl−-dependent, V-ATPase-driven H+ entry, which activates an acidic sphingomyelinase that generates ceramide. A ceramide-dependent PKCζ activation then occurs, which is followed by a PKCζ-dependent serine phosphorylation of the NOX2 regulatory subunit p47phox (164). This triggers NOX2 subunit self-assembly and subsequent NOX2-derived superoxide anion generation, which, in turn, activates Fyn (164). This mechanism was confirmed for Mrp2 and Bsep in p47phox KO mice, where neither hyperosmolarity-induced Fyn activation nor endocytosis of these canalicular transporters was recorded (25), and the same applies to Ntcp (195).
Inflammation-induced hepatocellular transporter internalization: role of inflammatory cytokine-induced NOX activation
Systemic and local hepatic inflammatory conditions are associated with increased ROS in hepatocytes. Both OS and inflammation, two processes that feed one another, play a key role in several hepatopathies, such as alcoholic (220) and nonalcoholic fatty liver disease (131), sepsis-induced cholestasis (177), autoimmune hepatitis (153), and hepatocyte injury during hypoxia/reoxygenation after hepatectomy (36) or liver transplantation (16).
Much of our present knowledge of the role of OS in inflammation-induced cholestasis emerged from experimental studies in sepsis, a model disease of liver inflammation. Sepsis is experimentally mimicked by administrating LPS. This endotoxin can act directly on hepatocytes (118) or, by proxy, through Kupffer cells (85, 217), which secrete inflammatory cytokines, such as interleukin (IL)-6, TNFα, and IL-1β, which, in turn, promote ROS generation (138).
Administration of LPS to rats induces endocytosis of both Mrp2 (18, 19, 41, 107, 160, 161, 176, 188, 204, 222, 225) and Bsep (160, 161, 208, 225); Mrp2 endocytosis was reversed by ROS scavengers, such as dimerumic acid (188, 222). Exposure of human slices to LPS also induces internalization and further downregulation of the expression of both transporters through post-transcriptional mechanisms (48), suggesting a key role of exacerbated degradation of the endocytosed transporters in human cholestatic hepatopathies (48). LPS seems to induce low levels of ROS, since F-actin remained unaltered 3 h after LPS administration (176).
The cholestatic action of LPS depends on cytokines secretion, since maneuvers that decrease cytokines synthesis or release, such as dexamethasone administration (107) and heat stress (18, 19), blocked LPS-induced Mrp2 internalization in rats. However, evidence to discern whether this effect depends on one cytokine in particular or on some combination of them is lacking.
From previous results, it is clear that the mechanism proposed for LPS-induced transporter internalization implies both the release of cytokines and ROS formation. The link between both phenomena seems to be NOX (Fig. 5).
Typically, a cytokine interacts with a hepatocyte receptor that couples to NOX to generate ROS. For TNFα, PKCα has been proposed to mediate the interaction between TNF receptor-1 (TNFR1) and NOX2 in fibroblasts (124), whereas both in HEK293 embryonic renal cells (205) and in HeLa cells (223), NOX1/3 and NOX1/2 activation, respectively, occurs by the interaction of the cytosolic domain of TNFR1 with RAC1/riboflavin kinase (RFK) and p22phox.
The mechanism probably depends on the cellular type involved. For IL-1ß, the link between the cytokine receptor and NOX2 is provided by ERK1/2 and p67phox in monocytes (190). Recently, our group confirmed that the sequence TNFα-NOX-ROS is implicated in Mrp2 endocytosis in rat hepatocytes, since TNFα-induced internalization of Mrp2 was blocked by both antioxidants and the NOX inhibitor apocynin, which also blocked TNFα-induced ROS generation (31). Apocynin is a NOX inhibitor that prevents the organizer NOX subunit p47phox from binding to p22phox; since p47phox is part of NOX1 and NOX2, either or both NOX isoforms could be involved.
The NOX2-generated ROS, predominantly superoxide anion, activate downstream signaling molecules that finally internalize canalicular transporters. One of them could be Fyn, since this tyrosine-protein kinase participates in hypertonicity-induced hepatocellular transporter internalization, an event that shares with TNFα both NOX2 activation and NOX2-mediated superoxide anion production (see previous item).
Other possible mediators are cPKC (42), p38MAPK, and ERK1/2, which are all known to mediate transporter internalization induced by t-BuOOH (203). Actually, TNFα activates different PKC isoforms (α, δ) (57, 93) and MAPKs (211), and the latter also occurs with IL-1β (173). Recently, we corroborated the involvement of ERK1/2 downstream of the TNFα-NOX-ROS pathway that leads to Mrp2 internalization in rat hepatocyte couplets (31). However, ERK1/2 inhibition and antioxidants only partially restored Mrp2 transport activity, indicating that other nonredox-sensitive pathways are involved. Whether, as described under other oxidizing conditions (186), ERK1/2 activates a downstream phosphatase that dephosphorylates radixin, thus disrupting Mrp2 membrane anchoring, remains to be confirmed.
Obstructive cholestasis-induced hepatocellular transporter internalization: role of BS-induced NOX activation
Biliary tree obstruction leading to acute BS parenchymal accumulation is a common trait in several cholestatic hepatopathies, with cholelithiasis, tumors, and strictures being the most common causes. BDL, an experimental model that mimics it, induces both Mrp2 and Bsep internalization 48 hs after this maneuver in rats (151), and a similar abnormal localization persists after 7 days of BDL (204). The altered localization and further degradation of Mrp2 may be the cause of the rapid post-transcriptional downregulation of this transporter (151, 204). In line with this, antioxidants improved hepatic Mrp2/Bsep protein levels without improving messenger RNA (mRNA) levels (224).
The mechanisms leading to this endocytic event in BDL remain uncertain. A disturbed radixin phosphorylation status followed by Mrp2 internalization has been reported (101). Accumulation of endogenous BSs secondary to the biliary blockage is likely involved, since BS overload to rats induces Mrp2 endocytosis, associated with disturbed localization of both actin and radixin (175), a finding most likely due to BS-induced ROS generation (192) (Fig. 6).
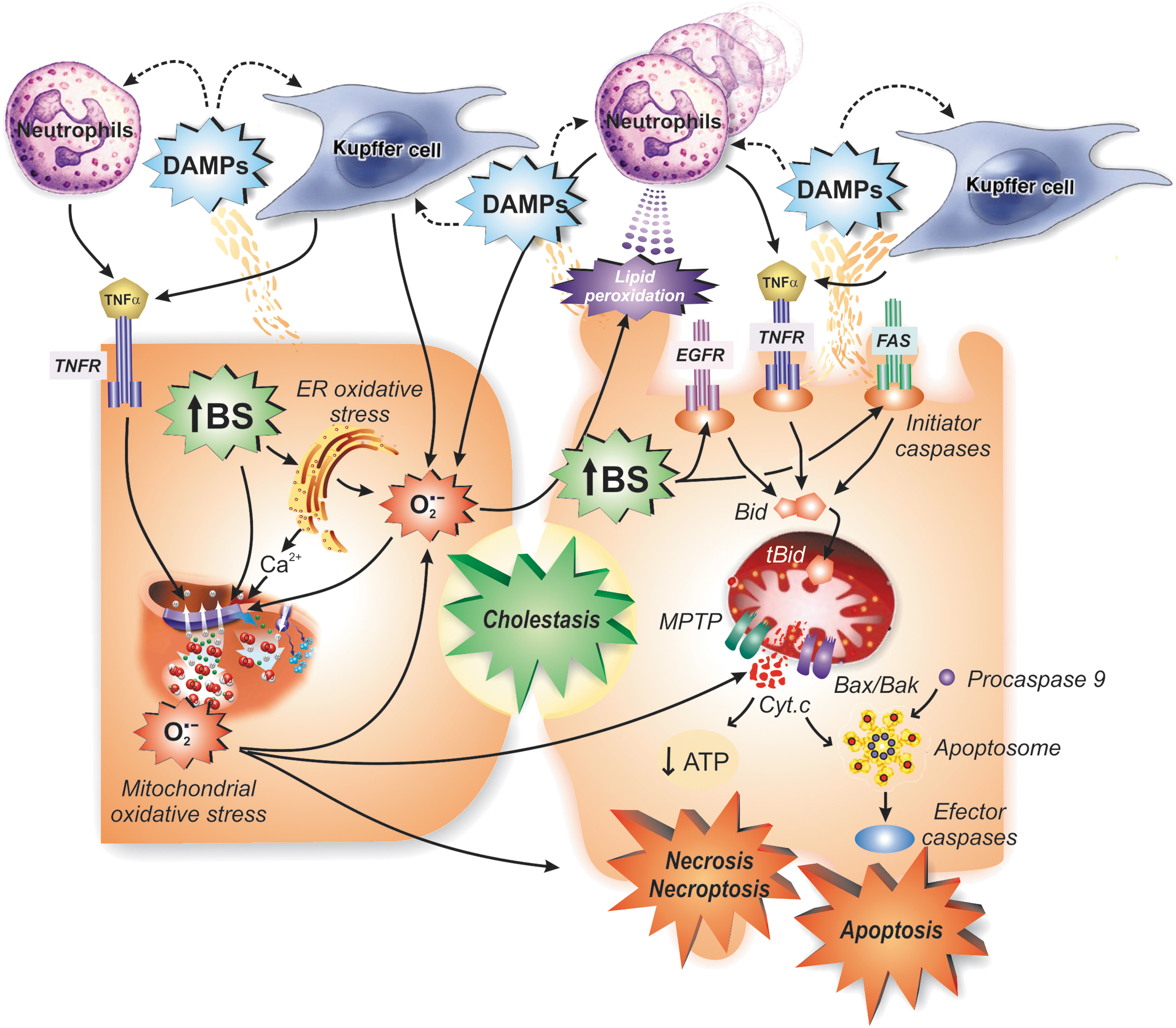
BSs uncouple oxidative phosphorylation, and inhibit both NADH:ubiquinone-1 oxidoreductase (complex I) and decylubiquinol:ferricytochrome c oxidoreductase (complex III) in hepatic mitochondria (103), and the superoxide anion thus generated and other parent oxidant species can cause further mitochondrial dysfunction (194). Accumulated BSs also produce endoplasmic reticulum (ER) stress, which causes superoxide and Ca2+ release that further affects, by contiguity, mitochondrial function and integrity (156). This triggers MPTP formation (1), which allows for the release into cytosol of cytochrome c, and further apoptosis (when ATP levels are still conserved) or necrosis and necroptosis (when ATP is depleted) (90).
Intracellular damage-associated molecular pattern molecules are released by death cells, and promote the release of superoxide anion and proinflammatory cytokines by Kupffer cells and neutrophils chemoattracted to the zone by lipid peroxidation products (86). These two immunological cells can exacerbate mitochondrial injury and cell death either directly via cytokine-dependent and NOX2-derived ROS-dependent damage mechanisms or, for proinflammatory cytokines, via the extrinsic pathway of apoptosis, after binding to their membrane receptors; accumulated BSs reinforce this apoptotic pathway by inducing epidermal growth factor receptor and Fas activation in a ligand-independent manner (128) (Fig. 6).
OS in BDL could be exacerbated by systemic endotoxemia (125), which arises in this model owing to the bacterial and endotoxin translocation into portal circulation caused, in turn, by the lack of endotoxin-neutralizing, bacteriostatic, and mucosal-trophic effects of gut luminal bile (149). Supporting this view, endotoxin-resistant mice did not downregulate Mrp2 protein expression after BDL (210). Both the selective periportal downregulation and the endocytosis of Bsep were associated with portal inflammation by neutrophils infiltrating this zone, and their subsequent TNFα and IL-1β release (42); perhaps, paracrine signaling of NOX2-derived superoxide anion, entering hepatocytes via anion channels, is also involved. Kupffer cells are also activated by endotoxemia in this model, thus representing a possible alternative source of both cytokines and NOX2-derived superoxide (115). Finally, retained BSs activate NOX2, via a signaling pathway involving sphingomyelinase, ceramide, and PKCξ (167) (Fig. 5).
Therefore, the mechanisms described above for NOX2-dependent hepatocellular transporter endocytosis associated with cytokines and BSs should also be in operation in the BDL model. Actually, in rats, BDL leads to a cPKC-dependent NOX2 activation by enhancing the assembly with the p47phox subunit, which was prevented by the antioxidant melatonin, via counteraction of cPKC activation (201).
Endogenous Protection Against OS-Induced Hepatocellular Transporter Internalization: Role of Bilirubin
Bilirubin is the end product of heme breakdown in mammals. Catabolism of hemoglobin, but also of other hemoproteins, yields high amounts of toxic heme, which is enzymatically cleaved into iron, carbon monoxide (CO), and biliverdin by heme oxygenase-1 (HO-1) (127). Rapidly afterward, biliverdin is reduced to bilirubin by biliverdin reductase (127) (Fig. 7).
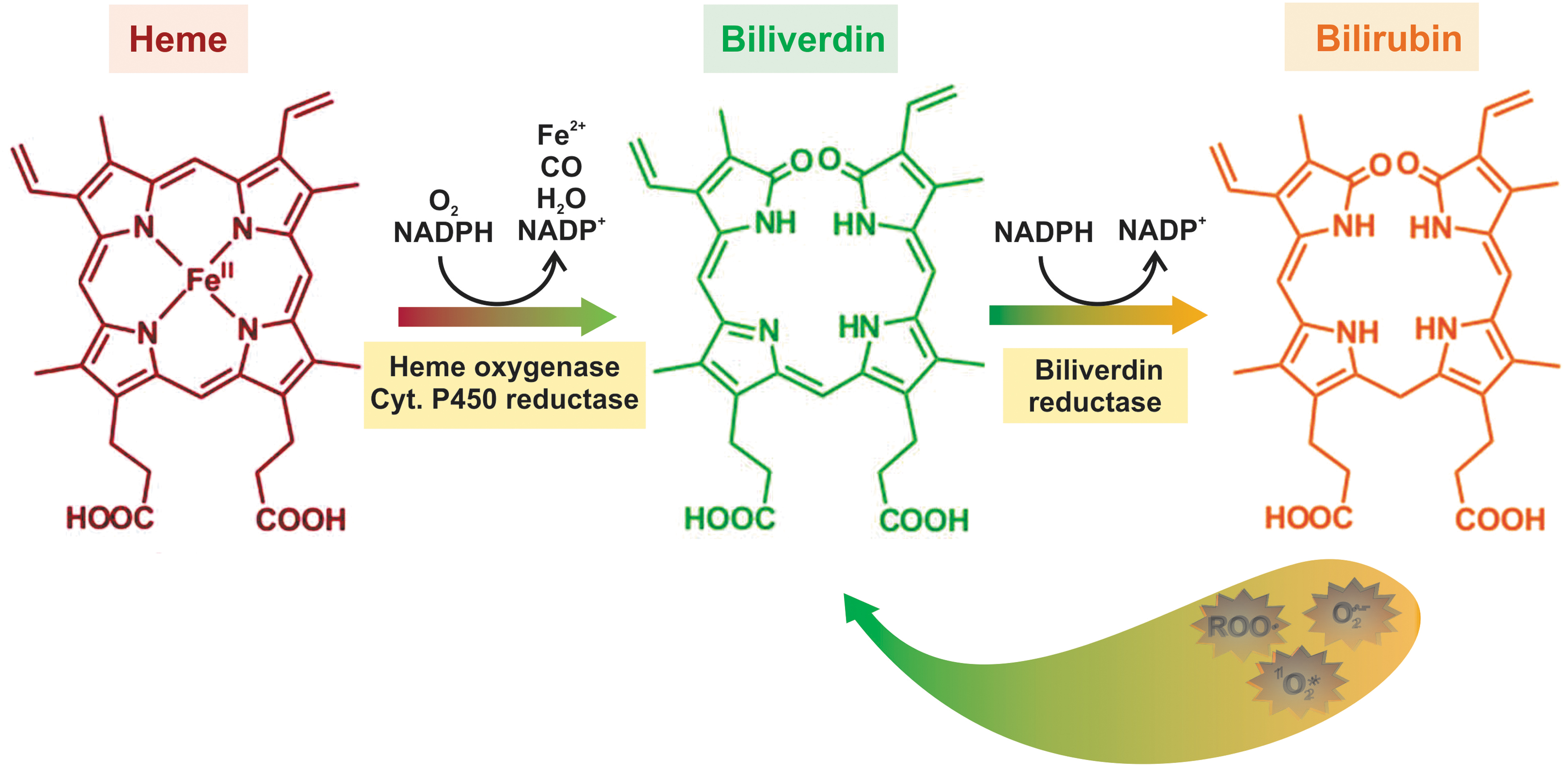
After being carried to the liver attached to albumin, bilirubin is taken up through not yet well-characterized and possible multiple transport mechanisms, followed by binding to intracellular proteins and uridine diphosphate (UDP)-glucuronosyl transferase-catalyzed conjugation with glucuronic acid in the smooth ER. The resulting bilirubin mono- and diglucuronides are excreted into bile via MRP2/Mrp2 (35) (Fig. 1).
Long considered a potentially harmful waste product and its serum elevations a sole sign of liver disease, the consensus on bilirubin nowadays has evolved to that of a biologically relevant molecule that exerts a myriad of beneficial effects (209). Prime among them are its antioxidant effects (196). It is a scavenger of reactive nitrogen species, superoxide anion, singlet oxygen, and peroxyl radicals (87), with the latter explaining its protective effects against membrane lipid peroxidation (218). The antioxidant properties of bilirubin are those expected from its chemical structure. Albumin-bound, unconjugated bilirubin has an extended network of conjugated double bonds and two reactive hydrogen atoms that are donated to ROS, thus exerting its antioxidant effects (196). By doing so, it is converted back into its metabolic precursor, biliverdin, which is then recycled back into bilirubin by the enzyme biliverdin reductase, using NADPH as substrate (12) (Fig. 7).
On this basis, our group investigated whether bilirubin exhibits antioxidant cytoprotective properties in the context of a cholestatic oxidative insult. Even at physiological concentrations (∼17 μM), bilirubin fully prevented t-BuOOH-induced rise in ROS formation in isolated rat hepatocytes, as well as the acute increase in GSSG output in IPRLs, a sensitive marker of hepatocellular OS (116). This cytoprotective effect was afforded by preventing actin-cytoskeleton disarrangement, and the resulting counteraction of Mrp2/Bsep endocytosis. This was associated with a striking prevention of the impairment of both bile flow and the biliary output of model substrates of Mrp2 and Bsep. Bilirubin also prevented t-BuOOH-induced cPKC activation (14), the signaling pathway that mediates both the cytoskeletal rearrangements and the canalicular transporter endocytosis induced by t-BuOOH (see The Ca2+-cPKC Signaling Pathway: Effects on the Actin Cytoskeleton and Actin-Binding Proteins section).
We confirmed these bilirubin cytoprotective effects in the whole animal model, by elevating endogenous bilirubin via induction of HO-1, the rate-limiting enzyme in bilirubin synthesis, by using hemin, an inducer and substrate of HO-1 (130); hemin induces HO-1 expression in both Kupffer cells and hepatocytes, but not in HSCs (56).
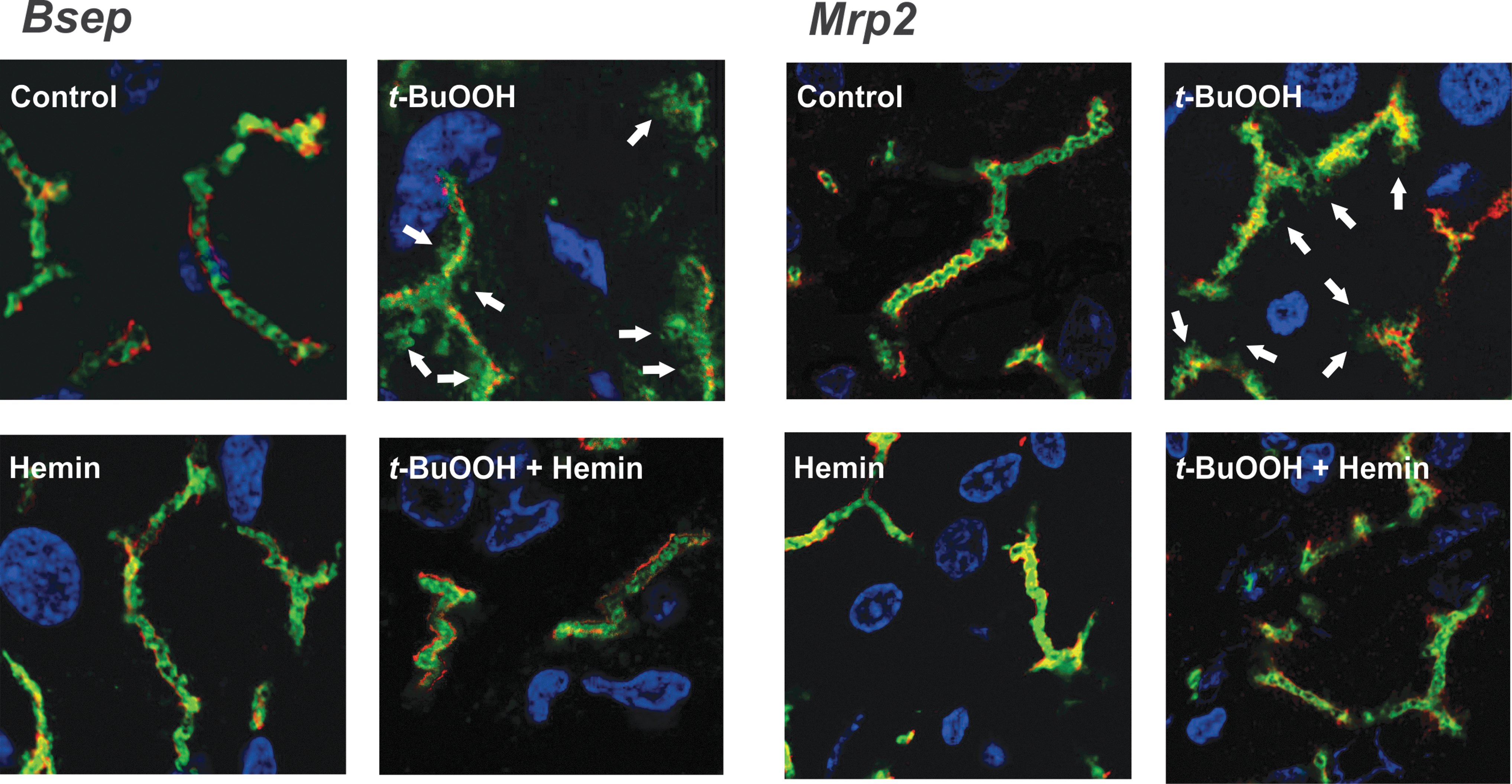
This compound counteracted the elevation of OS surrogate parameters induced by t-BuOOH in vivo, such as oxidation of intracellular proteins and membrane lipids, the increase of hepatic GSSG-to-total glutathione ratio, and the compensatory induction of catalase and superoxide dismutase (130). Accordingly, hemin fully prevented t-BuOOH-induced drop of both bile flow and the biliary output of BSs and glutathione, the major driving forces of BSDBF and BSIBF, respectively; this was accompanied by maintenance of the proper localization in membrane of the respective canalicular carriers of these substrates, Bsep and Mrp2, which were otherwise endocytosed by the oxidative insult (Fig. 8). A similar improvement in Mrp2 localization was afforded by HO-1 induction in a model of liver ischemia/reperfusion, another oxidative maneuver (43).
Clinical Implications of OS-Induced Hepatocellular Transporter Internalization
OS is present in most hepatopathies, including obstructive cholestasis, chronic cholestatic liver diseases (e.g., PBC and PSC), sepsis- and pregnancy-induced cholestasis, hepatic ischemia–reperfusion injury after liver transplantation or hepatectomy, alcoholic and nonalcoholic steatohepatitis, viral, toxic, and autoimmune hepatitis, and hepatopathies associated with hepatic accumulation of transition metals that promote ROS formation by the Fenton/Haber–Weiss reaction (91), such as copper (Wilson's disease) and iron (e.g., hemochromatosis, iron-loading anemia) (58, 121).
Some of them are considered “primary” forms of cholestatic disease, since the pathogenic factor leads primarily to cholestasis, as observed in obstructive cholestasis, PBC, PSC, and cholestasis by sepsis and pregnancy. In all these cases, transporter internalization is a common early event, and a well-recognized pathomechanism to explain the biliary transport dysfunction. Indeed, endocytosis of canalicular transporters has been reported in patients with (i) obstructive extrahepatic cholestasis (27, 100, 189); (ii) autoimmune hepatitis-induced inflammatory cholestasis (100); (iii) mixed (obstructive plus inflammatory) cholestatic hepatopathies, such as PBC (99) and PSC (100); and (iv) drug-induced acute cholestasis (100, 216). Since downregulation of the protein expression of these carriers is mainly post-transcriptional in human beings, the endocytosis and further degradation of canalicular transporters may therefore be a relevant mechanism that explains the hepatopathy (34, 169).
Patients with biliary obstruction who underwent percutaneous transhepatic biliary drainage showed differential levels of carrier relocalization, depending on the effectiveness of the biliary drainage. (100, 189); this suggests a key role for accumulated endogenous compounds in this pathogenic mechanism. The most likely candidates are accumulated BSs, since (i) Mrp2 internalization occurs after BS overload, associated with detachment of radixin from this transporter (175), and (ii) a similar lack of interaction with radixin was shown in human cholestatic diseases (99, 100). OS most likely mediates these deleterious BS effects, since (i) BSs elevate hepatocellular ROS (192), and (ii) OS causes radixin dissociation from Mrp2 (186).
The remaining hepatopathies occurring with OS listed above are considered “secondary” forms of cholestasis, since patients presenting cholestatic manifestations during the course of the disease are relatively common (89). Internalization of hepatocellular transporters has been also documented in many of these cases, as for example in alcoholic hepatitis and in other inflammatory diseases (100), such as nonalcoholic steatohepatitis (70). Therefore, it is tempting to propose that a causal link exists between the occurrence of OS in these hepatopathies and their cholestatic manifestations, with the relocalization of hepatocellular transporters playing a relevant pathogenic role. Cholestasis does not occur in all patients with these diseases. Therefore, a differential cholestatic-prone condition due to genetic or environmental factors should exist, with susceptible patients bearing a pre-existing borderline bile secretory dysfunction, which becomes apparent when a cholestatic oxidant insult is superimposed.
Apart from the cholestatic oxidative processes described above, the role for cholestasis of the redox-signaling mechanism triggered by NOX activation by proinflammatory cytokines is extremely relevant from the physiopathological point of view. Different levels of hepatic inflammation are always expected in cholestasis, either as the initial triggering factor of the hepatopathy or secondary to the accumulation of BSs; this activates an inflammatory innate immune response, with upregulation of cytokines that prompt the influx of neutrophils and the onset of a further oxidative cell injury (24).
In all these cases, the antioxidant effects of bilirubin, which accumulates in these hepatopathies due to the impairment in Mrp2 function and the HO-1 induction associated with OS and cytokines (158), may play a relevant anticholestatic role. In other words, a worse cholestatic outcome would be expected if bilirubin were not present to exert its antioxidant effects. If so, we can envisage hepatocyte accumulation of bilirubin in cholestasis as part of the adaptive response that the liver spontaneously evokes in cholestasis to attenuate the damaging effects of accumulated BSs, in this case, their oxidative deleterious effects. This may be crucial, since, in cholestasis, there is an impairment in enzymatic and nonenzymatic antioxidant defenses that favors the cholestatic liver injury (191). Therefore, reinforcement of the antioxidant defenses with formulations containing antioxidant compounds as an adjuvant therapy in cholestasis (133) would be expected to further contribute to minimize the BS-induced oxidant hepatocellular damage, as has been shown in different cholestatic animal models (122, 193).
Stimulation of the generation of endogenous antioxidants, particularly bilirubin via induction of HO-1, can be another therapeutic alternative. This approach has already shown promising effects in experimental cholestasis (130, 143), and HO-1 pharmacological inducers are already available for pharmacological use (119).
Conclusions and Perspectives
Since the late ‘90s, when internalization of hepatocyte transporters was first described (168), extraordinary advances have been made on the molecular biology of this phenomenon in experimental and clinical cholestasis, and in the understanding of the role for signaling pathways in this alteration. This has opened multiple opportunities to target therapeutically both the structures involved in this pathogenic mechanism and the transductional pathways involved in its regulation. The prompt implementation of pharmacological actions at this level, even in some cases with a prophylactic aim, is mandatory, given the relevance of this pathological mechanism as a very early event in the occurrence of the cholestatic injury, and its role in the aggravation and perpetuation of the disease.
The recognition of the key role that OS plays in this process, either as the very first triggering factor in hepatopathies whose pathomechanisms are primarily oxidative or as an aggravating factor in cholestatic hepatopathies where accumulation of pro-oxidant compounds occurs, opens additional therapeutic alternatives based upon its prevention/reversion. This includes not only the use of antioxidant agents but also the possibility to enhance bilirubin production via HO-1 inducers, two strategies that successfully counteracted the endocytosis of canalicular transporters in experimental settings.
An important role for NOX-derived ROS in cholestasis is also emerging. First described as part of physiological adaptive processes, it also seems to be pathologically relevant, since it is triggered by accumulated BSs and inflammatory cytokines. Different levels of hepatic inflammation always occur in chronic cholestasis, either as the initial triggering factor (inflammatory cholestasis) or secondary to BS toxicity. This opens new therapeutic possibilities based upon its specific control. Actually, NOX-derived ROS play a key role in cholestasis-associated fibrosis (123), and therefore, its therapeutic targeting may have multiple beneficial effects, including the counteraction of the cholestasis itself.
Based upon the crucial importance that OS has in the triggering and perpetuation of the cholestatic injury, we believe that the primary control of oxidative processes in cholestasis is the initial step in the difficult, long-term aim of discovering better therapeutic approaches targeting these processes, and offers a glimmer of hope for the cure of cholestatic diseases. Nowadays, we count with a limited number of anticholestatic drugs, with ursodeoxycholic acid being the more commonly used (172). Interestingly, we showed that this compound affords membrane stabilization of canalicular transporters after a cholestatic insult (160), and it bears antioxidant properties as well (114), in part by attenuating ER stress (147). Perhaps, both properties are causally linked.
Many significant questions remain to be addressed:
What is the actual role of OS in the complex net of events that trigger and perpetuate the cholestatic injury? We are nowadays positive that OS is a key factor, but other deleterious factors can collaborate as well, as for example cytokine-activated signaling cascades that are often turned on in cholestasis, and that may act, in part, via nonredox-dependent mechanisms.
i) Which are the ultimate molecular targets whose phosphorylation status is modified to trigger carrier endocytic internalization by OS-evoked transduction pathways? Many cellular structures known to be crucial for the proper localization of hepatocellular transporters, such as cytoskeletal components, endocytic proteins, and membrane lipid microdomains, as well as the transporters themselves, can be modulated by this mechanism, and the confirmation of the involvement of these structures will give us the possibility to target them with a therapeutic aim.
ii) Can changes in localization of these transporters not only be prevented but also reversed by pharmacological treatments counteracting these alterations? This is crucial, since most cholestatic hepatopathies need to be treated with a curative rather than a prophylactic aim. Fortunately, preliminary experimental evidence indicates that reinsertion of endocytosed transporters and cytoskeleton rebuilding occur spontaneously in a rapid and controlled form (154, 155, 185), thus opening the possibility of taking advance of or even accelerating this process with curative purposes.
v) Can antioxidants be safely used to treat cholestasis? The antioxidant doses to be used are expected to be relatively high, to compensate for the baseline low endogenous antioxidant levels that cholestatic patients have, even early in the disease course (58). Therefore, safety and effectiveness will need to be properly tested in well-designed clinical trials.
Answering to these questions will surely help to envisage therapeutic strategies in cholestasis that restore the normal redox status to ensure suitable transporter functional localization and to preclude exacerbated degradation. These therapeutic approaches may complement therapies designed to transcriptionally enhance carrier expression, by allowing proper localization and membrane stability to the de novo-synthesized transporters.
We hope progresses in experimental therapeutics based on these findings stimulate researchers and physicians to employ this knowledge to envisage more effective, innovative antioxidant therapeutic strategies for the treatment of human cholestatic liver diseases, thus tackling this “radical” way to stop making bile.
Footnotes
Authors' Contributions
M.G.R. conceived and designed this review article. M.G.R., C.L.B., and F.A.C. designed the illustrations. All the authors searched bibliographic databases, wrote different parts of the original draft, revised critically the whole article for important intellectual content, and gave the final approval of the version to be published.
Author Disclosure Statement
The authors declare that they have no competing financial interests.
Funding Information
All the authors are supported by the University of Rosario, and by research grants from CONICET (PUE-2016-22920160100089CO) and ANPCyT: PICT-2015-1242 (C.L.B.), PICT-2015-2714 (F.A.C.), PICT-2016-1613 (M.G.R.), PICT-2016-2166 (E.J.S.P.), and PICT-2017-2407 (E.J.S.P.).