
Abstract
Aims:
Myocardial fibrosis after myocardial infarction (MI) leads to heart failure. Nitration of protein can alter its function. cAMP-response element binding protein (CREB) is a key transcription factor involved in fibrosis. However, little is known about the role of nitrated CREB in MI-induced myocardial fibrosis. Meanwhile, downstream genes of transcription factor CREB in myocardial fibrosis have not been identified. This study aims to verify the hypothesis that nitrated CREB promotes MI-induced myocardial fibrosis via regulating the transcription of Col1a2 and Cxcl12.
Results:
Our study showed that (1) the level of nitrative stress was elevated and nitrated CREB was higher in the myocardium after MI. Tyr182, 307, and 336 were the nitration sites of CREB; (2) with the administration of peroxynitrite (ONOO−) scavengers, CREB phosphorylation, nuclear translocation, and binding activity to TORC2 (transducers of regulated CREB-2) were attenuated; (3) the expressions of extracellular matrix (ECM) proteins were upregulated and downregulated in accordance with the expression alteration of CREB both in vitro and in vivo; (4) CREB accelerated transcription of Col1a2 and Cxcl12 after MI directly. With the administration of ONOO− scavengers, ECM protein expressions were attenuated; meanwhile, the messenger RNA (mRNA) levels of Col1a2 and Cxcl12 were alleviated as well.
Innovation and Conclusion:
Nitration of transcription factor CREB participates in MI-induced myocardial fibrosis through enhancing its phosphorylation, nuclear translocation, and binding activity to TORCs, among which CREB transcripts Col1a2 and Cxcl12 directly. These data indicated that nitrated CREB might be a potential therapeutic target against MI-induced myocardial fibrosis. Antioxid. Redox Signal. 38, 709–730.
Introduction
Myocardial infarction (MI) is one of the most common adverse outcomes of coronary heart disease, for which the therapeutic strategies are still limited. Repair after MI is characterized by cardiomyocyte necrosis, inflammatory cell infiltration, and an abnormal deposition of a large amount of extracellular matrix (ECM) components (Anzai, 2013). In addition, residual cardiomyocyte hypertrophy, angiogenesis, and an abnormal accumulation of cardiac fibroblasts (CFs) are also specific manifestations of fibrous repair after MI (Jering et al, 2021). CFs play an essential role in the synthesis and secretion of ECM proteins. ECM proteins determine the degree of myocardial fibrosis, and are divided into structural proteins and nonstructural proteins (Bao et al, 2018; Ma et al, 2012; Wight and Vernon, 2019).
At present, the exact mechanisms involved in MI-induced myocardial fibrosis still remain unclear, and further efforts are needed to clarify this question so as to provide new ideas and potential targets for the detection and treatment in clinic. Studies have shown that nitrative stress was increased in the myocardium after MI (Moreno et al, 2002). Through electron transport, superoxide anion (O2 •−) reacts with nitric oxide (NO) to form peroxynitrite (ONOO−). Peroxynitrite increases the level of nitro-proteins through binding to tyrosine residues in proteins to produce 3-nitrotyrosine (3-NT), which is the footprint of peroxynitrite (Beckman, 2002).
Innovation
Myocardial infarction (MI)-induced nitrative stress leads to nitration of proteins. cAMP-response element binding protein (CREB) is a key transcription factor involved in myocardial fibrosis. However, little is known about the impact of nitrated CREB in MI-induced myocardial fibrosis. In this study, for the first time, we demonstrated that nitrated CREB promoted MI-induced myocardial fibrosis via regulating the transcription of Col1a2 and Cxcl12. These findings imply that the nitrated CREB might be a potential therapeutic target against MI-induced myocardial fibrosis.
Since containing phenolic ring structures, aromatic amino acids can be modified by peroxynitrite, in which tyrosine is the most common potential target for nitration. As one kind of post-translational modification, nitration may have an effect on protein's function, and the application of treatment based on protein nitration has been reported (Elshaer et al, 2018). However, the role of protein nitration in MI-induced myocardial fibrosis remains unclear.
cAMP-response element binding protein (CREB, CREB1) is a transcription factor participating in extensively pathophysiological processes, and is located downstream of multifunctional signaling pathways, suggesting a multifunctional role in cardiovascular diseases (Cheng et al, 2019; Pardali et al, 2017; Yao et al, 2019). Several studies have proved that CREB is correlated with the progression of cardiac fibrosis (Yi et al, 2019), nevertheless deeper insights are still needed for a better understanding of the basal mechanism. As we know, the transcriptional function of CREB is affected by multiple factors, such as phosphorylation, nuclear translocation, and binding to coactivators. Are there any novel factors regulating the transcriptional function of CREB in MI-induced myocardial fibrosis? CREB contains aromatic amino acids, which are potential targets of nitration. However, little is known about the impact of nitrated CREB on its transcriptional function in MI-induced myocardial fibrosis.
In this work, we try to explore the role of nitrated CREB in MI-induced myocardial fibrosis. First, we detected the nitration level of CREB after MI. Then we identified the nitration site of CREB, and illuminated nitrated CREB promoted its phosphorylation, nuclear translocation, and binding to transducers of regulated CREB (TORCs). CREB not only induced the proliferation and migration of CFs, but also promoted the synthesis and secretion of ECM proteins. To further verify the effect of nitrated CREB, downstream genes of transcription factor CREB were investigated. Chromatin immunoprecipitation (ChIP)-seq combined with RNA-seq results showed that CREB transcribed Col1a2 and Cxcl12 directly after MI. Meanwhile, nitrative stress enhanced MI-induced ECM protein synthesis and secretion. Finally, nitrated CREB promoted MI-induced myocardial fibrosis through improving the transcription of Col1a2 and Cxcl12.
Results
Nitrated CREB was higher in MI-induced myocardial fibrosis and Tyr182, 307, 336 were the nitration sites of CREB
To investigate the relationship between nitrated CREB and MI
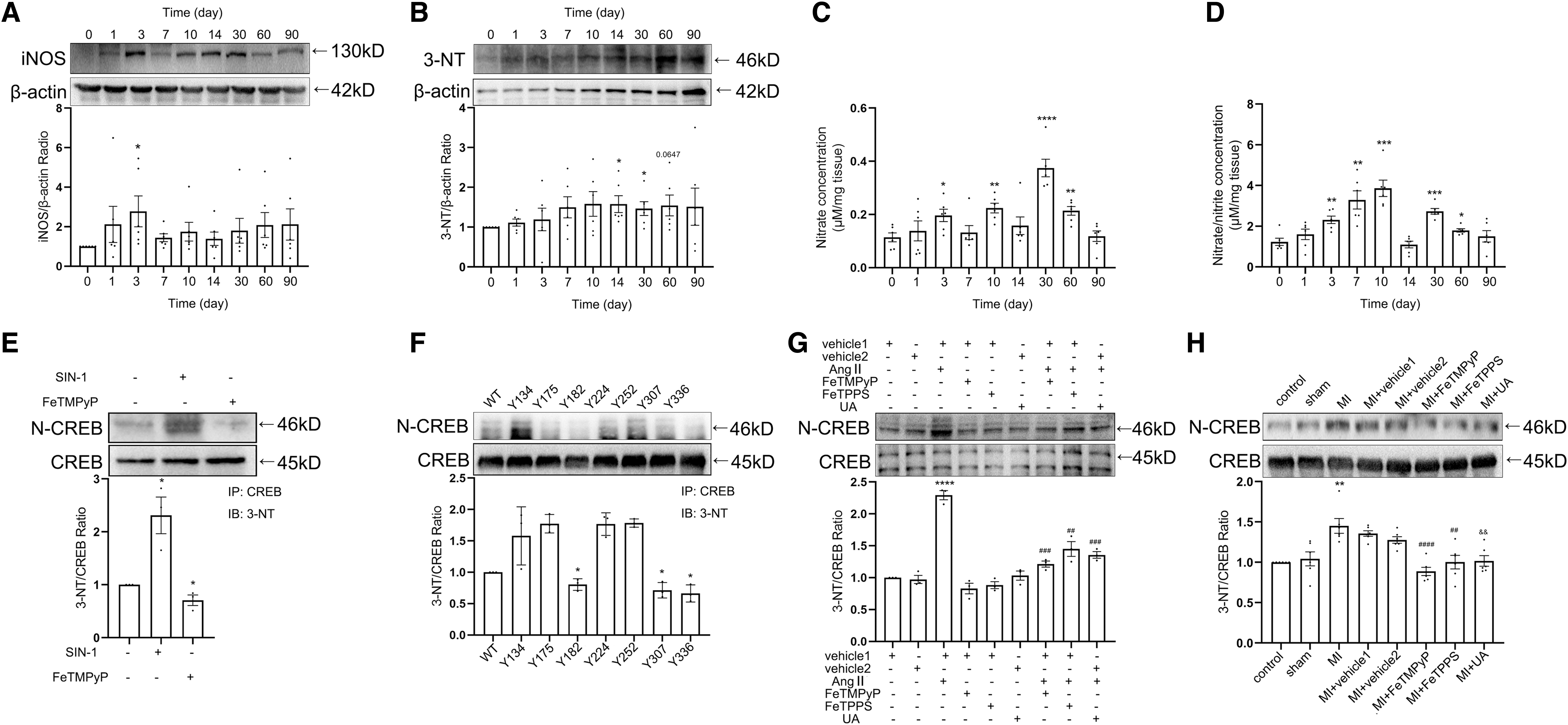
To investigate the effect of nitrated CREB on its transcriptional function, the donor of ONOO− 3-morpholino-sydnonimine (SIN-1), and three kinds of ONOO− scavengers, Fe (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride porphyrin pentachloride (FeTMPyP), 5, 10, 15, 20-tetrakis (4-sulfonatophenyl) porphyrinato iron (III) chloride (FeTPPS), and uric acid (UA) were applied to the fibroblasts, respectively (Aliena-Valero et al, 2021; Lamoke et al, 2015; Suofu et al, 2010; Suzuki et al, 2020; Vila et al, 2019; Yao et al, 2012; Zhou et al, 2008). The level of nitrated CREB was increased significantly with the administration of SIN-1, and was decreased with the administration of FeTMPyP compared with the control group (Fig. 1E and Supplementary Fig. S2C).
We then sought to identify the tyrosine residue(s) undergoing modification in CREB. There are seven tyrosine residues in CREB. The recombinant plasmid vectors were constructed by encoding a CREB mutant, in which tyrosine was replaced by alanine (CREB-Y→A). Tyr182, 307, and 336 were identified as the nitro-site according to immunoprecipitation (IP) (Fig. 1F and Supplementary Fig. S2D).
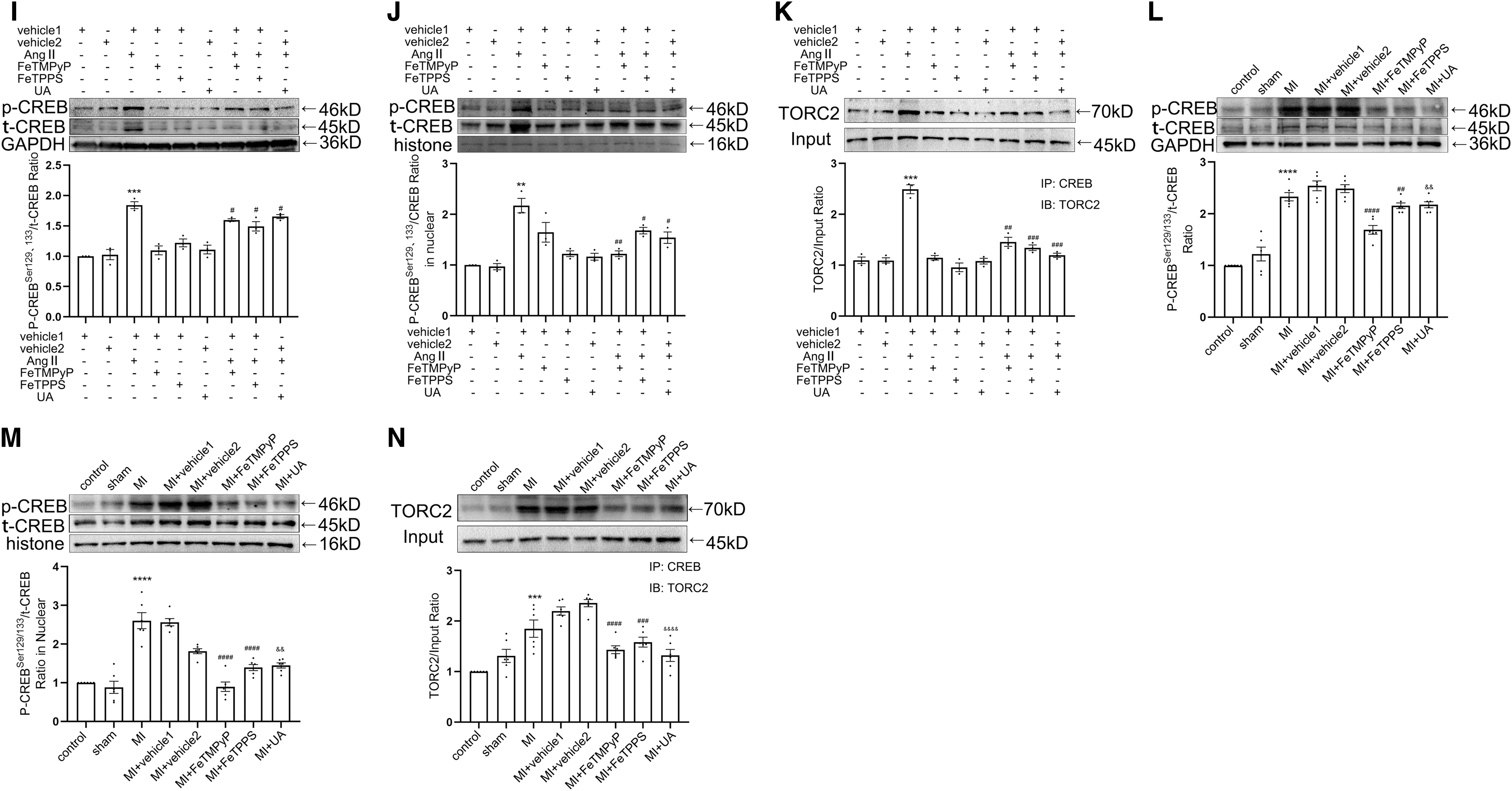
Angiotensin II (AngII) was used to increase the nitrative stress (Du et al, 2019). co-IP showed that the treatment of ONOO− scavengers attenuated AngII-enhanced nitrated CREB level in vitro (Fig. 1G and Supplementary Fig. S2E). To investigate the role of nitrated CREB in MI-induced myocardial fibrosis in vivo, we detected the nitration level of CREB in rat tissues 4 weeks after MI (Supplementary Fig. S1L). Results showed that the nitration level of CREB was increased after MI, and the nitration of CREB was decreased with the administration of ONOO− scavengers consistently (Fig. 1H and Supplementary Figs. S1N and S2F). Phosphorylation, nuclear translocation, and binding activity to transcription coactivators are all important factors affecting the transcription function of CREB.
Among the three main coactivators of CREB, TORCs bind to the leucine zipper region where CREB nitration is most likely to modulate rather than the other two coactivators. With the administration of ONOO− scavengers, the phosphorylation level of CREB, the expression of phosphorylation CREB protein (p-CREB) in nuclear, and the binding activity to TORC2 were decreased both in vitro and in vivo (Fig. 1I–N and Supplementary Fig. S2G–L). Taken together, these results suggested that nitration of CREB was elevated post-MI, which promoted transcriptional function of CREB.
CREB promoted ECM protein synthesis as well as proliferation and migration of CFs in vitro
CREB is involved in multiple cellular functions as a transcription factor, and the function of CREB in fibrosis was confirmed first. Primary CFs were harvested from Sprague-Dawley (SD) rats, which appeared as a long spindle shape (Supplementary Fig. S1A). CREB was overexpressed with pAV-CMV-CREB transfection and was knocked down with si-CREB (siRNA-CREB-homo-919) in rat CFs, whose efficiency was demonstrated by Western blot (Supplementary Fig. S1B, C). The significant upregulation and downregulation of p-CREB were observed in the pAV-CREB group and si-CREB group, respectively (Fig. 2A, B and Supplementary Fig. S3A). The expressions of fibronectin (FN1), collagen I (Col I), and osteopontin (OPN) were increased in pAV-CREB groups, and were decreased in si-CREB groups (Fig. 2C, D, and Supplementary Fig. S3B). The rat CF proliferation was enhanced or attenuated 24 and 48 h after the transfection with pAV-CREB or si-CREB correspondingly (Fig. 2E).

GO and KEGG analysis were used to evaluate the proliferation and migration of CFs (Figueiredo et al, 2020). RNA-seq showed that differential genes were enriched in the functional regions of proliferation (Fig. 2F, G). Meanwhile, the proliferation-related genes were analyzed, and the results showed that Ccnd1, Aurkb, Foxm1, Kifc1, Birc5, and Ccnb1 were decreased in the si-CREB group (Fig. 2H). The migration of CFs was detected using the same pattern with proliferation. KEGG analysis showed that differential genes were enriched in the focal adhesion regions of migration (Fig. 2I). Transwell showed that the rat CF migration was enhanced or attenuated 24 and 48 h after the transfection of pAV-CREB or si-CREB correspondingly (Fig. 2J, K). Together, these data suggested the multiple functional roles of CREB, including promoting ECM synthesis as well as the proliferation and migration of CFs in vitro.
CREB promoted the MI-induced myocardial fibrosis in vivo
To further clarify the multiple roles of CREB in MI

The above results suggested that CREB was involved in the process of myocardial fibrosis after MI by regulating the synthesis and secretion of ECM proteins in vivo.
CREB promoted the expressions of ECM proteins 14 days post-MI
Next, the specific role of CREB in MI-induced myocardial fibrosis was clarified. Histological staining results indicated that the collagen deposition appeared scattered 10 days after MI. On day 30, the collagens began to distribute regularly (Fig. 4A, B). The appearance of fibrosis is followed by a recession of the cardiac function. Ultrasound parameters showed a significant decline in EF (%) and FS (%) on days 1, 7, and 10, followed by a recovery at days 14–90. A decrease appeared in LVAW (mm) from days 1 to 14, accompanied with an increase in LVPW (mm) from days 1 to 7 after MI (Fig. 4C–F and Supplementary Fig. S1F). The level of fibrosis was elevated.

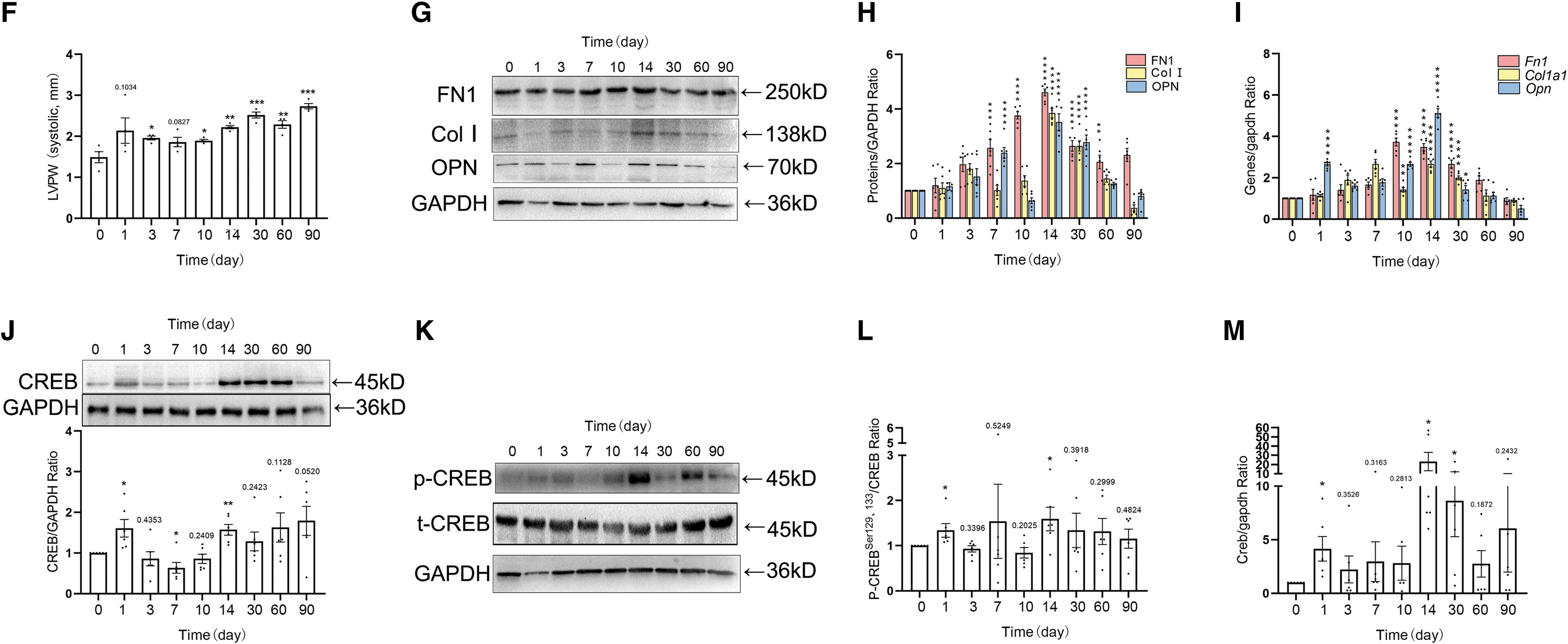
The expressions of FN1, Col I, and OPN were increased on day 14 at the protein and mRNA levels, respectively (Fig. 4G–I and Supplementary Fig. S3D). CREB is located downstream of various phosphokinases and is involved in multiple pathological processes. Both the total CREB protein, p-CREB and CREB mRNA, levels were elevated on days 1 and 14 (Fig. 4J–M and Supplementary Fig. S3E, F). Meanwhile, the peak of ECM protein expressions was 14 days after MI. These results suggested that CREB might be directly related to the expression of ECM proteins.
CREB transcribed Col1a2 and Cxcl12 directly
CREB is a well-known transcription factor with a leucine zipper structure. The above experimental results had proved that CREB participated in the process of myocardial fibrosis through regulating proliferation, migration of fibroblasts, and synthesis of ECM proteins. To further clarify the role of nitrated CREB in MI-induced myocardial fibrosis, we investigated the potential downstream genes of transcription factor CREB. Studies have shown that CREB is directly involved in the transcription of cyclins (Desdouets et al, 1996), however, whether CREB is directly involved in the transcription of these ECM proteins remains unclear. Using Etv2 as a positive internal control (Rasmussen et al, 2012), we isolated fibroblasts from heart tissues 14 days after MI for ChIP-seq, and the results showed that only 4.44% of the sites that CREB bound were located in the promoter region of gene structure (Fig. 5A).
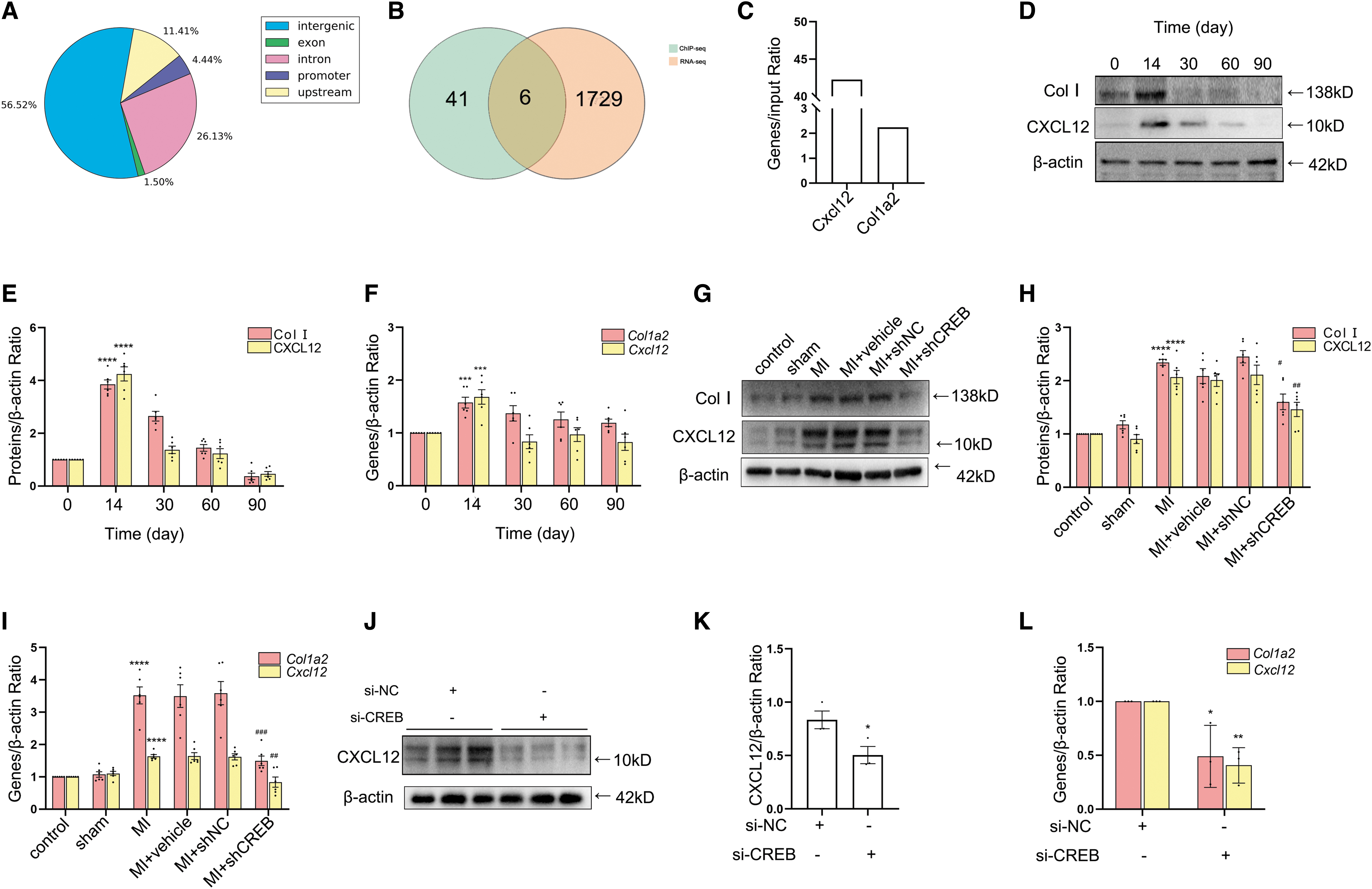
Among the ECM protein genes in ChIP-seq results, we found that CREB bind to the promoter region of Col1a2 directly. Collagen I is made by the assembly of three chains (two α1 chains and one α2 chain) into procollagen (Rittié, 2017). According to the intersection of ChIP-seq and RNA-seq analysis, we found that CREB could directly bind to the promoter region of Cxcl12 (Fig. 5A–C). CXCL12 (SDF-1) is a chemokine that participates in post-MI tissue repair released by chemotactic fibroblasts and endothelial cells (ECs) (Di Maggio et al, 2017; Guo et al, 2020). Fibroblasts extracted from heart tissues 14 days after MI were used to verify the actual binding activity of CREB to the Col1a2 and Cxcl12 promoter regions through ChIP assay, which indicated a positive result (Fig. 5C).
The tissues and cells mentioned in the above experiments were used to detect Col I and CXCL12 expressions at the protein and mRNA levels, respectively. Col I and CXCL12 expressions were significantly increased 14 days after MI at the protein and mRNA levels, respectively (Fig. 5D–F and Supplementary Fig. S3G). After downregulating of CREB expression either by small interfering RNA (siRNA) or by lentivirus, the expressions of Col I and CXCL12 were decreased at the protein and mRNA levels, respectively (Figs. 2C, 2D, 5G–L, and Supplementary Fig. S3H, I). These results suggested that CREB participated in the process of myocardial fibrosis via directly initiating the transcription of Col1a2 and Cxcl12 after MI.
Nitrated CREB participated in MI-induced myocardial fibrosis via accelerating transcription of Col1a2 and Cxcl12
We then clarified that nitrative stress inhibited the synthesis of ECM proteins in vitro. The protein and mRNA levels of FN1, Col I, and OPN were attenuated with the administration of ONOO− scavengers, respectively (Fig. 6A–C and Supplementary Fig. S3J). Together, the administration of ONOO− scavengers attenuated AngII-induced ECM protein synthesis. Next, we further studied the relationship between CREB nitrification and fibrosis in vivo. Histological staining showed less collagen deposition in the MI+ONOO− scavenger groups than in the MI+vehicle groups (Fig. 6D, E). Ultrasound parameter results showed that compared with the MI+vehicle group, there was no significant change of EF, FS, and LVAW in the MI+ONOO− scavenger groups. However, LVPW was decreased compared with the MI+vehicle groups (Fig. 6F–I and Supplementary Fig. S1M).
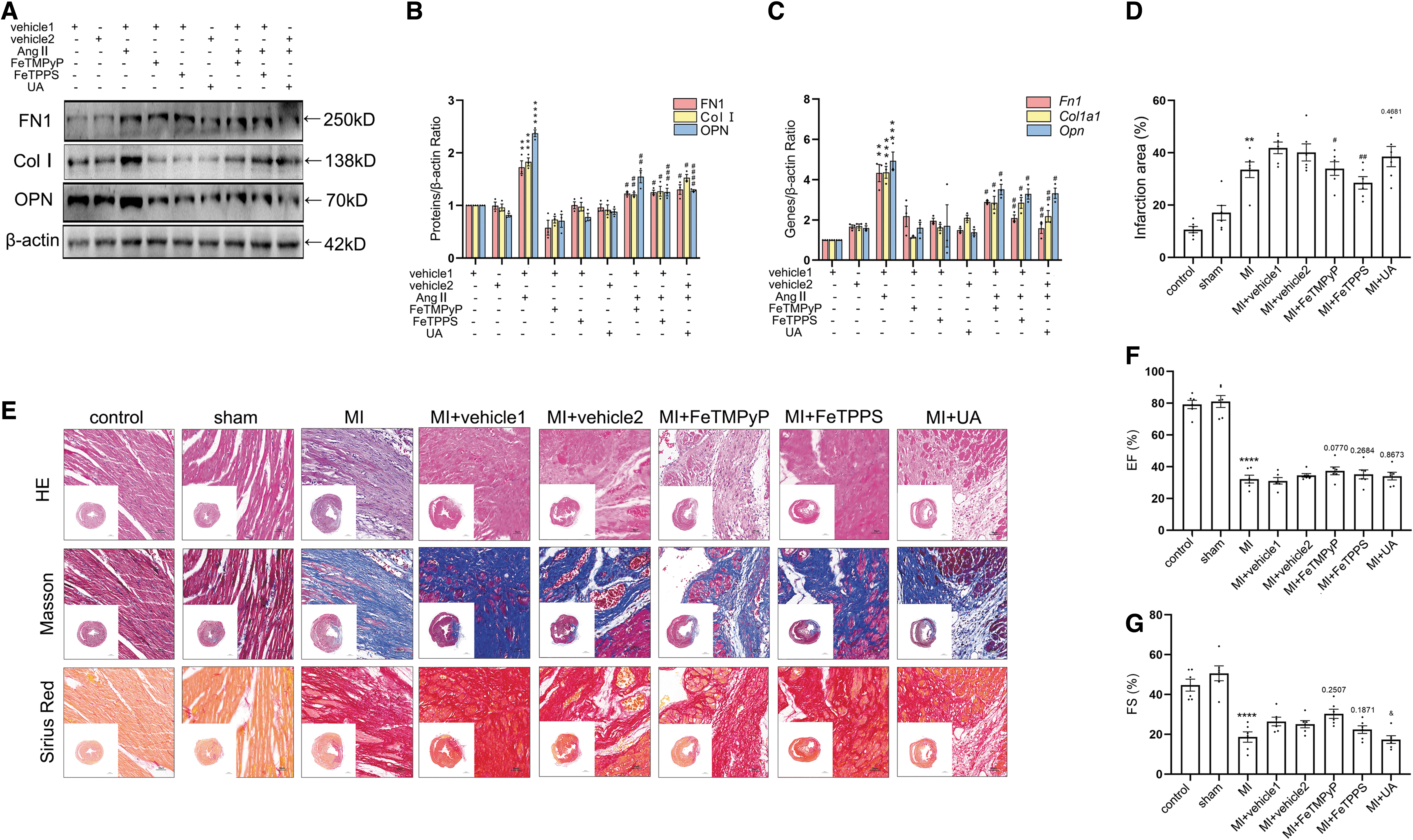
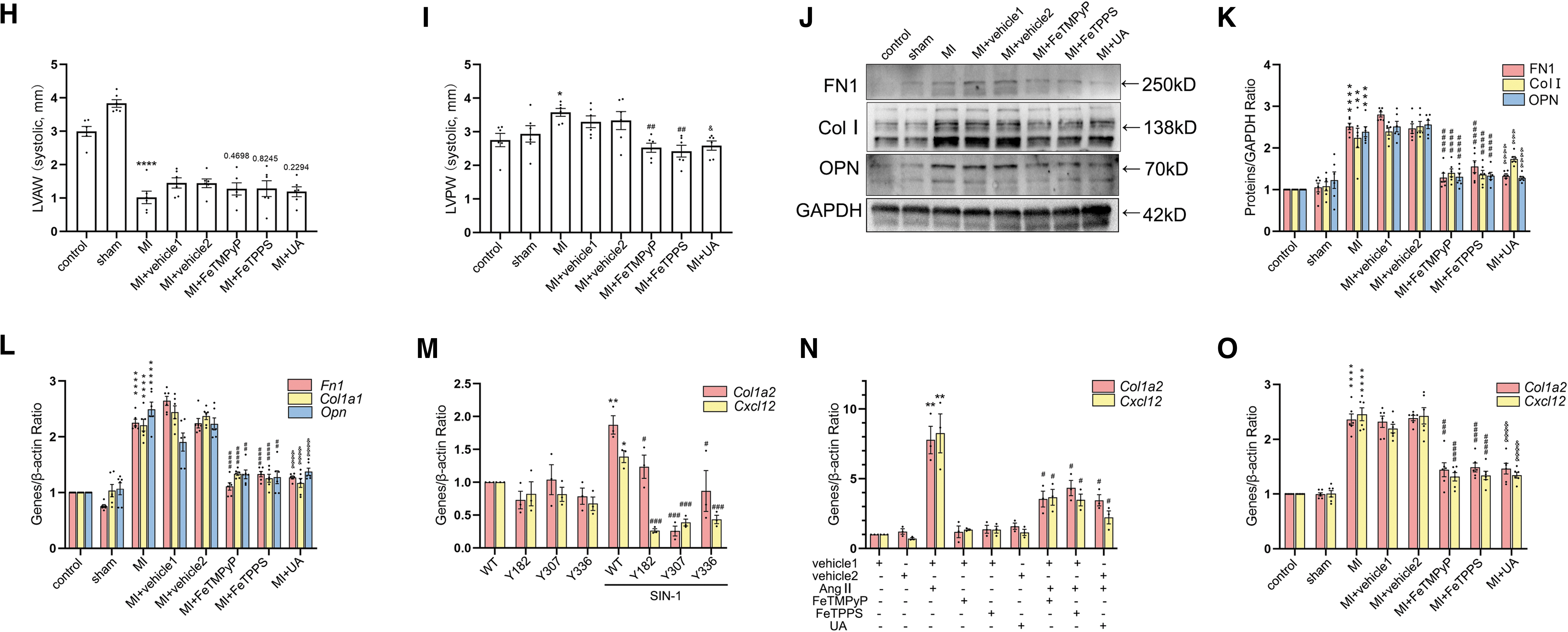
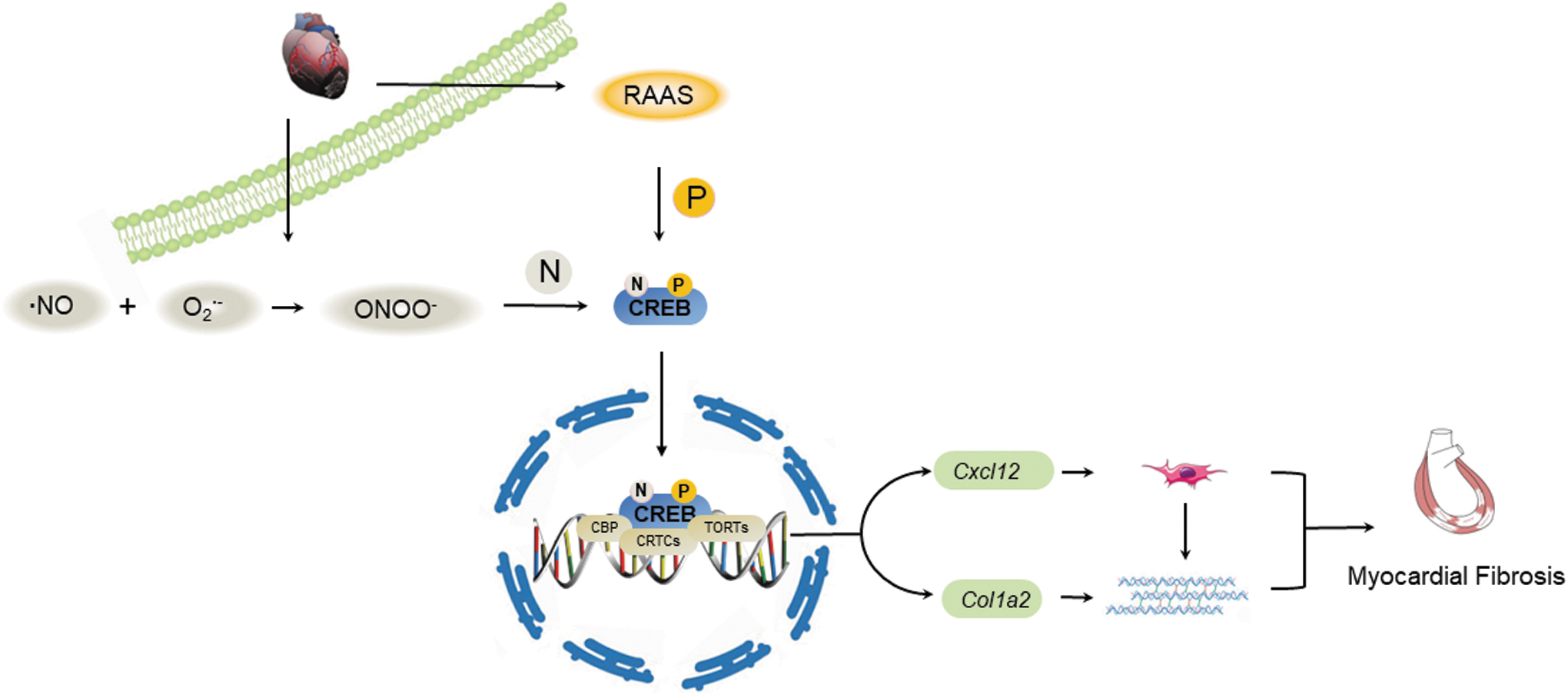
The levels of FN1, Col I, and OPN were decreased after the injection of ONOO− scavengers at the protein and mRNA levels, respectively (Fig. 6J–L and Supplementary Fig. S3K). mRNA levels of downstream genes were used to detect the transcriptional function of nitrated CREB. Transfection with CREB-Y182A, CREB-Y307A, and CREB-Y336A significantly attenuated SIN-1-induced increasing of Col1a2 and Cxcl12 mRNA levels (Fig. 6M). Concurrently, our results showed that the mRNA levels of Col1a2 and Cxcl12 were decreased with the administration of ONOO− scavengers in vitro and in vivo (Fig. 6N, O). Taken together, the above results suggested that nitrated CREB exacerbated MI-induced myocardial fibrosis via accelerating transcription of Col1a2 and Cxcl12 (Fig. 7).
Discussion
Our results demonstrated a novel mechanism connecting the increased protein nitration in the myocardium with myocardial fibrosis after MI. In this study, for the first time we revealed that CREB, as a key transcription factor located downstream of multiple signaling pathway, could be nitrated after MI and nitration of CREB participated in MI-induced myocardial fibrosis.
In our work, it is interesting to find a promotive effect of nitrated CREB occurrence in MI-induced myocardial fibrosis. Since the early 1990s, the nitration of proteins has been established as one kind of post-translational modification and has been widely studied. Quite a few of diseases are summarized into an inflammatory reaction process, for example, atherosclerosis is exudative inflammation and chronic hepatitis is hyperplastic inflammation. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are products of inflammatory reaction, and the nitration of proteins is mainly implemented by RNS. More and more proteins have been found undergoing nitration in different disease models. The nitration of proteins mainly occurs in tyrosine and other aromatic amino acid residues. Nitration of proteins may undergo a gain-of-function or loss-of function.
Most studies have shown that nitration of proteins can inhibit protein function in general (Alvarez et al, 2011). However, the nitration of sarcoendoplasmic reticulum Ca2+-ATPase (SERCA2) was positively associated with myocardial fibrosis in diabetic heart disease (Liang et al, 2016). In some experiments, the administration of metallothionein (MT), an inhibitor of nitration, attenuated myocardial fibrosis (Bowers et al, 2004; Cai et al, 2006), suggesting that the process of nitration possibly promoted the function of certain proteins, however, the underlying mechanism has not been clarified.
ONOO− is a strong oxidant in living organisms, produced by the reaction of NO with O2 •−. ONOO− can react with most biomolecules such as proteins, lipids, and nucleic acids, promoting the nitration of biomolecules. Excessive ONOO− leads to the accumulation of 3-NT in vivo, which changes the protein structure and function. Lots of ONOO− scavengers have been wildly used to inhibit nitrative stress, including FeTMPyP, FeTPPS, UA, and MT. However, whether these scavengers affected fibrosis via their nonspecific effects is unclear. To avoid the nonspecific effects of a single scavenger, we used three of the commonly used scavengers (FeTMPyP, FeTPPS, and UA) for better understanding the role of nitration in myocardial fibrosis.
Our results showed that, compared with the vehicle groups, although the administration of FeTMPyP and FeTPPS had little influence on some of signaling pathways including the expressions of ECM proteins and Cxcl12, there was no statistical significance in most parameters. Meanwhile, the administration of UA had no significant influence on these signaling pathways. Therefore, the suppression of FeTMPyP, FeTPPS, and UA on fibrosis is mainly due to their common effects, that is, scavenging of ONOO−.
As mentioned above, as a footprint of ONOO−, the occurrence of 3-NT reflected nitration of protein. However, the level of 3-NT was different from that of iNOS in our experiment due to the existence of ONOO−-independent nitration and nitric oxide synthase (NOS)-independent NO generation (Balint et al, 2001; Sabadashka et al, 2021). Through site-directed mutation assay, we have verified Tyr182, Tyr307, and Tyr336 as the nitro-sites of CREB. Tyr182 is located in the Q2 domain of CREB, which is a glutamine-rich constitutive activation domain. The Q2 domain is necessary and sufficient for the recruitment of the RNA polymerase II transcription complex. Mutations in Q2 abrogated the basal transcriptional activity of CREB (Johannessen et al, 2004). Nitration of Tyr182 may play its role by inhibiting the binding of CREB to RNA polymerase. Tyr307 and 336 are located in the alkaline leucine zipper domain of CREB, where CREB binds to DNA.
The key to CREB selective binding to DNA is the hydrophobic domain between basic region residue Gln277 and leucine zipper residue Asp341. There are three main coactivators that regulate the transcription function of CREB, among which CBP (CREB-binding protein) binds to kinase-inducible domain (KID), CRTCs (CREB regulates transcriptional) binds to Arg314 in DNA binding domain, and TORCs binds to DNA binding domain (Altarejos and Montminy, 2011; Conkright et al, 2003; Richards et al, 1996). Specifically, the residues that CBP binds are Ser109, 111, 114, 117, 121, 142, and 143. The effect of TORCs is independent of Ser133 phosphorylation (Smith et al, 2021). Our work showed that nitration of CREB occurred at Tyr307 and 336, both of which were located in the bZIP domain. The binding residue site of TORCs to CREB has not been identified, which suggested that the nitration of CREB may affect the binding process between TORCs and CREB.
We have found that the administration of ONOO− scavengers attenuated the binding activity between CREB and TORC2. However, the nitration of TORCs has not been elucidated, and simultaneously, whether the interaction between nitrated CREB and nitro-TORC2 affects the final fibrosis process still needs to be further studied.
In our experiments, the nitration of CREB enhanced its transcriptional function by promoting its phosphorylation level. Nitration and phosphorylation are both post-translational modifications of proteins, and phosphorylation is the active form of CREB. CREB owns three main phosphorylation sites in KID, in which Ser129 and 133 play a positive role (Swarthout et al, 2002) and Ser142 plays a negative role (Niederberger et al, 2007). We have found that the administration of ONOO− scavengers attenuated the phosphorylation of CREB at Ser129 and 133. We also confirmed that the active nitration residue sites were far apart from the phosphorylation sites in the sequence. However, the distance between them is relatively close in the three-dimensional structure. Possibly there may be a certain interaction between these two post-translational modifications and thus affected the final effect.
In addition to phosphorylation, the nitration of CREB enhanced its transcription function by promoting its nuclear translocation in our research. Cardiac diseases, associated with increased oxidative–nitrative stress, perform increased transcription factor activation, such as mitogen-activated protein kinase (c-Jun N-terminal kinase and p38) (Rajesh et al, 2010). Transcription factor NFκB shares many similarities in functions with CREB, including their facilitating effects on angiotensin and ROS (Haack et al, 2013). Similarly, nitro-NFκB showed an enhanced nuclear translocation (Suzuki et al, 2020; Xu et al, 2018). This promotional regulation of nuclear translocation may be the effect of nitration on certain transcription factors that have similar functions. Singer et al have found that CREB-Ser142 phosphorylation correlated with transient nucleocytoplasmic translocation of CREB (Liu et al, 2013). The enhanced nuclear translocation of CREB by nitration modification may be related to its Ser142 phosphorylation. We have elucidated the relationship between nitration and Ser129 and 133 phosphorylation.
In the future, the relationship between nitration and Ser142 phosphorylation needs to be further explored to clarify the effect of nitration modification on CREB nuclear translocation.
Multiple functions of CREB were examined to determine the ultimate effect of its nitration. We demonstrated that CREB could regulate the proliferation and migration of fibroblasts, and promote the secretion of ECM proteins to induce myocardial fibrosis after MI. Several downstream genes of CREB, such as c-Fos, Etv, Muc2, and Zo-1, have been reported in fibrosis diseases. However, its promotive role mainly occurs in macrophages, smooth muscle cells (SMCs), and epithelial cells (Huang et al, 2020; Zhang et al, 2019). The main effector cells of fibrosis are fibroblasts, and it is essential to study the downstream genes of CREB in fibroblasts. Thus, we explored other genes whose promoter domains CREB might bind to in CFs.
Transcriptionally, CREB participates in the transcription of cyclin D1 and Nectin-2 to promote cell proliferation and migration (Lui et al, 2006; Perrault et al, 2021). Besides, other proliferation and migration-related genes are controlled via the functional cooperation between their transcription factors and CREB. Nevertheless, little has been reported about the role of CREB in ECM protein transcription. In our study, Col1a2 was the only CREB-regulated transcript ECM protein gene in ChIP-seq result. Among the other ECM proteins, OPN was unique for its multiclass transcription factor regulation effect (Sawaki et al, 2018; Shirakawa et al, 2020). OPN is involved in the transition of SMCs from a synthetic phenotype to a secretory phenotype, in which c-Fos played an important role (Guo et al, 2021).
CREB is one of the most important transcription factors of c-Fos (Coulon et al, 2010), that is, in SMCs, CREB may also regulate the expression of OPN by regulating the expression of c-Fos. These inconsistencies suggest that OPN expression after MI may be a multicellular and multifunctional interaction manifestation in which CREB is involved.
According to ChIP-seq, CREB bound to the Cxcl12 promoter domain. Simultaneously, Cxcl12 could be transcribed by CREB directly according to ChIP analysis. As reported, Cxcl12 was regulated by multiple transcription factors. Transcription factor c-MYB and NR2F1 bind to the Cxcl12 promoter domain in cancer cells (Chen et al, 2010; Gao et al, 2019). After ischemia, HIF-1α regulates CXCL12 expression by binding to its promoter (Hu et al, 2007). Treatment with CREB antisense oligonucleotide significantly attenuated the expression of HIF-1α, but not vice versa (Lu et al, 2015). Thus, there may be a more complex relationship among CREB, HIF-1α, and CXCL12. CXCL12 plays its relevant roles through binding to its receptors CXCR4 and CXCR7. Human genome-wide association studies have identified novel loci downstream of the CXCL12 gene associated with angiogenesis after MI (Farouk et al, 2010).
CXCL12 has been reported by its chemotaxis in several cell types of the heart such as vascular ECs, SMCs, cardiomyocytes, fibroblasts, and pericytes (Ghadge et al, 2021), among which the migration of fibroblasts and inflammatory cells occurred before 14 days during the development of myocardial fibrosis. The formation of collateral circulation is the key step of repair after injury. Attracted by proangiogenic signals, ECs step into sprouting to form new vessels, recruitment of pericytes, and vascular SMCs promote vessel stabilization (Eelen et al, 2020; Potente et al, 2011). It is inferred that CXCL12 could chemotaxis other types of cells to participate in angiogenesis 14 days after MI, such as vascular ECs, SMCs, and pericytes. Spontaneous reperfusion (SR) based on the formation of collateral circulation was associated with a lower appearance of 90-day composite of death, heart failure, or cardiogenic shock (Shavadia et al, 2020).
The activation of CXCL12 by CREB may be involved in the formation of SR clinically. Meanwhile, nitration highly affected the function of the protein, and the function of CREB is reflected in its transcriptional facilitation. In this study, both the mRNA levels of Col1a2 and Cxcl12 treated with ONOO− scavengers or mutation plasmid in vitro and in vivo were assessed, and nitrated CREB showed promotive effects in the transcription of Col1a2 and Cxcl12.
In conclusion, we found that nitration of CREB participated in MI-induced myocardial fibrosis through reinforcing its regulation function by enhancing p-CREB, CREB nuclear translocation, and binding to TORCs, among which CREB transcripts Col1a2 and Cxcl12 directly. These data indicated that nitrated CREB might be a potential target against MI-induced myocardial fibrosis and provided a novel therapeutic strategy for myocardial fibrosis based on protein nitration.
Materials and Methods
Cell culture, plasmid, siRNA, and transfection
Adult male SD rats were used for the harvest of primary CFs (Ma et al, 2020). Briefly, the ventricle was obtained aseptically, cleaned, rinsed, and minced into small pieces with saline. Saline should be precooled in advance. The heart pieces were digested at 37°C with a digestive fluid, which contained trypsin (0.05%, 0458; Lablead Biotechnology) and collagenase II (0.05%, V900892-1; Sigma-Aldrich), 5 min each time until the tissue fragments disappeared. The digestive juices mixed with the cells were sucked into the high-glucose Dulbecco's minimal essential medium/Ham's F12 (DMEM/F12, glucose, 4.5 g/L, 10-092-CV; Corning) medium with 10% fetal bovine serum (FBS, 10091; Gibco) and 1% penicillin/streptomycin (KGY0023). The released cells from the digestions were pooled and filtered through a 200-μm sieve to remove the heart debris. Then, the obtained cells were separated by a 10-min centrifugation at room temperature.
Primary mouse CFs were cultured in DMEM/F12 with 10% FBS and 1% penicillin/streptomycin supplemented at 37°C in 5% CO2. The unattached cells were removed, and the plates were washed with DMEM/F12 several times. Under these conditions, 95% of the resulting attached cells were CFs (Lorenzen et al, 2015; Ma et al, 2019).
CFs were isolated from rat ventricle and cultured in high-glucose DMEM/F12 supplemented with 10% FBS and 1% penicillin streptomycin. For directing cell proliferation, cells were digested with pancreatin (KGY0012) and seeded onto 96-well plates (Corning) at a density of 8 × 103 cells/well. The cells were unfolded about 2 days later, and plasmid and siRNA were transfected with lipo6000 (C0526; Beyotime, Shanghai, China). Specific siRNA oligonucleotides targeting rat CREB and plasmid were purchased from GenePharma and WZ Biosciences, Inc (Table 1). For ectopic expression in primary CF cells, the full-length rat CREB complementary DNA (cDNA) and gene site-directed mutation (Try→Ala) full-length rat CREB cDNA were subcloned into the pAV-CMV-P2A-GFP vector (NM_031017.2; HanBio, Shanghai, China). After 6 h, the FBS-free medium was replaced with an FBS-contained medium for another 18 and 42 h.
Primer Sequences for Cell Transfection
Rat CF migration in vitro was assayed using a Transwell chamber with a polycarbonic membrane (6.5 mm diameter and 8 μm pore size, 24-well plates; Corning). CFs were trypsinized and resuspended at a density of 5 × 104 cells/well. The cells were unfolded about 2 days later. Plasmid (pAV-CMV-NC and pAV-CMV-CREB) and siRNA (si-NC and si-CREB) were transfected with lipo6000 in the upper chamber. After 6 h, the FBS-free medium was replaced with an FBS-contained medium in the lower chamber and the medium in the upper chamber was replaced with transfection reagent-free DMEM/F12. Cells were cultured for another 18 and 42 h. The cells that migrated to the lower surface of the membrane were fixed with 4% methanol and stained with 10% Giemsa (20200825; Solarbio, Beijing, China). Nonmigrating cells on the top surface of the membrane were removed with cotton swabs.
The cell number was measured within three randomly chosen fields at 200 × magnification, and the average number was calculated with ImagePro. Overexpression silencing efficiencies were analyzed using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) 24 h after the transfection and Western blot assay 48 h after the transfection.
Primary rat heart fibroblasts were divided into two batches to detect the effect of nitrative stress after MI in vitro. In the first batch, cells were divided into the control, SIN-1 (Y27730; Yuanye Bio-Technology, Shanghai, China), and FeTMPyP (75854; Cayman Chemical) groups. In the second batch, cells were divided into the control, AngII (s25704; Yuanye Bio-Technology), FeTMPyP, FeTPPS (17187; Cayman Chemical), UA (16219; Cayman Chemical), AngII+FeTMPyP, AngII+FeTPPS, and AngII+UA groups. Cells were incubated with 20 ng/mL of SIN-1, 20 ng/mL of FeTMPyP, 25 μM of FeTPPS or 50 μM of UA for 6 h (Jung et al, 2009). Cells were cultured with AngII at 100 nM for 24 h (Wang et al, 2020).
Rats and treatment
All animal procedures were approved by the Institutional Animal Care and Use Committee at the Capital Medical University according to the Guide for the Care and Use of Laboratory Animals (Eighth edition, 2011; License: AEEI-2019-125). Adult male SD rats of 4–6 weeks were used in this study. Experimental group rats were subjected to permanent left coronary artery ligation (LAD). The expressions of related genes were downregulated by in situ injection of lentivirus. Specific small-hairpin RNA (shRNA) oligonucleotides targeting rat CREB in which lentivirus was used as the vector were purchased from GenePharma.
Animal model
Adult male SD rats, aged 4–6 weeks, weighing between 200 and 300 g were anesthetized with chloral hydrate (10%, 0.3 mL/kg, intraperitoneally), and intubation was provided at 60 breaths per minute. Through a left anterior thoracotomy, the left anterior descending coronary artery was identified and permanently ligated 2 mm below the left atrium along the lower margin of the left auricle using a 6-0 Prolene monofilament polypropylene suture. Successful coronary artery occlusion was confirmed by visible blanching of the myocardium distal to the coronary ligation and ST segment elevation or QRS inversion in Electrocardiogram. The thorax and muscle layers were closed with the 3-0 sutures. Once spontaneous respiration resumed, the endotracheal tube was removed. Rats were placed on a warm pad maintained at 37°C and were closely supervised until full consciousness was regained. Sham surgery was performed exactly as above without coronary artery ligation.
At different days post-MI and following echocardiographic assessment, the hearts of chloral hydrate (10%, 0.3 mL/kg, intraperitoneally) were excised and fixed in 4% paraformaldehyde for histological analysis. Left coronary artery ligation and transthoracic echocardiography were performed and analyzed severally. Transthoracic two-dimensional M-mode echocardiography was performed with Vevo2100 system. The ultrasound system was used to collect hemodynamic parameters, and through software analysis, EF%, FS%, LVAW (mm), and LVPW (mm) were calculated.
Grouping
Rats were divided into three batches. In the first batch, rats were divided into 0, 1, 3, 7, 10, 14, 30, 60, and 90 days after MI. In the second batch, rats were divided into control, sham, MI, MI+vehicle, MI+shNC (shRNA with negative control), and MI+shCREB groups. In MI+vehicle, MI+shNC, and MI+shCREB groups, after LAD ligation, 20 μL of saline, an amount of 1 × 108 unit/20 μL of shNC, and an amount of 1 × 108 unit/20 μL of shCREB (shRNA with CREB, 5′-TTCTCCGAACGTGTCACGT-3′, 5′-GGTACTACCATTCTACAAT-3′) were injected at five sites below the ligated spot. Lentiviruses were used as vector for shRNA. Lentivirus carrying nonsense RNA sequence was used as negative contro1. Four weeks after MI, the transfection efficiency of lentivirus was evaluated by Western blot and RT-qPCR.
In the third batch, rats were divided into control, sham, MI, MI+vehicle1, MI+vehicle2, MI+FeTPPS, and MI+UA groups. Four weeks after MI, the rats were sacrificed for heart samples (Kompa et al, 2021). The ONOO− scavenger-administration groups received FeTMPyP (3 mg/kg every 3 days), FeTPPS (10 mg/kg every 3 days), or UA (16 mg/kg every 3 days) through intraperitoneal injection for five-time administrations (Sun et al, 2016). Saline administration or Locke's buffer administration was used as negative control. According to the introduction by the manufacturer, FeTMPyP was stored at −20°C for a long term and dissolved into normal saline before performing experiments with a concentration of 1 mg/mL, which could be stable for more than 24 h. We made a fresh preparation every time. According to the experimental design, we started FeTMPyP injection 2 weeks after MI. Four weeks after MI, the rats were sacrificed for heart samples.
Histological staining
Tissues were fixed with 4% paraformaldehyde, and after dehydration by gradient alcohol and xylene, tissues were embedded in paraffin and cross-sectioned (4 μm). After dewaxing with gradient alcohol and xylene, histological staining was used to confirm the fibrosis levels in 0-, 1-, 3-, 7-, 10-, 14-, 30-, 60-, and 90-day groups; control, sham, MI, MI+vehicle, MI+shNC, and MI+shCREB groups; and control, sham, MI, MI+vehicle1, MI+vehicle2, MI+FeTPPS, and MI+UA groups. Vehicle1 stands for saline, and vehicle2 stands for Locke's buffer.
Hematoxylin–eosin staining
A hematoxylin–eosin (HE) staining was performed to detect the degree of fibrosis following the manufacturer's instructions (RY-IHC002a; ROBY, Beijing, China). The sections were observed under an optical microscope. The tissues were dyed pink to indicate the cytoplasm, blue-violet to indicate the cell nuclei, and pale pink to indicate the collagen fiber.
Masson staining
A Masson staining was performed to detect the fibrosis level following the manufacturer's instructions (1027A18; Leagene, Beijing, China). The sections were observed under an optical microscope. The muscle fibers appeared as red stain and the collagen fibers appeared as blue stain.
Sirius red staining
A Sirius red staining was performed to detect the fibrosis level following the manufacturer's instructions (G1471; Solarbio). The sections were observed under an ordinary optical microscope. The muscle fibers appeared as yellow stain and the collagen fibers appeared as red stain.
Immunoblotting
Tissues and cells were lysed (sc-2003; Santa), and proteins in the supernatant extracts were quantified using a BCA Protein Assay Kit (23227; Thermo Scientific). Total cell lysates containing 100 μg of protein were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF membranes (ISEQ00010; Millipore). After blocking with 5% nonfat dry milk in Tris-buffered saline-Tween-20 (TBST) for 2 h, the membranes were incubated with the primary antibodies (β-actin, 1:2000, A2319, ABclonal; GAPDH, 1:3000, ym3029, ImmunoWay; FN1, 1:2000, 66042-1-Ig, Proteintech; collagen I, 1:1000, 66761-1-Ig, Proteintech; OPN, 1:1000, 22952-1-AP, Proteintech; CREB, 1:1000, 12208-1-AP, Proteintech; p-CREB, 1:1000, ab10564, Abcam), which were prepared by TBST at 4°C overnight. The membranes were washed by TBST three times (10 min each time), and then, the membranes were incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000, ZB-2301-Rabbit, ZB-2305-Mice; ZSGB-BIO) at 37°C for 1.5 h.
After washing three times, the protein bands were visualized with enhanced chemiluminescence (SQ202; Thermo Scientific) and detected using a ChemiDocTM MP imaging system (BIO-RAD). The protein bands were then scanned using Image Lab™ Software Version 4.1
Reverse transcription-quantitative polymerase chain reaction
A reverse transcription kit (AT311; Transgene, Beijing, China) was applied to synthesize the cDNA. RT-qPCR was accomplished using SYBR Green PCR Master Mix (Q211-01; Vazyme, Nanjing, China). Fold changes in mRNA expression were calculated using the 2−ΔΔCt method. The sequences of the primers used in RT-qPCR are listed in Table 2; the primers were purified by PAGE.
Primer Sequences for Reverse Transcription-Quantitative Polymerase Chain Reaction
Sequencing
RNA-seq (BGI Genomics, Beijing, China) and ChIP-seq (KANGCHENG BIOTECH, Beijing, China) were used to understand the mechanisms of CREB-involved myocardial fibrosis. The fibroblasts were extracted from 4- to 8-week normal SD rat heart tissues for RNA-seq, which were divided into si-NC and si-CREB groups, n = 3 distinct samples for each group. The fibroblasts were extracted from eight rat heart tissues 14 days after MI for ChIP-seq. The results were taken intersection by p value (p < 0.0001) and fold change (FC) value (Log2FC >10).
Proliferation and migration
CCK-8 (FC101-02; TransGen, Beijing, China) and Transwell (G1015; Solarbio) were used to detect the proliferation and migration capacity of primary CFs, which have been described before (Li et al, 2019).
Immunoprecipitation and co-IP
IP was used to trace the nitration level of CREB, and co-IP was used to detect the integration of CREB and TORC2. Tissues and cells were lysed without ultrasonic treatment, and proteins in the supernatant extracts were quantified using a BCA Protein Assay Kit. First, the lysate was precipitated by affinity beads conjugated by protein A/G (sc-2003; Santa Cruz) to remove the nonspecificity integration. The use of CREB antibodies for IP and co-IP could cause the spontaneous precipitation of antigen–antibody complexes formed by the interaction of CREB polyclonal antibodies with nitro-Tyr and TORC2.
Second, the lysate was precipitated by affinity beads conjugated by protein A/G for 4 h at room temperature to purify the antibody–antigen–agarose compound. Purified antigens (proteins) were then visualized by SDS-PAGE, in which 3-NT (1:1000, ab110282; Abcam) and TORC2 (1:2000, ab109081; Abcam) were used as primary antibodies (Lin and Lai, 2017). Especially, TORC2 was not proved to be suitable for binding antibodies in IP experiments.
Chromatin immunoprecipitation
ChIP was used to detect protein-DNA interactions. Standard ChIP assays were performed in rats' primary CFs as previously described (Ballestar et al, 2003). Eight adult rat CFs were isolated 14 days after MI. Cells were treated with 1% formaldehyde for 15 min and sonicated to produce chromatin fragments (0.5 kb on average) by micrococcal nuclease (BioLabs, M02475). Predicted CREB responsive element DNA was immunoprecipitated with an anti-CREB Ab (9197; Cell Signaling Technology, Boston, MA). Magnetic beads (9006; Cell Signaling Technology) were used to bind the protein-DNA complexes. DNA purification (14209S; Cell Signaling Technology) and qPCR were performed by using SYBR Green PCR Master Mix (Q211-01; Vazyme). Sensitivity of PCR amplification was evaluated on serial dilutions of total DNA that was collected after sonication (input) (DeCaprio and Kohl, 2020). The oligonucleotide primer sequence is given in Table 3.
Primer Sequences for Chromatin Immunoprecipitation
Determination of total nitrate–nitrite
Total nitrate–nitrite levels were measured by the nitrate–nitrite colorimetric assay kit (780001; Cayman Chemical) based on Griess method, in which a chromophore with a strong absorbance at 545 nm was formed. NO values obtained by this method represent the total amount of nitrate and nitrite expressed in pmol/mg tissue.
Statistical analyses
All data were expressed as mean ± standard error of the mean. Comparisons between two or more groups were evaluated by Student's t-test (unpaired t-tests) or one-way analysis of variance Tukey's multiple comparisons test, respectively. p Values <0.05 were considered significant. Statistical analyses were performed using GraphPad Prism 8.2.1.
Footnotes
Acknowledgments
We are thankful for the technical instruction about permanent left coronary artery ligation of rats from Ms. Lingqiao Lu. We are grateful to Ms. Qing Xu for performing the cardiac echocardiogram of rats in the Central Lab of Capital Medical University.
Authors' Contributions
W.W. and K.X.: Conceptualization, methodology, and software. K.X.: Data curation and writing—original draft preparation. K.X. and S.C.: Investigation and resources. W.W. and H.L.: Supervision. K.X., S.C., J.C., W.Y., X.Z., D.J., and Y.W.: Writing—reviewing and editing.
Author Disclosure Statement
The authors have no conflict of interest to declare.
Funding Information
This work was supported by the National Natural Science Foundation of China (Nos. 91839107, 32071134 to W.W.).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.