
Abstract
Significance:
As the central metabolic organ, the liver is exposed to a variety of potentially cytotoxic, proinflammatory, profibrotic, and carcinogenic stimuli. To protect the organism from these deleterious effects, the liver has evolved a number of defense systems, which include antioxidant substrates and enzymes, anti-inflammatory tools, enzymatic biotransformation systems, and metabolic pathways.
Recent Advances:
One of the pivotal systems that evolved during phylogenesis was the heme catabolic pathway. Comprising the important enzymes heme oxygenase and biliverdin reductase, this complex pathway has a number of key functions including enzymatic activities, but also cell signaling, and DNA transcription. It further generates two important bile pigments, biliverdin and bilirubin, as well as the gaseous molecule carbon monoxide. These heme degradation products have potent antioxidant, immunosuppressive, and cytoprotective effects. Recent data suggest that the pathway participates in the regulation of metabolic and hormonal processes implicated in the pathogenesis of hepatic and other diseases.
Critical Issues:
This review discusses the impact of the heme catabolic pathway on major liver diseases, with particular focus on the involvement of cellular targeting and signaling in the pathogenesis of these conditions.
Future Directions:
To utilize the biological consequences of the heme catabolic pathway, several unique therapeutic strategies have been developed. Research indicates that pharmaceutical, nutraceutical, and lifestyle modifications positively affect the pathway, delivering potentially long-term clinical benefits. However, further well-designed studies are needed to confirm the clinical benefits of these approaches. Antioxid. Redox Signal. 35, 734–752.
The Heme Catabolic Pathway
The heme catabolic pathway belongs to a phylogenetically old and conserved system (23, 42, 200) long believed to represent the only means of eliminating potentially toxic and pro-oxidant heme molecules.
The entire heme degradation pathway (Figs. 1–2) is activated by the heme oxygenase (HMOX) enzyme. Containing two isoforms (HMOX1, OMIM *141250 and HMOX2, OMIM *141251) (141, 200), this ubiquitous microsomal enzyme is found throughout the human body. HMOX produces biologically significant amounts of bilirubin (∼300 mg [500 μmoles] in a 70 kg human per day), mostly from hemoglobin originating in senescent erythrocytes (16, 187). HMOX1, believed to be the most inducible enzyme in the human body (104, 110), plays a major role in protecting against exaggerated oxidative stress. HMOX1 gene expression falls under the control of nuclear factor erythroid 2-related factor 2 (Nrf2). This evolutionarily conserved, multifunctional transcription factor is responsible for cellular redox balance regulation, antioxidant protection, and phase II detoxification responses in mammals (100).
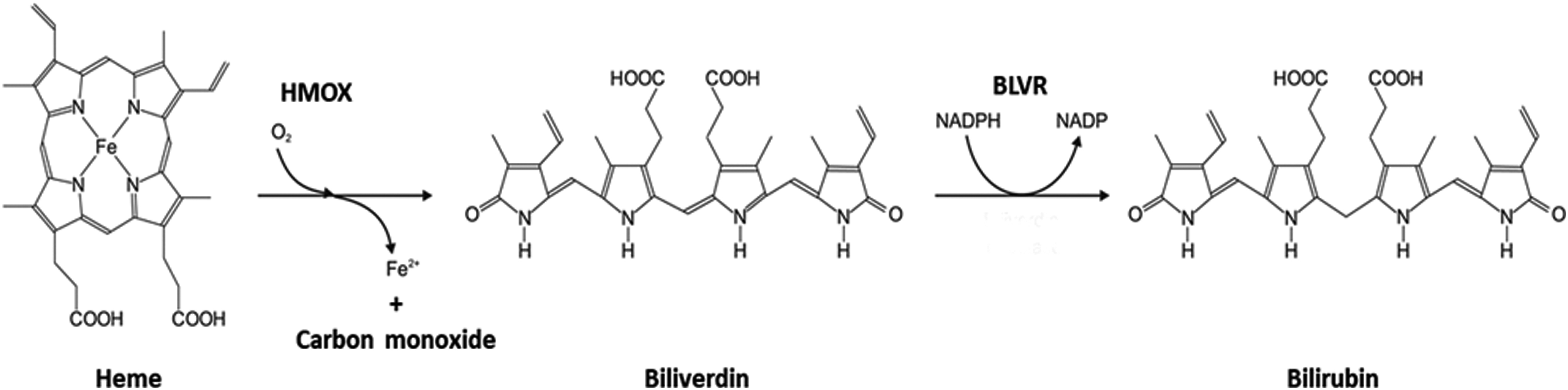
Although not widely distributed in the body, HMOX2 is highly expressed in hepatocytes (141). HMOX1 is overexpressed in Kupffer cells, liver-specific fixed macrophages (15), and, surprisingly, also in hepatic stellate cells (HSC) (207). These topological characteristics have pathophysiological consequences, particularly for iron metabolism in the liver and for protection against liver inflammation (15) and hepatic fibrogenesis (207). The basic function of HMOX is to split the cyclic tetrapyrrole heme into the linear tetrapyrrole structure of biliverdin, while simultaneously releasing ferrous iron and carbon monoxide (CO). Heme also induces ferritin, which sequesters released iron and prevents iron-mediated oxidative damage (9). The CO generated significantly contributes to hepatic sinusoidal tone and proper vascular functioning in liver tissue (167).
Like its immediate precursor bilirubin, biliverdin boasts potent antioxidant properties. Whether these effects are produced under in vivo conditions in the human body is debatable, since biliverdin is reduced to bilirubin almost immediately by ubiquitous biliverdin reductase (BLVR). Biliverdin has direct antioxidant effects, preventing both lipoperoxidation (162, 165) and hypochlorite-mediated pro-oxidation (164) and, similar to bilirubin, inhibiting NADPH oxidase-induced superoxide generation (43).
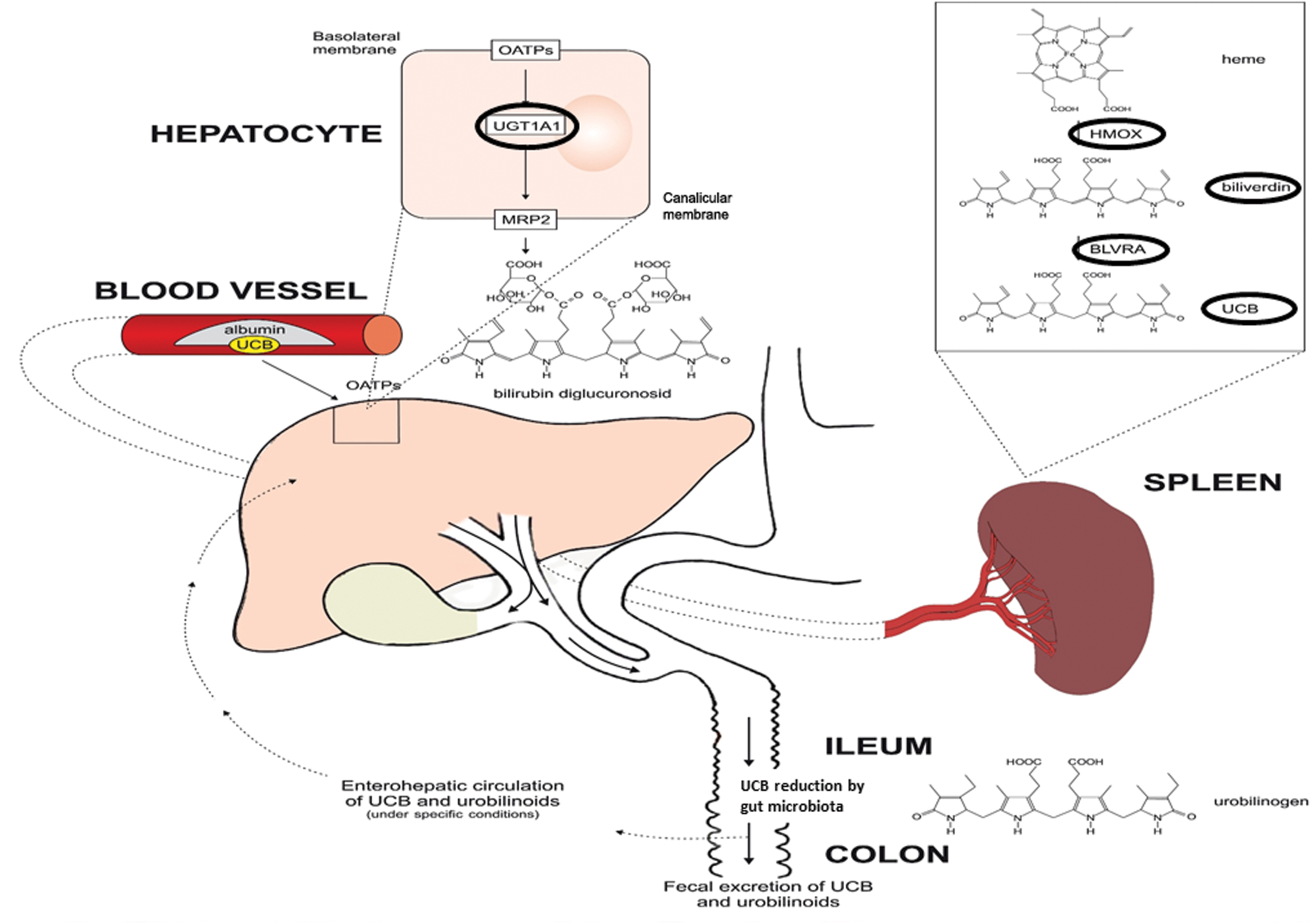
BLVR in the human body consists of two isoforms: BLVR A and B (BLVRA/B, OMIM Nos. *109750 and *600941, respectively). Apart from its enzymatic activity, BLVR also has an important signaling function. This key enzyme acts as a rare multispecific kinase (serine/threonine/tyrosine) that is particularly active in the insulin signaling pathway (91), posing considerable metabolic consequences (119). Among the most potent of the endogenous antioxidants (44), bilirubin is conjugated in liver cells with glucuronic acid by microsomal bilirubin UDP-glucuronosyltransferase 1A1 (UGT1A1), another member of the heme catabolic pathway. Its partial deficiency results in mild elevation of serum unconjugated bilirubin concentrations typically observed in Gilbert's syndrome (187).
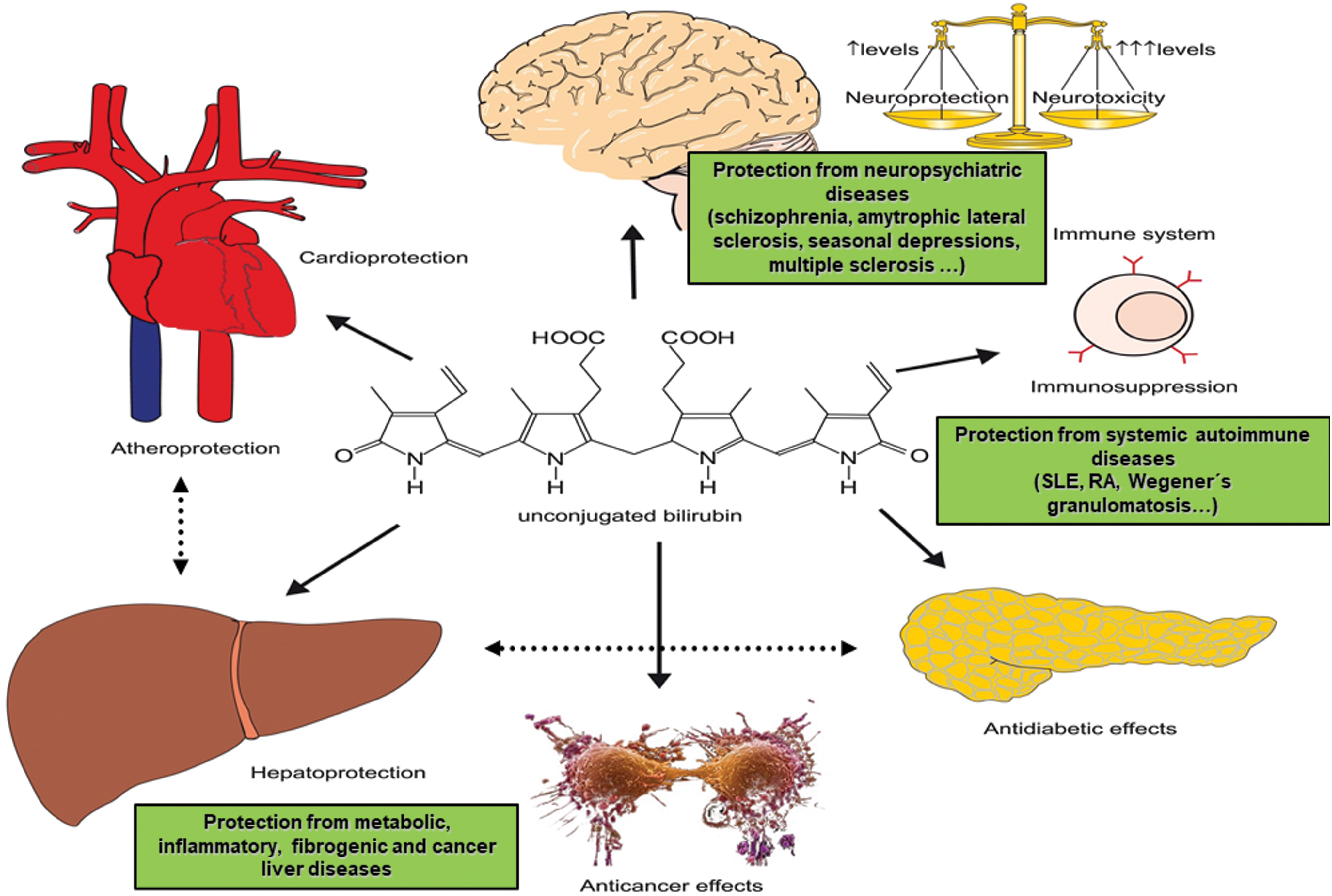
Surprisingly, bilirubin was recently found to act as a ligand of several specific cell receptors, including the aryl hydrocarbon receptor (AhR) and peroxisome proliferator-activated receptors (PPAR) α and γ, which are implicated in the development of a variety of metabolic diseases such as obesity, diabetes, and metabolic syndrome (183). Based on this evidence, bilirubin seems to function like an endocrine molecule, as previously suggested (182) (Fig. 3).
The Heme Catabolic Pathway and Regulation of Oxidative Stress in Liver Pathology
The liver is a major organ attacked by reactive oxygen and nitrogen species (ROS and RNS, respectively). Hepatic cell subpopulations, which include hepatocytes, Kupffer cells, HSC, endothelial cells, cholangiocytes, and hepatic progenitors, are all functionally affected by increased oxidative stress (93, 112). ROS and RNS induce HMOX1 expression in the liver, at least partially, by Nrf2 (7), a key modulator of HMOX1 expression (100). Most liver diseases are associated with oxidative stress, which plays a pivotal role in the following complications: pathogenesis of alcoholic liver disease, inflammatory hepatic conditions, drug-induced liver injury, autoimmune liver disorders, metabolic dysfunction-associated fatty liver disease (MAFLD), liver fibrosis, liver cirrhosis, and liver cancer (53, 93, 144, 194). In mitigating the progression of a variety of liver diseases, the enzymes and intermediates of the heme catabolic pathway, best exemplified by HMOX1, are understood to have significant protective effects (60, 122, 148).
Heme oxygenase 1
HMOX1 is expressed mainly in Kupffer cells of the liver, and also in hepatocytes in response to various noxious stimuli (122). HMOX1 plays important roles in protecting hepatocytes from oxidative damage, as documented in previous studies of hepatic ischemia/reperfusion injury (IRI) (29, 92, 97; see also section “Products of the Heme Catabolic Pathway As Protective Factors in Hepatic IRI and Liver Transplantation” below) and viral infections of the liver (216).
The central protective role of HMOX1 against pathogenic pathways in liver diseases has been documented also in clinical studies of HMOX1 promoter gene polymorphisms. Those gene variations resulting in higher HMOX1 activity are associated with improved outcomes in liver transplant rejection (20; see also section “Products of the Heme Catabolic Pathway As Protective Factors in Hepatic IRI and Liver Transplantation” below), pediatric MAFLD (24), and liver cirrhosis progression (38).
BLVRA-biliverdin-bilirubin
BLVRA plays a crucial role in the regulation of oxidative stress in the liver. An in vitro study of various liver cell types revealed a negative association between BLVRA expression and ROS production (49). This finding is consistent with animal data by Chen et al., who demonstrated exaggerated systemic oxidative stress in Blvra knockout mice (26). Similarly, a recent in vitro study of a murine Blvra-deficient hepatocyte cell line reported increased ROS production and oxidative stress (50). Complementing the negative association between Blvra expression and oxidative stress, it has been shown that biliverdin has a similar protective effect on bile acid-induced oxidative stress (49). These findings correspond with experimental models of neurodegenerative diseases, which confirm the accumulation of oxidatively modified proteins in the brain cortices of BLVRA-deficient mice (88).
Previous comprehensive studies have documented the antioxidant activities of bilirubin (188) and the effects of biliverdin on bile acid-induced ROS generation (49). With potent antioxidant effects on liver tissue, bilirubin probably functions as the major endogenous buffer against oxidative stress induced by bile acids accumulated during cholestasis (111). In addition to unconjugated bilirubin, its conjugates such as bilirubin ditaurate (used as a surrogate for bisglucuronosyl bilirubin accumulated during cholestasis) (163 –165) and delta bilirubin (a bilirubin covalently bound to albumin accumulated in circulation during cholestasis) (203) have similar antioxidant effects.
The protective effect of unconjugated bilirubin is most pronounced in the lipid membranes of liver cells and cell organelles. While the lipid-binding properties (219) of bilirubin help to counteract membrane lipid peroxidation, water-soluble antioxidants such as glutathione are better at protecting water-soluble proteins (153). Studies have shown that the antioxidant effects of bilirubin are strongly potentiated by the bilirubin/biliverdin redox cycle, protecting cells against 10,000-fold higher concentrations of hydrogen peroxide (10, 11). It should be noted, however, that not all authors support the biological relevance of this cycle, a subject that has provoked much debate (71, 103, 107).
In correlation with systemic bilirubin levels, intracellular concentrations of bilirubin reflect the various conditions of oxidative stress in the liver and other organs, as demonstrated in HepG2 hepatoblastoma cells and a rat animal model (212). In one in vitro study performed using various cell types (including those of hepatic origin), different pro- and antioxidant thresholds were observed for intracellular bilirubin concentrations, indicating that the antioxidant functions of bilirubin are highly varied (18). The antioxidant and potentially cytotoxic effects of bilirubin and lumirubin (a photo-oxidation product generated during phototherapy for hyperbilirubinemia) were recently investigated. Using HepG2 hepatoblastoma cells, the study demonstrated a dependent relationship between the potential toxicity of high bilirubin concentration and the glycolytic reserve in these cells (36).
In addition to this mechanism, several additional pathogenic pathways have been implicated in the toxicity associated with severe hyperbilirubinemia. These effects include neuron dysfunction due to the binding of bilirubin to myelin-rich membranes (196), bilirubin-induced DNA fragmentation (52), and decreased mitochondrial respiration, which alters membrane potential and permeability, leading to apoptosis (117, 138).
Conversely, it has been reported that under reduced and nontoxic concentrations, both pigments have beneficial biological impacts on mitochondria, substantially affecting lipid and glucose metabolism (36). Our animal study of aged hyperbilirubinemic Gunn rats found that mildly elevated serum bilirubin concentrations had the following positive impacts on various biological functions: attenuation of oxidative stress, improved anthropometric parameters, decreased inflammatory status, increased glucose tolerance, fewer signs of cellular senescence, and enhanced mitochondrial function (211). These findings also apply to the anti-inflammatory activity of bilirubin, as confirmed in primary hepatocytes exposed to the compound. Hyperbilirubinemic Gunn rats treated with bacterial lipopolysaccharide (LPS) were found to exhibit lower levels of liver injury and inflammatory markers compared with their normobilirubinemic littermates (178).
Carbon monoxide
Endogenously produced CO is an important regulator of the liver vasculature. CO also interacts with pathways responsible for protecting against oxidative stress, including the Nrf2 system, antioxidant-responsive (ARE) genes, and glutathione metabolism (81). These observations are supported by a number of experimental studies demonstrating the protective effects of CO gas and CO donors on the following: hepatic IRI (89; see also section “Products of the Heme Catabolic Pathway As Protective Factors in Hepatic IRI and Liver Transplantation” below), hemorrhagic shock (120), systemic inflammation-associated liver injury (19), and steatohepatitis (177). All of the above studies found that CO actively suppressed the generation of ROS.
Products of the Heme Catabolic Pathway As Protective Factors Against MAFLD/Metabolic Dysfunction-Associated Steatohepatitis Development
Key pathogenic aspects
MAFLD/metabolic dysfunction-associated steatohepatitis (MASH) is a complex metabolic disease that covers a spectrum of conditions ranging from simple steatosis to steatohepatitis, the latter potentially progressing to liver fibrosis, cirrhosis, and ultimately, liver cancer (Fig. 4). Increasing in global prevalence, MAFLD/MASH substantially affects life quality and expectancy, with severe social and economic impacts (94). The complex pathogenesis of this metabolic disease is rooted in deteriorating lipid metabolism, hepatic overflow of free fatty acids, increased de novo hepatic lipogenesis, and impaired free fatty acid elimination (94). These processes initiate the generation of ROS, development of low-grade inflammation, induction of proinflammatory pathways (such as nuclear factor kappa B [NF-κB]), activation of specific toll-like receptors (such as TLR2 and 4), and production of inflammatory cytokines (34). These phenomena further impair coexistent insulin resistance (94).
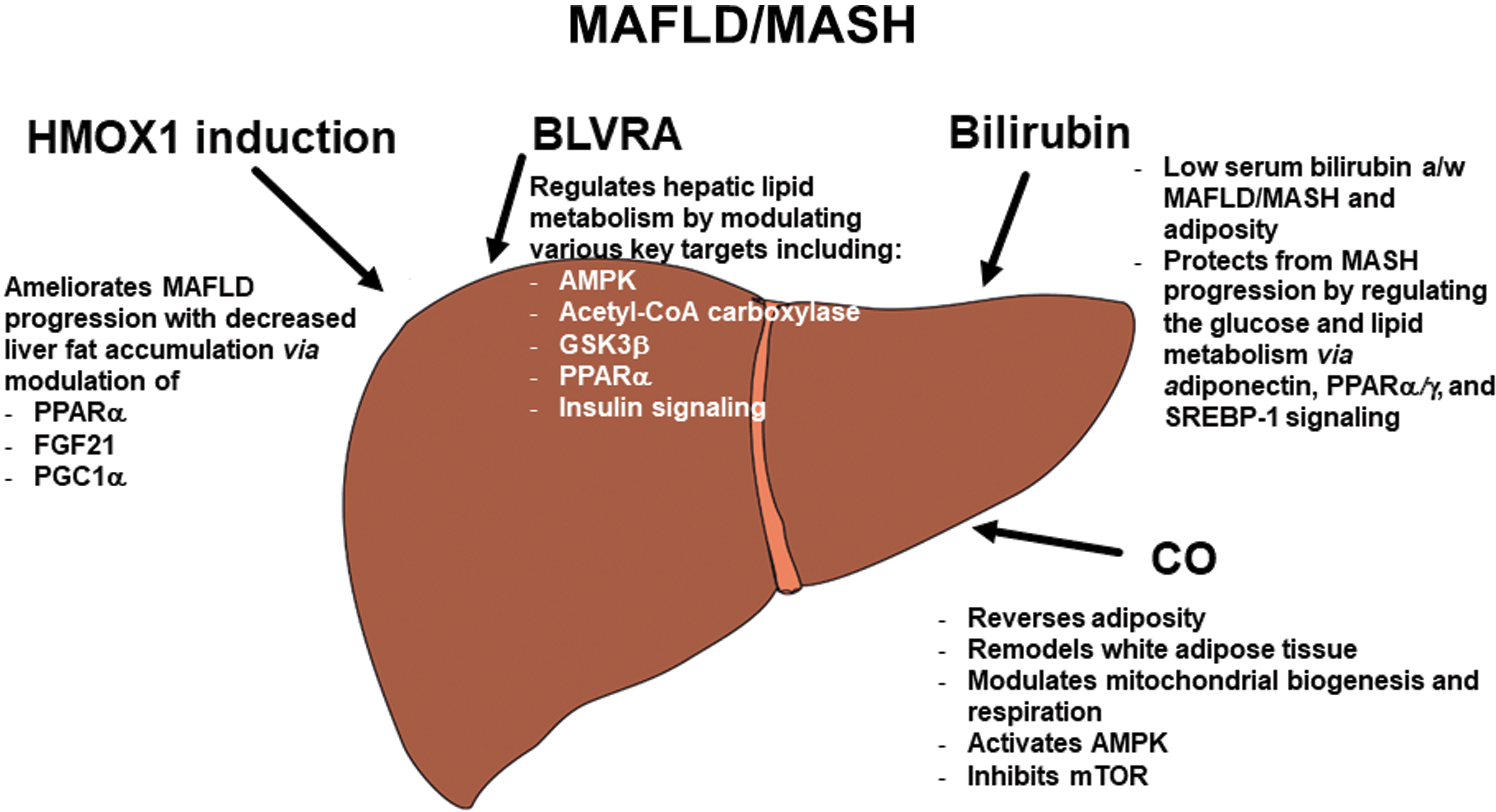
On the cellular level, the master regulatory effectors of energy metabolism—which include PPAR-γ coactivator-1α (PGC1α) (activated by free fatty acid-induced AMP-activated protein kinase [AMPK]) and the PPARs—are all dysregulated in MAFLD/MASH (34). Combined with the impact of impaired adipokine signaling (129), the vicious circle of this disease is perpetuated.
Heme oxygenase 1
Due to its effects on the metabolism of glucose and lipids, HMOX1 is understood to significantly affect the pathogenesis of liver steatosis and its subsequent transition to steatohepatitis and fibrosis (149, 159). Although most of the available reports are derived from experimental data, they clearly show the impacts of HMOX1 and its products on the pathogenesis of MAFLD/MASH. HMOX1 upregulation has been shown to ameliorate disease progression in animal models of MAFLD. A study by Wang et al. found that HMOX1 induction not only reduced hepatic steatosis and inflammation, but also prevented fibrotic changes in mice placed on a methionine-choline-deficient (MCD) diet (192).
Similarly, Salley et al. documented the MASH-ameliorating effects of HMOX1 induction, observing improvements in both inflammatory and fibrotic lesions in a rat model of diabetes (142). Another study reported decreased fat accumulation following HMOX1 induction in a murine model of obesity. Importantly, the authors also detailed the modulation of cellular metabolic targets, including PPARα, fibroblast growth factor 21 (FGF21), and PGC1α (59). The key role of PGC1α was also confirmed in an animal study by Raffaele et al. (136). These data tally with the results of a clinical study on pediatric MAFLD patients by Chang et al. They found that children with reduced activity of HMOX1 promoter gene polymorphisms were more susceptible to the development of MAFLD (24).
Biliverdin reductase A
As proven in a liver-specific Blvra knockout mouse model (55), BLVRA regulates hepatic lipid metabolism by directly affecting the key enzymes implicated in lipid metabolism. These include AMPK, an important cellular energy sensor (181), acetyl-CoA carboxylase, an essential and rate-limiting enzyme in fatty acid metabolism (25), glycogen synthase kinase-3β (GSK3β), one of the most active cellular kinases and boasting more than 100 known targets, involved in the regulation of multiple cellular functions, including lipid and glucose metabolism, among others (1, 17).
In addition, BLVRA has been shown to regulate nuclear receptor PPARα and its downstream effectors (55), key elements in the energy homeostasis pathways of various metabolic diseases (35). The metabolic role of BLVRA has been recently confirmed. In vitro study by Miralem et al. (108) proved the effect of BLVRA on regulation of GSK3β. Lanzillota and colleagues confirmed the dysregulation of both AMPK and mammalian target of rapamycin (mTOR), an evolutionarily conserved nutrient-sensing protein kinase that regulates metabolism (157) hyperactivation in Blvra-deficient mice (88). The essential role of Blvra in the lipid metabolism and pathogenesis of obesity was verified in mice with adipocyte-specific deletion of Blvra demonstrating multiple cellular and systemic metabolic effects (158). These results were confirmed by an in vitro study (by the same group) of a murine BLVRA-deficient hepatocyte cell line. The authors reported increased ROS generation, reduced numbers of mitochondria, and deterioration of mitochondrial metabolism (50).
BLVRA, which acts as a potent multispecific kinase, significantly modulates the insulin signaling pathway (47, 91) implicated in the pathogenesis of MAFLD/MASH (33). In a recent human clinical study, low BLVRA expression was associated with MASH, adipose tissue inflammation, and insulin-signaling dysregulation (30). As demonstrated in recent animal studies (13, 156), insulin resistance, also present in Alzheimer's disease (172), falls under the direct control of BLVRA, which is impaired in this brain disorder (14).
Bilirubin
Serum bilirubin concentrations are negatively associated with metabolic syndrome, type 2 diabetes mellitus (115, 118), and MAFLD/MASH in both pediatric and adult patients (86, 96, 134). A large study by Hjelkrem et al. found that MASH occurrence was 16 times less likely in subjects with Gilbert's syndrome (benign hyperbilirubinemia) (61). In another study, the presence of unconjugated hyperbilirubinemia was associated with a much milder course of MAFLD, with lower rates of progression to MASH and fibrosis (84). This finding was replicated in a study by Salomone et al. They reported serum unconjugated bilirubin concentration to be an independent predictor of advanced inflammation and fibrosis in MASH patients (143).
These data were further confirmed by Anwar et al. in their systemic review of seven clinical studies examining the association between serum bilirubin concentrations and MAFLD risk (6). However, not all results support the protective effect of bilirubin on MAFLD, as recently demonstrated by two Mendelian randomization studies (85, 102). Therefore, to elucidate all of the potential factors affecting this putative association, further well-designed controlled studies are required.
It should also be noted that hyperbilirubinemic subjects with Gilbert's syndrome have substantially lower BMI, glucose, insulin, C-peptide, and triacylglycerol serum concentrations, most likely due to the activation of AMPK, PPARα/γ, and PgC1α (109). Improved metabolic health was reported in a study by Seyed Khoei et al. (155) and in other studies reviewed by Bulmer and colleagues (21).
The largely positive clinical data on the protective effects of bilirubin are supported by several important experimental studies on hyperbilirubinemic and bilirubin-treated animals. In a diet-induced obesity mouse model, bilirubin treatment led to improved cholesterol and glucose metabolism and increased adiponectin production. Enhancements were also observed for mRNA expression of PPARγ, sterol regulatory element-binding protein-1 (SREBP-1), and the insulin receptor, which likely contributed to increased insulin sensitivity and glucose tolerance in this animal model (98). Amelioration of liver steatosis in mice fed a high-fat diet was clearly demonstrated in genetically engineered hyperbilirubinemic mice phenotypically resembling Gilbert's syndrome (58). This improvement was mediated via bilirubin-induced activation of PPARα (58), a selective intracellular target of bilirubin previously identified by the same authors (160).
Bilirubin has been shown to remodel white adipose tissue in mice via modulation of mitochondrial activity (51); a similar effect on mitochondrial metabolism was observed in our recent study (36). One research group recently identified lower levels of hepatic steatosis, MASH, and hepatic fibrosis in humanized transgenic hyperbilirubinemic mice fed a high-fat diet (87). In another animal study of mice with diet-induced MAFLD treated with pegylated bilirubin nanoparticles, a marked improvement in liver disease was observed (57).
As discussed above, in MASH patients, mitochondria are typically overloaded with free fatty acids, principally due to increased fat intake and de novo lipogenesis. This situation is further worsened by the presence of insulin resistance, which in turn leads to mitochondrial dysfunction and overgeneration of ROS (106). Vitamin E, which prevents lipid oxidation, has been proposed as a potentially powerful therapeutic agent (37). The synergistic interaction between bilirubin and vitamin E has also been demonstrated (165). Although yet to receive specific attention, the antioxidant effects of bilirubin can be assumed to mitigate the pathophysiological mechanism behind MASH development.
The association between physical inactivity and the risk of MAFLD/MASH is widely accepted (139). Analogously, increased energy expenditure due to both aerobic and resistance exercises is associated with the prevention and amelioration of fatty liver disease (179). The negative correlation between body mass index and serum bilirubin concentrations is also acknowledged (4, 155). In obese subjects, bilirubin is most likely overconsumed due to obesity-induced oxidative stress (131). In this context, notable data from a recent animal study revealed exercise-induced elevation of plasma bilirubin concentration, which was accompanied by substantial improvements in glucose and lipid metabolism (56). These findings tally with observations by Swift et al., who documented significant increases in serum bilirubin concentration in previously sedentary postmenopausal women placed on an exercise regimen (171). Similar bilirubin elevations have been found in Polish (201) and elite Czech athletes (202).
Carbon monoxide
According to recent experimental research, CO (whether endogenously produced or administered as a therapeutic agent) has considerable therapeutic potential for inhibiting MAFLD/MASH progression. In dietary-induced obese mice treated with a CO donor (carbon monoxide releasing molecule A1 [CORM-A1]), chronic treatment with this therapeutic substance reversed adiposity, led to remodeling of white adipose tissue, and normalized insulin resistance independently of changes in food intake or activity (63, 64). The most likely mechanism is modulation of mitochondrial metabolism, since CO is a potent regulator of both mitochondrial biogenesis and mitochondrial respiration (99, 145, 169).
Similarly, the therapeutic activities of CO inhalation on hepatic steatosis have been reported in mice placed on an MCD diet; activation of AMPK and inhibition of mTOR complex I were identified as the cellular mechanisms responsible for these effects (78). In another study of obese mice administered a high-fat diet and exposed to CO inhalation, mitochondrial metabolism substantially improved. This treatment resulted in reduced hepatic lipid accumulation as well as enhanced mitochondria biogenesis and metabolism (77). More recently, another animal model of MASH documented similar benefits of CO (in the form of CORM-A1) on mitochondrial functions in mice fed a high-fat/high-fructose diet (177).
Products of the Heme Catabolic Pathway As Protective Factors Against Liver Inflammation
Key pathogenic aspects
The anti-inflammatory role of the heme catabolic pathway is well-documented. Its complex impact on the inflammatory response enlists a number of mechanisms (Fig. 5). These include regulation of cytokine gene expression via TLRs and the control of inflammasome-dependent cytokine maturation and macrophage polarization [for a review, see Ryter (140)]. Specific inflammatory pathways affected by BVRA and bilirubin, a potent immunosuppressant, are discussed in detail below. It should be noted that the anti-inflammatory and metabolic activities of the heme catabolic pathway are interlinked. These mechanisms, which comprise joint signaling pathways such as TLR4, the regulation of mTOR target genes including protein kinase B (Akt), protein kinase C (PKC), and AMPKα, and the modulation of mitogen-activated protein kinases (MAPKs) and NF-κB (discussed in detail below), are important in counteracting the development of metabolic syndrome and MAFLD (34; see also section “Products of the Heme Catabolic Pathway As Protective Factors Against MAFLD/MASH Development” above).
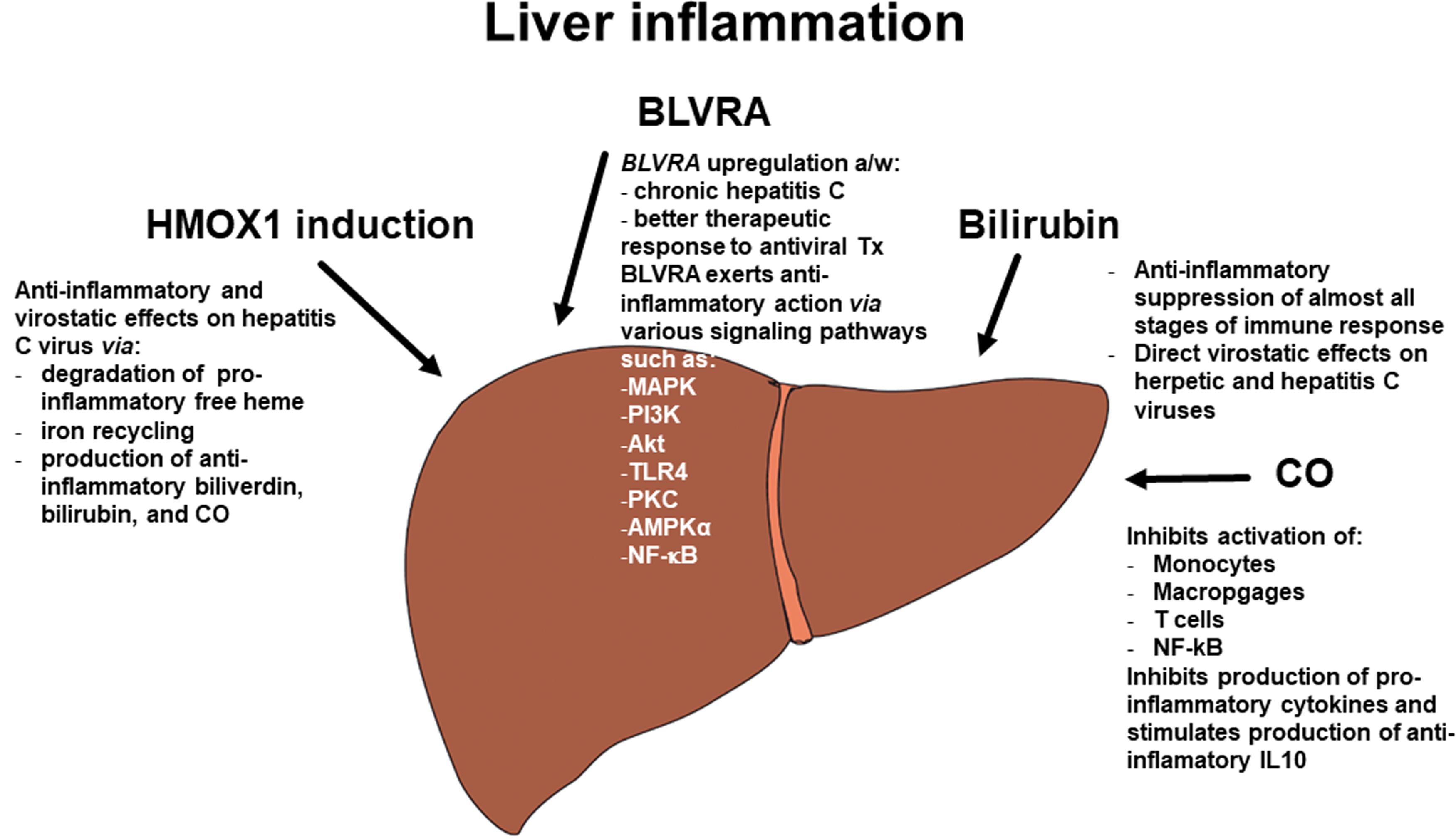
Heme oxygenase 1
HMOX1 is the most important inflammation regulator in the heme catabolic pathway (125). In addition to its anti-inflammatory potential (127), the gene has a virostatic function, as confirmed in a study of viral hepatitis C (216). It is involved in the degradation of proinflammatory and pro-oxidant free heme, iron recycling, and the production of the anti-inflammatory molecules bilirubin and CO (69).
Biliverdin reductase A
BLVRA, the second enzyme in the heme catabolic pathway, significantly contributes to the control of inflammation, as documented in our own study. We demonstrated the protective role of BLVRA in hepatitis C viral infection. Compared with healthy control subjects, patients with chronic HCV infection displayed increased mRNA levels of BLVRA in peripheral white blood cells. Interestingly, individuals who responded to standard antiviral therapy had significantly upregulated BLVRA expression in peripheral blood leukocytes, whereas those without BLVRA overexpression were nonresponsive. In addition, mRNA levels of BLVRA in peripheral white blood cells closely correlated with levels in liver tissue, indicating that peripheral blood cells could be used as a surrogate tissue to access the heme catabolic pathway in the liver (166).
Significantly, it is not only enzymatic activity per se, but also the specific signaling of BLVRA expressed on the cytoplasmic membranes of macrophages—mediated via phosphatidylinositol 3-kinase and Akt signaling pathways—that has potent anti-inflammatory effects on the inhibition of proinflammatory cytokine production (198). Upon exposure to biliverdin, BLVRA translocates to the nucleus of mouse macrophages, from where it blocks specific gene expression.
It also acts as a transcription factor in a variety of signaling pathways, including TLR4 (124). In a study by Zhang et al., biliverdin inhibited TLR4 signaling in leukocytes and triggered the phosphorylation of mTOR-specific targets, including Akt, PKC, and AMPKα (213). These findings support the modulatory function of biliverdin and BLVRA in the TLR4 signaling pathway. The critical role of BLVRA in macrophage polarization and anti-inflammatory cytokine production has been demonstrated both in vitro and in an animal model of kidney disease (65). The effects of BLVRA on various signaling pathways, including MAPK, Akt, and NF-κB activation, have been documented in LPS-induced inflammation in macrophage cells (79).
As previously mentioned, low BLVRA expression in peripheral blood leukocytes is associated with inflammation of adipose tissue in patients with MASH (30), representing yet another function of BLVRA.
Bilirubin
Bilirubin has immunosuppressive effects on almost all effectors of both innate and adaptive immune responses. The compound also protects against inflammation-induced liver injury, the extensive clinical consequences of which have been reviewed in detail (22, 70). In agreement with these findings, our recent study of hyperbilirubinemic Gunn rats exposed to bacterial LPS confirmed the ameliorative effect of bilirubin on liver injury (178). Like biliverdin and BLVRA, bilirubin inhibits TLR4 signaling (67), an important factor in the pathogenesis of liver disease. In a mouse model of colitis, bilirubin was shown to inhibit TLR4, suppress the production of proinflammatory cytokines, and regulate changes in the intestinal microbiome and gut barrier (214).
Based on a recent review by Corral-Jara et al., all components of the heme catabolic pathway, including heme, HMOX, biliverdin, bilirubin, and CO, are likely to be involved in the pathogenesis of various types of viral hepatitis (31). In addition to regulating specific immune responses against hepatitis viral infections, both biliverdin and bilirubin have direct antiviral effects, as demonstrated in herpetic and hepatitis C viruses (113, 146, 215).
According to one study, bilirubin considerably alleviated sterile inflammation in mice fed a high-fat diet (87), a finding consistent with the profound effects of the pigment on immune system responses being in play also in MASH development (22, 70).
Carbon monoxide
CO is not only a powerful modulator of immune responses, but it also affects many inflammatory signal transduction pathways [for a review, see Hoetzel et al. (62)]. As well as having direct effects on immune-competent cells, CO has been shown to inhibit the activation of monocytes, macrophages, leukocytes, and T cells (126, 147). Previous studies have highlighted CO-induced suppression of proinflammatory cytokine production and NF-κB activation through the modulation of several important cellular proinflammatory signaling pathways (123, 147). Yan et al. confirmed that these anti-inflammatory effects also apply to liver pathology. In a murine model of acute liver failure induced by LPS/D-galactosamine, treatment with CORM-3 was found to suppress proinflammatory cytokine production, increase anti-inflammatory IL10 cytokine secretion, and inhibit nuclear expression of NF-κB (205).
Products of the Heme Catabolic Pathway As Protective Factors in Hepatic IRI and Liver Transplantation
Hepatic IRI is a multifactorial process triggered by transient tissue deprivation of oxygen and subsequent reoxygenation. This complication can occur as a result of various clinical conditions such as shock of any etiology, hepatic resection, and liver transplantation. The mechanisms of hepatic IRI particularly involve dysregulation of redox status and activation of the innate immune system (176). During the initial ischemic phase, oxygen in liver cells is depleted, with ROS generation increasing during the second reperfusion phase. The consequent massive flux and activation of inflammatory cells—including macrophages, Kupffer cells, neutrophils, and T cells—perpetuate a number of apoptotic processes [for a review, see Hirao et al. (60)].
Pointing to its important therapeutic potential, the heme catabolic pathway is understood to be implicated in protection against IRI as well as transplant rejection in various organs and tissues (29, 121). The pathway comprises a complex protective system that counteracts inflammation, apoptosis, cell proliferation, and pro-oxidation, and also modifies other immune responses, as discussed above (121).
Heme oxygenase
HMOX expression and activity seem to be of key importance in preventing IRI (97), driven primarily by HMOX expressed on both infiltrating macrophages and resident Kupffer cells (114). A number of experimental studies have documented the essential role of HMOX1 in alleviating IRI in both cold-preserved (2, 74) and aged livers (193). A large study by Buis et al. involving more than 300 liver transplant patients highlighted the beneficial effects of HMOX. Subjects with high expression of the HMOX1 genotype were protected from unfavorable outcomes and had better graft survival rates than individuals with low expression (20). As reported in a clinical study by Geuken et al., the ability to upregulate HMOX1 activity during liver transplantation seems to be more important than steady expression in protecting liver grafts (46). In another recent study, conducted by Kadono et al., the human biopsies of liver transplant recipients were evaluated. The authors found negative associations between innate (represented by CD68, cathepsin G, TLR4, CXCL10), adaptive (such as CD4, CD8, IL17), and costimulatory (CD28, CD80, CD86) immune system molecules. Most importantly, liver transplant recipients with high HMOX1 expression exhibited a trend toward improved overall survival, indicating the direct effect of the HMOX system on immune responses in liver transplantation (72).
Biliverdin/bilirubin
The protective function of biliverdin was documented in an experimental rat model of IRI by Fondevila et al. Following cold ischemia of liver grafts and syngeneic orthotopic liver transplantation, biliverdin therapy extended animal survival, increasing from 50% in untreated controls to 90%–100%. This effect correlated with improved liver functions and preserved hepatic histology (41). The beneficial effects of biliverdin therapy observed in both donors and recipients were observed in a swine model of liver transplantation (5). Interestingly, a significant improvement in liver function was reported in one model of a rat liver exposed to cold ischemia following bilirubin rinse. This rinsing of liver grafts with bilirubin before reperfusion substantially improved the survival rate (75). Significantly, the cytoprotective effects of biliverdin and bilirubin seem to apply to other organ transplantations, as reviewed by Ollinger and colleagues (121).
Carbon monoxide
Boasting impressive vasoactive properties, CO exerts an important protective influence on IRI, as evidenced by a number of experimental animal studies (97). These include models of rats with CO-enriched blood and rat livers perfused either with CO or different CORM compounds (3, 68, 132, 161, 170, 199).
The collective experimental and clinical data strongly support the beneficial role played by the entire heme catabolic pathway in hepatic IRI and liver transplantation.
Products of the Heme Catabolic Pathway As Protective Factors Against Liver Fibrogenesis
The heme catabolic pathway significantly contributes to the process of fibrogenesis (Fig. 6). Inhibition of the TLR4 signaling pathway seems to be important not only in the immune response but also in liver fibrogenesis (133, 197). Accordingly, biliverdin, BLVRA, and bilirubin may protect against liver fibrogenesis by blocking HSC activation, a finding reported in a study by Tang et al. Their in vitro study documented the inhibition of HSC activation and a decreased mRNA ratio for tissue inhibitor of metalloproteinase-1/matrix metalloproteinase-2 (TIMP-1/MMP-2) upon exposure to only moderately increased bilirubin concentrations (173). Apart from hepatic fibrogenesis, MMPs are also involved in hepatocellular cancer (HCC) development and the pathogenesis of other liver diseases (45). Bilirubin was shown to inhibit MMPs in other pathological conditions such as nasopharyngeal cancer (32) and wound healing in diabetic rats (137). The same effects of bilirubin were observed in a murine model of UVB-induced skin photodamage (8) and in an experimental model of breast cancer involving HMOX1 induction (95). The HMOX gene itself, which is expressed on HSC, inhibits hepatic fibrogenesis via proapoptotic effects on cells mediated by NF-κB (207). As mentioned above, the protective effects of HMOX1 induction on hepatic fibrogenesis were reported in MASH animal models (142, 192).
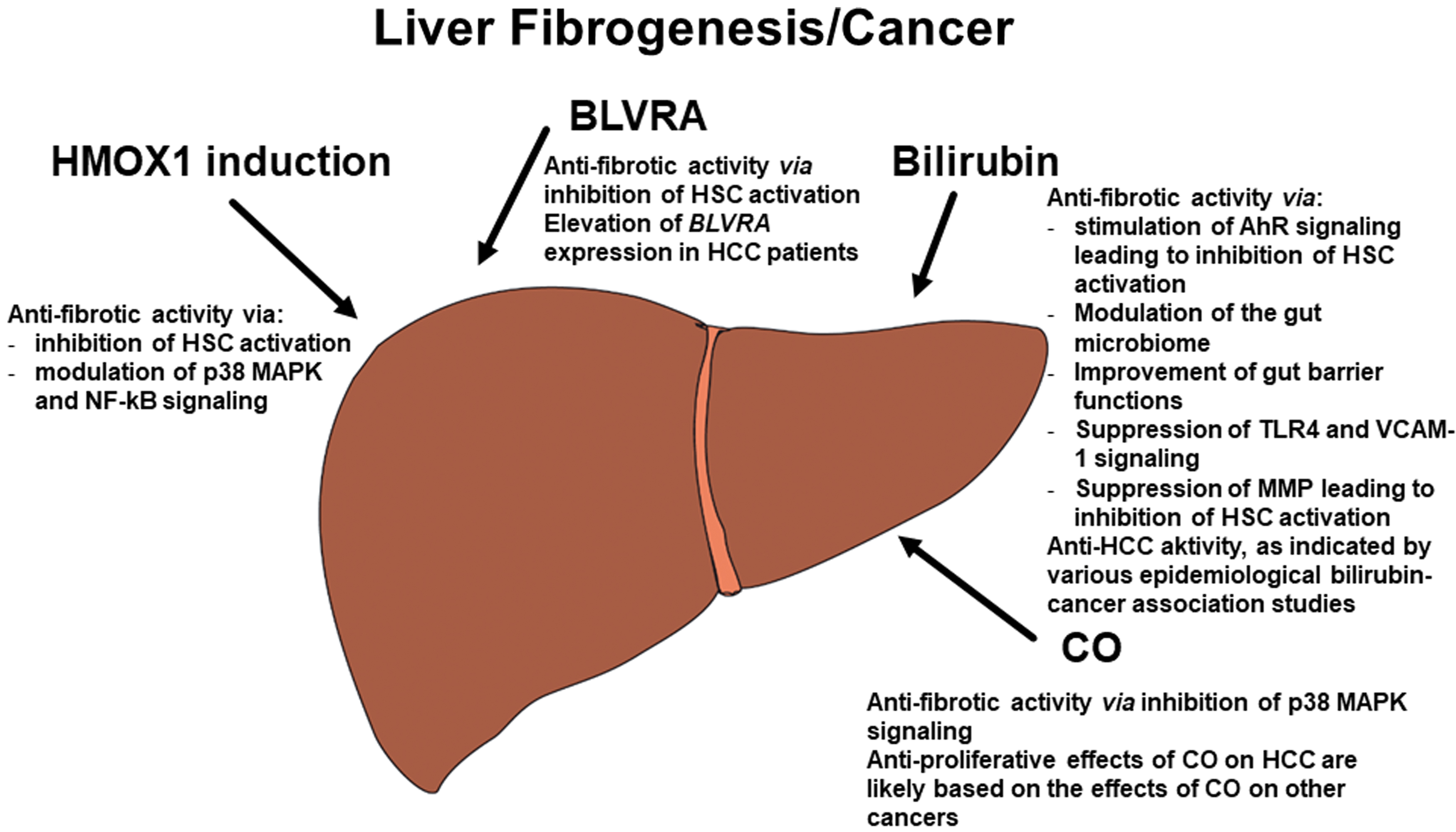
CO also boasts a number of antifibrotic effects, as demonstrated in an experimental model of kidney disease (116). Heme is a potent biomolecule that acts as a molecular switch, since different concentrations of free heme generate different cellular responses during both inflammation and fibrosis [for a review, see Lundvig et al. (101)]. Although exposure to high levels of heme has been shown to induce hepatic fibrosis (175), an experimental Mdr2 knockout mouse model with reduced HSC activation in liver tissue demonstrated that pharmacological induction of HMOX1 prevents and even reverts liver fibrosis (12). Interestingly, similar effects of HMOX1 induction have been observed in the pancreas. Nutraceutical upregulation of HMOX1 was found to prevent pancreatic stellate cell activation and development of pancreatic fibrosis (151), with these effects principally driven by the HMOX1/CO-modulated p38 MAPK pathway (152). Based on a recent review, CO also significantly contributes to fibrogenesis in pancreatic tissue (150).
AhR-mediated fibrogenetic pathways are among the other potential cell targets affected by bilirubin, a potent ligand of this nuclear receptor (130). AhR is significantly involved in the process of hepatic fibrogenesis, as confirmed by fibrotic changes in the liver tissue of Ahr-deficient mice (39). This may be at least partially due to the beneficial role of AhR in the activation of HSC, as recently described in an in vitro study on primary mouse and human HSC (206).
As previously mentioned, bilirubin was shown to improve both the gut microbiome and barrier functions in an animal model of colitis (214), with similar results reported for treatment of murine acute colitis with hyaluronic acid bilirubin nanoparticles (90, 189). In a murine study by Vogel and Zucker (190) and in another work by the same authors (221), bilirubin ameliorated dextran sodium sulfate-induced colitis, helping to preserve colonic functions and prevent trafficking of leukocytes by inhibition of vascular cell adhesion molecule-1 (VCAM-1)-mediated signaling. Bilirubin has also been shown to inhibit VCAM-1-dependent airway inflammation (76).
It is widely accepted that dysfunctional intestinal permeability increases hepatic exposure to gut bacteria products, which act as TLR4 ligands that trigger hepatic fibrogenesis (154) and ultimately the development of liver cirrhosis (73).
In summary, the above collective data clearly demonstrate the profound impact of the entire heme catabolic pathway on hepatic fibrogenesis.
Products of the Heme Catabolic Pathway As Protective Factors Against Liver Carcinogenesis
Heme oxygenase
The inconclusive role of HMOX in carcinogenesis is the subject of conflicting reports in the literature (195) (Fig. 6). A study by Park et al. found that HMOX1 expression did not seem to influence the prognosis of HCC patients (128). However, another clinical study revealed low expression of Nrf2 and HMOX1 in HCC biopsies, pointing to the beneficial role of HMOX1 in this cancer (28). Supporting these clinical data, an experimental in vitro study by Zou et al. found that HMOX1 inhibited HepG2 hepatoblastoma cell growth mediated via microRNA pathways (218). In another experimental in vitro animal study by the same authors, the inhibitory effects of HMOX1 on HepG2 cell growth and migration were observed, with CO identified as an important contributing factor (217).
Biliverdin reductase A
BLVRA mRNA expression was found to be a predictor in our small study of patients with HCC. Compared with controls, mRNA levels of BLVRA in the livers of HCC patients increased by up to threefold. This finding correlated with BLVRA expression in peripheral leukocytes, a result perhaps attributable to a feedback mechanism for controlling increased oxidative stress associated with HCC progression (83). Our results were corroborated by an in vitro study on human HCC cell lines by Huan et al., who found that RNA interference of BLVRB suppressed HCC cell growth, despite BLVRB overexpression having an opposite effect (66).
Bilirubin
To date, there have been no epidemiological studies on the association of bilirubin with HCC. However, data in relation to overall cancer prevalence indicate a clear negative association between serum/plasma bilirubin concentration and cancer risk. Yamamoto et al. found a negative correlation between high serum bilirubin and cancer development in a large cohort of Japanese adults (n = 78,000) with no hepatobiliary disease (204). In our own study of the Polish arm of the HAPIEE cohort (n = 1700), bilirubin was associated with lower mortality, driven particularly by cancer mortality (186). The large NHANES study by Zucker et al. (covering 176,748,000 individuals) reported a negative association between serum bilirubin and the risk of nondermatological cancers, particularly gastrointestinal cancer (220). Interestingly, a study by Han et al. found a negative association between liver donor preoperative total bilirubin concentration and risk of HCC recurrence in transplanted patients (54).
Carbon monoxide
Although no specific research has confirmed the involvement of CO in HCC development, recent data from research on different cancers, recently summarized in a review article by Kourti et al. and corroborated by an experimental study by Zou et al. (217), suggest that CO may interfere with HCC pathogenesis (82), This hypothesis is consistent with our own in vitro and animal data on the therapeutic effect of CO on pancreatic cancer (185). Although promising results regarding CO treatment in MASH (HCC precarcinosis) have recently been published, further detailed research into the effects of CO on liver carcinogenesis is warranted.
Therapeutic Potential of Bilirubin Metabolism Modulation
To improve metabolic and liver health outcomes, there have been concerted efforts to explore therapeutic options aimed at modulating the heme catabolic pathway as well as its specific intermediates and products. Recently, several comprehensive reviews on the topic have highlighted the potential of various treatments (44, 159, 184, 191).
Several promising methods have been developed, with the possibilities of HMOX1 induction receiving the most attention of researchers (159). Many natural products commonly used as nutraceuticals have been shown to upregulate HMOX1 and improve MAFLD according to a number of experimental and clinical reports [comprehensively reviewed in Stec and Hinds (159)]. For example, silymarin flavonolignans are widely acknowledged to protect against a wide range of liver diseases (174). These agents also act as partial inhibitors of UGT1A1, mildly elevating the hepatic (168) and systemic (40) concentrations of bilirubin. Therefore, partial inhibition of UGT1A1 using either medical xenobiotics or better nutraceutical substances (aimed at potentiating bilirubin signaling activities) represents a promising therapeutic approach for patients with liver diseases (184).
A similar chemopreventive effect is offered by naturally occurring tetrapyrroles, chemical compounds that resemble the structures of biliverdin and bilirubin. The most promising of these agents include pulverized bovine gallstones, which have been used for centuries in Traditional Chinese Medicine (135, 210), green-plant chlorophylls (180), and phycocyanobilin from blue-green algae (80, 105). Modulation of the signaling and immunomodulatory functions of BLVRA has also been investigated, especially in the treatment of metabolic diseases such as obesity, diabetes, and MAFLD (47, 48).
Finally, with the advent of modern nanotechnologies, incorporation of bilirubin into various forms of nanoparticles has demonstrated remarkable biological benefits in a number of clinical pathologies, as recently reviewed in several comprehensive studies (27, 189, 208, 209). The same nanomedical approach has been notably applied to BLVRA-based peptides to successful effect, indicating the broad potential of this therapeutic modality in metabolic disease prevention (48).
Conclusion
The accumulating body of evidence indicates that the heme catabolic pathway, including its key proteins and pigments, has significant impacts not only on heme degradation but also on the metabolic functions of the human body. Primarily localized in the liver, a central metabolic organ, the pathway unsurprisingly plays a significant role in modulating the pathogenesis of a variety of liver diseases. However, further clinical studies are needed to elucidate the full range of pathogenetic mechanisms that drive this therapeutic pathway.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants NV18-07-00342 and RVO-VFN64165/2021 from the Czech Ministry of Health, and Progress Q25/LF1 from Charles University.