
Abstract
Significance:
The significance of ferroptosis in cancer therapeutics has now been unveiled. Specific ferroptosis inducers are expected as a promising strategy for cancer treatment, especially in cancers with epithelial mesenchymal transition and possibly in cancers with activated Hippo signaling pathways, both of which cause resistance to traditional chemotherapy but tend to show ferroptosis susceptibility.
Recent Advances:
Ferroptosis is a new form of regulated non-apoptotic cell death, which is characterized by iron-dependent lipid peroxidation, leading eventually to plasma membrane rupture. Its core mechanisms have been elucidated, consisting of a driving force as catalytic Fe(II)-dependent Fenton reaction and an incorporation of polyunsaturated fatty acids to membrane phospholipids via peroxisome-dependent and -independent pathways, and suppressing factors as prevention of lipid peroxidation with glutathione peroxidase 4 and direct membrane repair via coenzyme Q10 and ESCRT-III pathways.
Critical Issues:
Developments of ferroptosis inducers are in progress by nanotechnology-based drugs or by innovative engineering devices. Especially, low-temperature (non-thermal) plasma is a novel technology at the preclinical stage. The exposure can induce ferroptosis selectively in cancer cells rich in catalytic Fe(II).
Future Directions:
We also summarize and discuss the recently uncovered responsible molecular mechanisms in association with iron metabolism, ferroptosis and cancer therapeutics. Targeting ferroptosis in addition to the current therapeutic modalities would be important to cure advanced-stage cancer. Antioxid. Redox Signal. 39, 206–223.
Introduction
Despite the recent rapid development of precision cancer therapeutics, cancer remains the leading cause of death, accounting for ∼10 million deaths worldwide in 2020 (Sung et al, 2021). Anticancer drugs, mostly inducers of apoptosis, are one of the major approaches of cancer treatment. However, the effects of anticancer drugs are limited, owing to the intrinsic or acquired resistance to apoptosis-inducing drugs (Hassannia et al, 2019; Holohan et al, 2013; Okada and Mak, 2004; Su et al, 2016). Accordingly, establishing therapeutic strategies to induce nonapoptotic cell death is expected to open a new avenue by overcoming chemoresistance.
Recently, a growing number of research efforts suggest ferroptosis, a novel mode of regulated necrosis, as an ultimate candidate for therapeutic target of cancer (Conrad et al, 2016; Hassannia et al, 2019; Vanden Berghe et al, 2014; Yang and Stockwell, 2016). In this review article, we first describe the importance of iron metabolism in cancer. We then explain the molecular mechanisms and involvement of ferroptosis in cancer therapy. Finally, we provide a perspective on this expanding research area.
Classification of Cancer Therapy
Cancer therapy is classified into local therapies (e.g., surgical therapy, radiation therapy, and ablation therapy), systemic therapies (e.g., chemotherapy, targeted therapy, hormone therapy, and immune therapy), and their combination (Baskar et al, 2012; Ramos et al, 2021; Wang et al, 2019b). Local therapies are effective for the local control by eliminating tumor cells from the targeted area, achieving complete cure of cancer at an early stage or symptom relief at the advanced stage. In contrast, systemic therapy plays a pivotal role in controlling advanced or recurrent cancer to extend survival.
Among the systemic therapies, chemotherapy with apoptosis-inducing agents is still a major scheme of the systemic therapy. Thus, intrinsic or acquired resistance to chemotherapeutic agents is a serious problem that causes recurrence and poor survival outcomes in patients (Ramos et al, 2021). Hence, it is expected to develop novel therapeutic options to induce cell death other than apoptosis to overcome the chemoresistance and improve the prognosis of cancer patients.
Chemoresistance
Resistance to chemotherapeutic agents has been observed in nearly all sorts of anticancer drugs (Galluzzi et al, 2014; Murray et al, 2012; Sladek et al, 2002; Smith et al, 2006). Multiple mechanisms are involved in the resistance. One of the molecular mechanisms is the modification of pharmacokinetics in cancer patients, such as via acquired mutations in the genes metabolizing direct target molecules, absorption, and excretion of the drug or its metabolites (Alfarouk et al, 2015). Another mechanism is the mutation or activation of certain signaling pathways, such as the Kelch-like ECH-associated protein 1 (KEAP1)/nuclear factor-erythroid 2-related factor 2 (NRF2, or also known as NFE2L2)/Cullin3 (CUL3) pathway or phosphoinositide 3-kinase (PI3K)/AKT/mitogen-activated protein kinase (MAPK)/nuclear factor-κB (NF-κB)/notch pathway.
KEAP1/NRF2/CUL3 pathway is a master regulator of transcription in the genes associated with glutathione (GSH) synthesis/reduction, reactive oxygen species (ROS)/xenobiotics detoxification, thioredoxin-based antioxidant system, and heme/iron metabolism to gain chemoresistance (Barrera-Rodriguez, 2018; Jeong et al, 2020). PI3K/AKT/MAPK/NF-κB/notch pathway regulates angiogenesis, cellular proliferation, and drug concentration/distribution, which eventually contribute to chemotherapy resistance (Rajabpour et al, 2017).
In addition, tumor microenvironments, including hypoxia, acidity, collagen, hyaluronan, and fibronectin, and the interaction between tumor cells and the surrounding cells, such as cancer-associated fibroblasts, cancer-associated macrophages, or cytotoxic T lymphocytes (CTLs), mediate resistance to the chemotherapeutic drugs (Baghban et al, 2020). Furthermore, characteristics of each cancer of the patients, such as fraction of cancer stem cells and the degree of epithelial–mesenchymal transition (EMT), also determine the chemoresistance. Here we stress that cancer cells even with high chemoresistance inevitably require iron to survive.
Overview of the Role of Iron in Carcinogenesis and Cancer Metabolism
Iron in life
Iron is the most fundamental metal for life. No independent cells, including cancer cells, can live without iron because iron is adopted in the functional catalytic portion of various enzymes for its redox activity, in DNA replication (ribonucleotide reductase), adenosine triphosphate (ATP) synthesis (cytochrome oxidases), and antioxidant activity (catalase).
Iron is the most abundant element by mass inside the Earth, occupying 80% of the core and 6.3% of the surface (Frey and Reed, 2012). Furthermore, 3.8 billion years ago, when the first life was born on the Earth, the atmospheric environment was in a reducing condition and a high concentration of Fe(II) was dissolved in the primordial sea (Gutteridge and Halliwell, 2018). In addition to the abundance, iron holds an affinity to bind a broad range of ligands, thus contributing to redox reaction or electron transfer by changing status mainly between Fe(II) and Fe(III) (Frey and Reed, 2012).
Therefore, iron has been selected in many important catalytic reactions for electron transfer or acid–base reactions in life (Hosseinzadeh and Lu, 2016; Sanchez et al, 2017). After the origination of Cyanobacteria in ∼2.2 billion years ago, Fe(II) was rapidly oxidized to Fe(III) by oxygen released with photosynthesis, resulting in precipitation in the sea by forming insoluble complexes (Gutteridge and Halliwell, 2018). Consequently, as low as 2–3 × 10−9 kg/L of iron is contained in the sea water today.
Iron metabolism
Iron mainly takes two valences in life, catalytic soluble Fe(II) and noncatalytic nearly insoluble Fe(III) (Sanchez et al, 2017). Iron is stored in the form of Fe(III) to reduce cytotoxicity, bound to transferrin (Tf) or lactoferrin extracellularly, and encapsulated in ferritin core intracellularly to prevent precipitation. When iron goes through iron transporters to cross any biomembrane, Fe(III) needs to be reduced to Fe(II) by the corresponding specific reductase (Fig. 1).
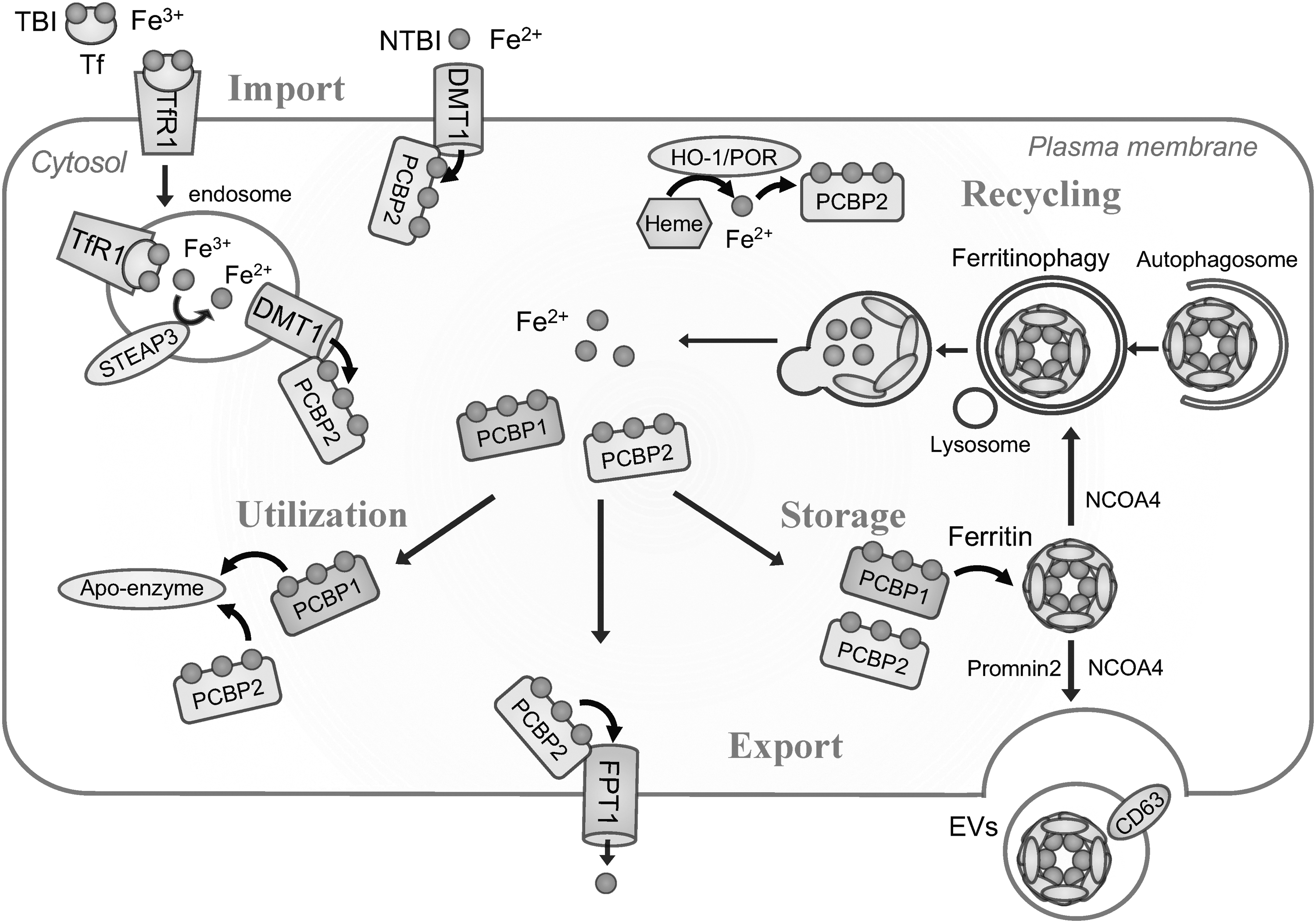
Cellular iron uptake
There are two major pathways for cellular iron uptake. One is through Tf receptor-mediated uptake of transferrin-bound iron (TBI) and the other is nontransferrin-bound iron (NTBI) uptake (Canonne-Hergaux and Gros, 2002; Dunn et al, 2007; Gunshin et al, 1997; Kautz et al, 2014). Major types of cells, including erythroblasts and cancer cells, take up iron in the form of TBI via binding to and internalization of the transferrin receptor 1 (TfR1) on the plasma membrane (Fillebeen et al, 2019).
After the endocytosis of TfR1, STEAP3 metalloreductase on the endosomal luminal membrane reduces released Fe(III) to soluble Fe(II) (Mancias et al, 2014) and transports Fe(II) into the cytosol through divalent metal transporter 1 (DMT1; solute carrier family 11 member 2, SLC11A2), which is a pH-dependent proton-coupled Fe(II) transporter (Gunshin et al, 1997). In contrast, uptake of NTBI is physiologically observed in duodenum or kidney during the absorptive process of NTBI from foods or reabsorption of NTBI from raw urine, respectively.
Furthermore, NTBI uptake is observed under iron overloaded conditions, where serum iron has exceeded the capacity of Tf, and direct uptake of NTBI into cytosol is mediated by DMT1, ZIP8 (zinc transporter 8, SLC39A8) or ZIP14 (SLC39A14) (Knutson, 2019). ZIP8 and ZIP14 have been originally identified as zinc transporters on the plasma membrane.
Intracellular iron delivery
The understanding of molecular mechanisms on cytoplasmic iron delivery has been revolutionized in the past decade. In the cytoplasm, most of Fe(II) is coordinated by reduced GSH to form Fe(II)-GS, possibly to reduce the oxidation and reactivity of Fe(II) (Philpott et al, 2020). In addition, significant population of Fe(II) or Fe(II)-GS is physiologically escorted by iron chaperones, poly r(C)-binding protein 1–4 (PCBP1–4), which are originally reported as RNA/DNA binding molecules (Swanson and Dreyfuss, 1988). Among the PCBPs, PCBP1 and PCBP2 are most abundantly expressed in a wide range of cells and tissues, and are present in the cytoplasm and nucleus.
PCBP3 and PCBP4 are expressed at low levels only in some tissues and at some periods of development (Leidgens et al, 2013; Yanatori et al, 2020). Though each PCBP escorts Fe(II), they function differently as chaperones. Regarding PCBP1 and PCBP2, major types of PCBPs, both of them deliver Fe(II) to ferritin, a major iron storage protein (Leidgens et al, 2013; Philpott et al, 2020; Shi et al, 2008) and to mononuclear iron enzymes prolyl hydroxylase (Nandal et al, 2011) and dinuclear iron enzyme deoxyhypusine hydroxylase (Frey et al, 2014).
Furthermore, PCBP2 can receive Fe(II) directly from DMT1 (Yanatori et al, 2014), can recover Fe(II) after disassembly of heme via heme oxygenase 1 (HO-1) with NADPH cytochrome P450 reductase (POR) (Yanatori et al, 2017), and can transfer Fe(II) to ferroportin 1 (FPT1, SLC40A1), the only iron exporter (Yanatori et al, 2016). In contrast, PCBP1 does not bind to DMT1 nor FPT1 (Frey et al, 2014; Yanatori et al, 2020; Yanatori et al, 2017; Yanatori et al, 2016), but appears as the only molecule to be able to upload excess cytosolic iron to ferritin cores with the help of ferritin heavy chain subunit (FTH1) (Philpott et al, 2020; Shi et al, 2008). The activity of PCBP1 and PCBP2 may be affected by cell type, expression of iron transporters or intracellular iron level, which requires further investigation.
Iron storage
Ferritin is a universal cellular protein for iron storage. Ferritin makes a hollow complex called apoferritin in cytosol by the assembly of 24 polypeptide units to store iron as Fe(III) in the core and reduce cytotoxity (Lopez-Castro et al, 2012). Ferritin consists of two subunits, H subunit to store iron safely by oxidizing Fe(II) with ferroxidase center and L subunit to mineralize for long-term storage of iron (Lopez-Castro et al, 2012). Under iron overload, due to overwhelming the capacity of ferritin, excess iron deposits as hemosiderin (Hino et al, 2021). In contrast, under cellular iron deficiency, degradation of ferritin is induced via nuclear receptor coactivator 4 (NCOA4)-mediated autophagy, named ferritinophagy.
Cellular iron export via transporter
Ferroportin (FPT1, SLC40A1) is the only known cellular Fe(II) exporter in higher species, mainly expressed in intestinal, splenic, and hepatic cells to transport iron from diets, senescent erythrocytes, and hepatic storage to serum, respectively (Ganz and Nemeth, 2012). FPT1 is post-transcriptionally regulated by two specific mechanisms, hepcidin and iron-responsible element (IRE)–iron regulatory protein (IRP) system.
Hepcidin, a 25 amino acid peptide hormone and a ligand for FPT1, is secreted mainly from hepatocytes, which promotes, after binding to FPT1 as a receptor on the plasma membrane, internalization and lysosomal degeneration of FPT1 to inhibit cellular export of iron (Nemeth et al, 2004; Park et al, 2001; Pigeon et al, 2001). IRE–IRP system as a post-transcriptional regulation plays an important role to maintain cellular iron homeostasis, which is described hereunder.
Cellular iron export via extracellular vesicles
As reported recently, extracellular vesicles (EVs) are implicated in iron export. Prominin2 (PROM2), a member of the prominin family of pentaspan membrane glycoproteins, stimulates the formation of ferritin-containing multivesicular bodies and exosomes to transport iron out of the cells (Brown et al, 2019). In addition, iron loading to cells induces the expression of CD63 and increases secretion of CD63+ EVs containing iron-loaded ferritin via the IRE–IRP systems, which indicates that secretion of CD63+ EVs is another strategy to export excess iron from iron-sufficient cells to iron-deficient cells within the same individual (Yanatori et al, 2021). Moreover, NCOA4 is also involved in iron-loaded ferritin export via CD63+ EVs by mediating the loading of ferritin to CD63+ EVs (Yanatori et al, 2021).
Iron regulation via IRE–IRP system
Cellular iron homeostasis is maintained by balanced expression of proteins involved in iron uptake, chaperoning, storage, and export. This balance is post-transcriptionally regulated by the binding of IRP1 and IRP2 to IRE within the messenger RNAs (mRNAs) of genes associated with iron metabolism. IRP1 and IRP2 are RNA-binding proteins, the amounts of which are controlled via iron-dependent regulation (Wang et al, 2007). In iron-starved cells, IRE–IRP interactions stabilize mRNA of genes for iron uptake (TfR1 and DMT1) and simultaneously inhibit translation of genes for iron storage (FTH1 and ferritin light chain subunit [FTL]) and export (FPT1 and CD63) (Kim et al, 1996; Yanatori et al, 2021).
In contrast, in iron-sufficient cells, inactivation of IRPs promotes mRNA degradation of genes for iron uptake (TfR1 and DMT1) and activates translation via releasing block at the 5′ untranslated region of genes for iron storage (FTH and FTL) and export (FPT1 and CD63) (Kim et al, 1996; Yanatori et al, 2021) (Fig. 2).
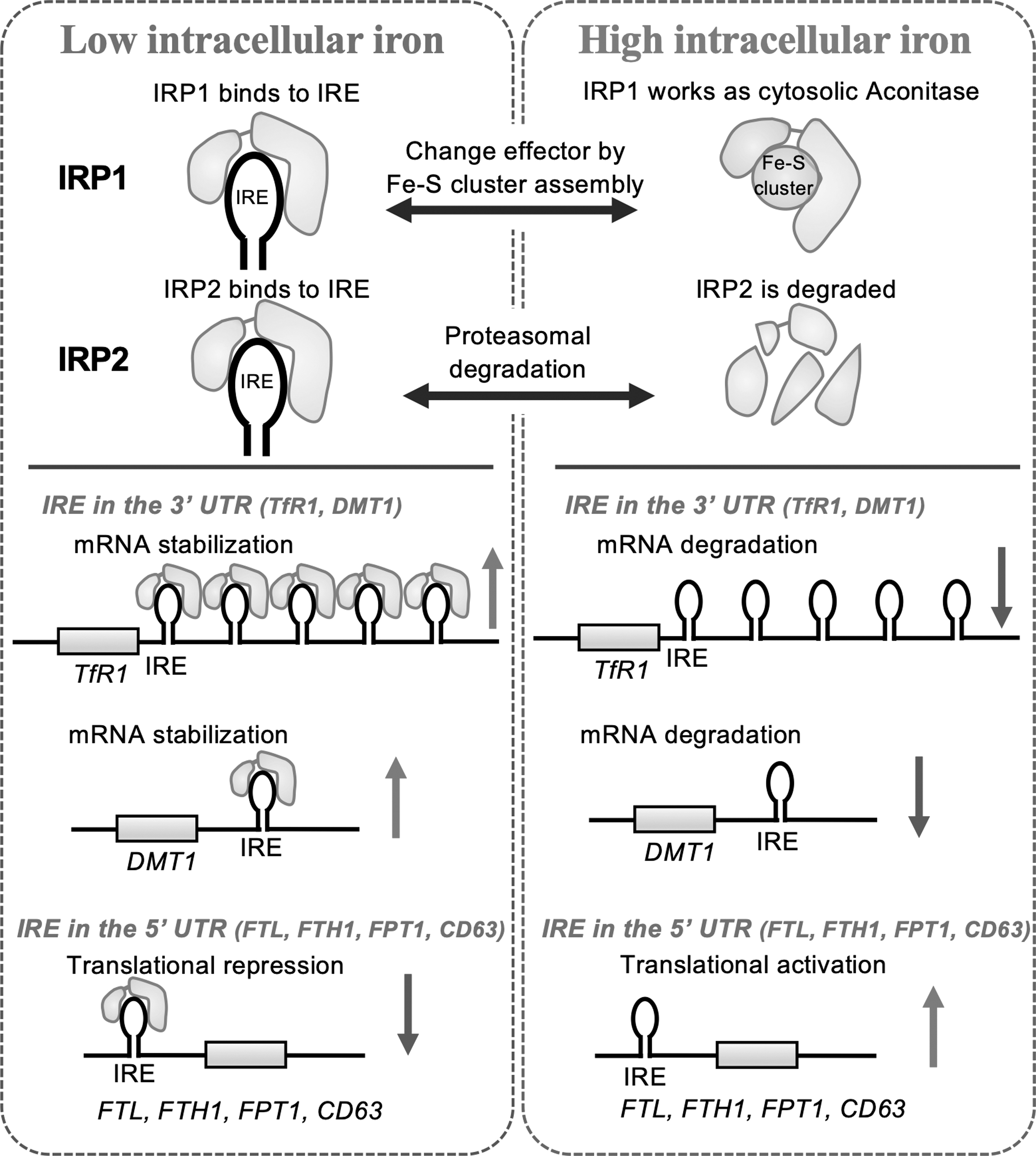
Systemic iron regulation
The total amount of iron in normal adult humans is 2.5–4 g, which is the highest among heavy metals in the body. Approximately 60% of iron is involved in erythropoiesis, utilized as the heme of hemoglobin in red blood cells and recycled by destruction of senescent red blood cells (Andrews, 1999). Most of the remaining iron is stored in the liver or spleen as ferritin or hemosiderin. With no active excretion, the body iron metabolism is a semiclosed system, and the amount of body iron is regulated by absorption. Since as low as 1 mg of iron is lost from peeling off or death of surface/intestinal epithelial cells, 1 mg is absorbed daily from duodenum to serum in the portal system under the regulation of hepcidin to stop dietary iron uptake via ferroportin (Harigae, 2018; Toyokuni et al, 2020).
Whereas the amount of iron is regulated through the ways already described, some pathological conditions may induce iron overload. Iron overload can be classified into hereditary hemochromatosis and secondary iron overload. Hereditary hemochromatosis is a disease that causes excess absorption and systemic deposition of iron via the mutations of HFE (Feder et al, 1996), hemojuvelin, HAMP (FPT1), or transferrin receptor 2 (Franchini, 2006).
Secondary iron overload is caused by chronic inflammation (e.g., chronic bacterial infection and asbestos exposure), increased erythroid cell death, including ineffective erythropoiesis (e.g., thalassemia, sideroblastic anemia, and myelodysplastic syndrome), excess iron administration (e.g., iron-rich diet, supplemental drugs, and repeated whole blood transfusion), and liver dysfunction (chronic viral hepatitis, alcoholic liver, nonalcoholic steatohepatitis, etc.) (Kohgo et al, 2008; Yanatori et al, 2020). In addition, local iron overload is developed by ovarian endometriosis (Yamaguchi et al, 2008).
Iron in carcinogenesis
Though iron is essential for every cell and the life, iron behaves as a double-edged sword. Excess iron generates ROS via Fenton reaction (Fe2+ + H2O2 → Fe3+ + •OH + OH−), leading to oxidative damage in various organs, including liver, heart, kidney, pancreas, and the central nervous system (Andersen et al, 2014; Toyokuni, 2002). Moreover, excess iron is also implicated in carcinogenesis by ROS generation to cause oxidative DNA damage resulting in genomic alterations (Poetsch, 2020). The human epidemiological and clinical observations have been described in detail in other publications.
For example, a significantly high incidence of hepatocellular carcinoma develops in the patients of genetic hemochromatosis (Kowdley, 2004), β-thalassemia (Finianos et al, 2018) or chronic HCV hepatitis (Miyanishi et al, 2019); malignant mesothelioma is induced long after asbestos exposure (Toyokuni, 2019); and ovarian clear cell carcinoma is frequently associated with ovarian endometriosis (Kajiyama et al, 2019; Pearce et al, 2012; Toyokuni, 2016; Toyokuni, 2002; Yamaguchi et al, 2008).
Overview of Ferroptosis, Catalytic Fe(II)-Dependent Regulated Necrosis Accompanied by Lipid Peroxidation
Ferroptosis
After intensive research on cellular death from various aspects, a novel iron-dependent programed necrosis called ferroptosis was coined in 2012. Ferroptosis is characterized by excessive accumulation of catalytic Fe(II) followed by lipid peroxidation, which is prevented by redox-inactive iron chelators (Dixon et al, 2012). Ferroptotic cells are morphologically characterized by shrinking mitochondria with normal or increased membrane density, mitochondrial membrane disruption, or reduced mitochondrial cristae (Dixon et al, 2012; Yagoda et al, 2007), and incomplete plasma membrane with release of intracellular contents, including damage-associated molecular patterns (DAMPs) (Wen et al, 2019), whereas apoptotic cells show membrane blebbing, nuclear/cytoplasmic fragmentation, chromatin condensation, cellular shrinkage, and swollen or ruptured mitochondria (Elmore, 2007).
Ferroptosis was first described on the process of cell death induced by erastin, which inhibits cysteine/glutamate antiporter, SLC7A11 (xCT, system Xc −), resulting in GSH depletion to inactivate glutathione peroxidase 4 (GPX4) (Dixon et al, 2012; Stockwell et al, 2017). Recent research revealed that ferroptosis is regulated by multiple systems, including iron metabolism, sulfur-dependent antioxidants, as well as peroxisome-dependent and peroxisome-independent pathways. Peroxidized phospholipids (PLs) in plasma membrane, especially PLs with polyunsaturated fatty acids (PUFAs) (Hadian and Stockwell, 2020; Jiang et al, 2021b; Li and Li, 2020), are recognized as the possible common end-stage effector.
The incorporation of PUFA onto membrane PLs is mediated by multiple enzymes. PUFAs, such as arachidonic acid (AA) and adrenic acid (AdA), are esterified at first by acyl-CoA synthetase long-chain family member-4 (ACSL4) to PUFA-CoA and are further processed by lysophosphatidylcholine acyltransferase-3 (LPCAT3) before the integration to the cell membrane (Doll et al, 2019). PLs containing PUFA (PL-PUFA) can be oxidized to PUFA-containing-phospholipid hydroperoxide (PL-PUFA-OOH) nonenzymatically by ROS generated via catalytic Fe(II)-dependent Fenton reaction, or enzymatically by lipoxygenases (LOXs), a nonheme iron containing enzyme or by POR (Stockwell et al, 2017; Tang and Kroemer, 2020a; Tang and Kroemer, 2020b; Zou et al, 2020b).
In addition, it was recently revealed that biosynthesis of plasmalogen via peroxisome is another mechanism for lipid peroxidation. Plasmalogen is produced by the collaboration of peroxisomes, a membrane-bound oxidative organelle, and endoplasmic reticulum (ER). Peroxisomal enzymes, such as fatty acyl-CoA reductase 1 (FAR1), glycerone-phosphate O-acyltransferase (GNPAT), and alkylglycerone phosphate synthase (AGPS), contribute to the synthesis of plasmalogens. Distinct from common PLs, plasmalogen uses an ether bond, instead of an ester bond, to connect the glycerophospholipid. Their precursor 1-O-alkyl-glycerol-3-phosphate (AGP) is synthesized by FAR1 and AGPS, and then transported to ER for further biosynthetic reactions involving 1-acylglycerol-3-phosphate O-acyltransferase 3 (AGPAT3).
Finally, the ER-resident enzyme plasmanylethanolamine desaturase 1 (PEDS1) mediates the production of PUFA plasmalogen, which is susceptible to lipid peroxidation (Zou et al, 2020a). Moreover, it was recently suggested that some kinds of selective autophagy are involved in triggering ferroptosis, including ferritinophagy retrieving iron from ferritin, and lipophagy and clockophagy, defined as “the selective degradation of the core circadian clock protein, aryl hydrocarbon receptor nuclear translocator-like (ARNTL), by autophagy,” forming lipid peroxidation (Jiang et al, 2021a; Liu et al, 2020).
Such lipid peroxidation of PUFAs produces a wide variety of oxidation products. Lipid hydroperoxides (LOOHs) are the initial products of peroxidation. Secondary products are aldehydes, among which malondialdehyde and 4-hydroxy-2-nonenal (HNE) are the most abundant and thought to inactivate proteins through crosslinking (Feng and Stockwell, 2018). Recently, our laboratory established that a mouse monoclonal antibody (HNEJ-1) to recognize equally His/Lys/Cys Michael adducts of HNE modification is ideal for the detection of ferroptotic cells in formalin-fixed paraffin-embedded (FFPE) pathological specimens, which demonstrated that fetal erythropoiesis and aging exploit ferroptotic process as physiological phenomena (Zheng et al, 2021).
In contrast, xCT (SLC7A11)-Cys/GSH-GPX4 axis is widely accepted as intracellular suppressive mechanism of ferroptosis, which repairs phospholipid hydroperoxide (PL-OOH) by the use of GSH (Brigelius-Flohe and Maiorino, 2013; Ursini et al, 1982). In addition, it was revealed that ferroptosis suppressor protein 1 (FSP1)/ubiquinone (CoQ10)/NAD(P)H pathway suppresses ferroptosis by repairing cell membrane directly and works as a standalone parallel system in harmony with the GPX4 system (Doll et al, 2019). Endosomal sorting complexes required for transport-III (ESCRT-III) pathway also work in membrane repair for ferroptosis suppression (Dai et al, 2020b) (Fig. 3).
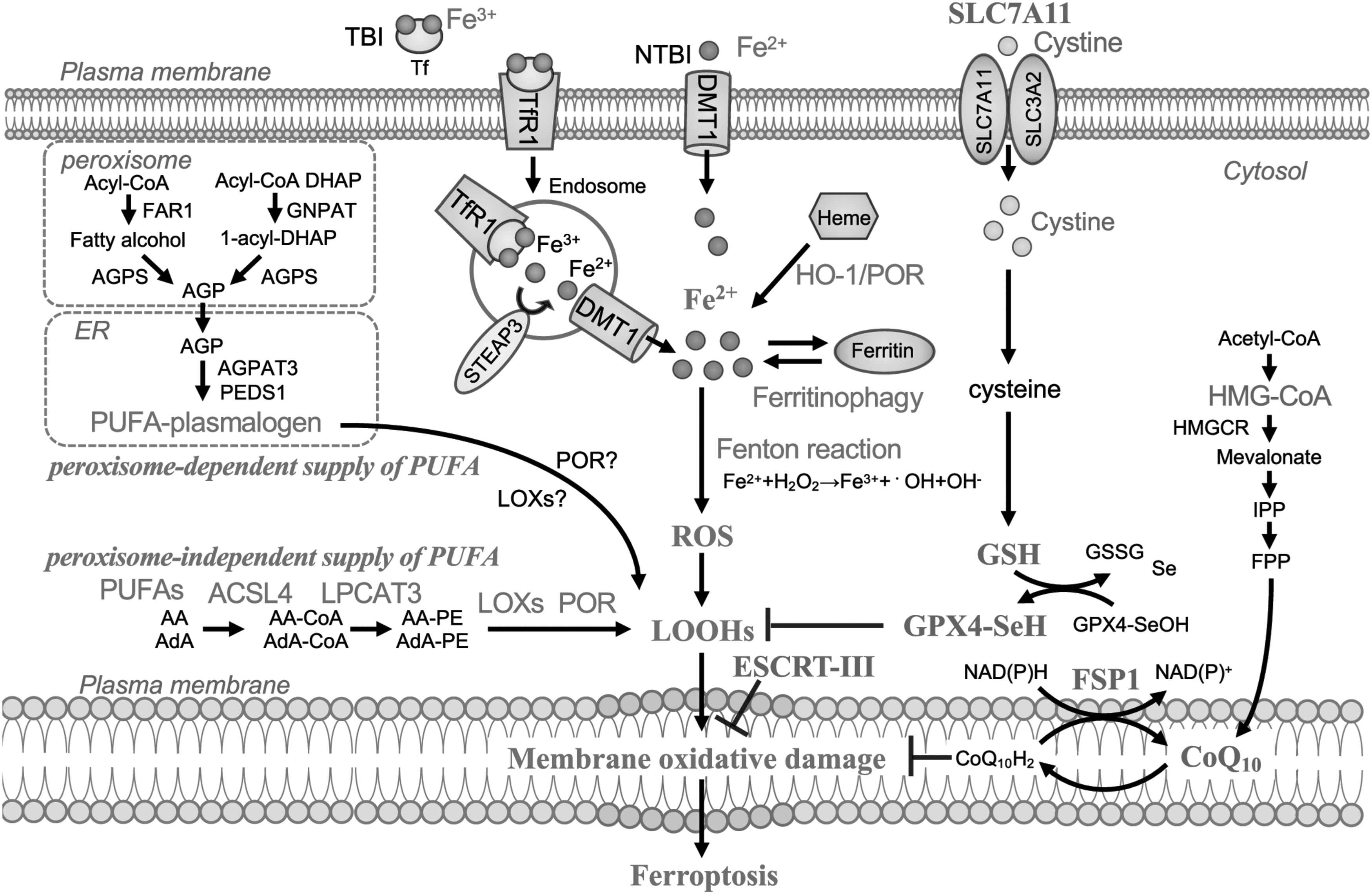
As already described, molecular mechanisms of ferroptosis are starting to be unveiled. However, the factor that makes cancer cells sensitive to ferroptosis is still elusive. One of the factors that determine the ferroptosis susceptibility is the origin and type of cancer. A sensitivity profiling of 177 cancer cell lines has revealed that cell lines of renal cell carcinoma and diffuse large B cell lymphoma are more sensitive to erastin-induced ferroptosis than cell lines from the other types of cancer (Yang et al, 2014).
In addition, intertumoral heterogeneity is another possible factor to determine ferroptosis sensitivity in cancer cells. In three-dimensional culture of lung cancer cells, the inner cells exhibited vulnerability to ferroptosis than surface cells, which is rescued by hyperactivation of NRF2 (Takahashi et al, 2020). Some transcriptional factors, including NRF2 and Hippo signaling pathway, or tumor differentiation, such as EMT, are reported as described hereunder to affect ferroptosis susceptivity in cancer cells.
NRF2 and ferroptosis
NRF2, which is regulated by KEAP1 by ubiquitination-proteasomal degradation, is a key transcriptional factor in response to oxidative stress and ferroptosis resistance (Ma, 2013; Mohammadzadeh et al, 2012). NRF2 decreases oxidative stress by the expression of proteins involved in the antioxidant systems, such as glutamate-cysteine ligase catalytic subunit (GCLC), HO-1, SLC7A11, and GPX4, and the expression of proteins implicated in the decrease of cellular iron, such as FTH1, FTL, and FPT1 (Anandhan et al, 2020). Thus, either KEAP1 knockdown or NRF2 activation makes the cells ferroptosis resistant (Fan et al, 2017; Sun et al, 2016), whereas NRF2 suppression enhances susceptibility to ferroptosis (Shin et al, 2018).
NRF2 is also modulated by ARF, which is an alternative reading-frame product of the CDKN2A locus. ARF suppresses the cAMP-responsive element binding protein (CBP)-dependent acetylation of NRF2, which is critical for sequence-specific DNA binding and expression of downstream target genes, including SLC7A11 (Chen et al, 2017; Sun and Brodsky, 2017). Therefore, ARF deletion leads to ferroptosis resistance.
EMT and ferroptosis
EMT is a process in which carcinoma cells lose their cellular polarity or adhesion, and acquire mesenchymal properties. During this process, tumor cells achieve more invasive nature and chemoresistance (Brabletz et al, 2018). Conversely, cells with EMT are reported to be vulnerable to ferroptosis inducers (FINs) (Bi et al, 2019; Daher et al, 2019; Hangauer et al, 2017; Viswanathan et al, 2017). Multiple factors for EMT are implicated in ferroptosis susceptibility. Zinc finger E-box-binding homeobox 1 (ZEB1) is a transcriptional factor, which is a major inducer of EMT.
ZEB1 modifies lipid metabolism to increase PUFAs, leading to ferroptosis susceptibility (Gubelmann et al, 2014). As another example, overexpression of ACSLs and stearoyl-CoA desaturase-1, which are associated with EMT of colon cancer, is also known to enhance ferroptosis sensitivity (Sanchez-Martinez et al, 2015). In addition, metadherin (MTDH), also known as Lyric, is also one of the factors to induce EMT, which increases the susceptibility to ferroptosis by inhibiting expressions of GPX4 and SLC3A2 (a system Xc − heterodimerization partner) (Bi et al, 2019). In contrast, integrin α6β4, which is predominantly expressed in epithelial cells, induces ferroptosis resistance by repression of ACSL4 via activation of the Src-STAT3 pathway (Brown et al, 2017).
Hippo-Yes-associated protein 1/transcriptional coactivator with PDZ-binding motif signaling pathway and ferroptosis
Hippo signaling pathway is an evolutionally conserved pathway that regulates organ size by modulating cellular proliferation and apoptosis (Lai et al, 2005; Tao et al, 1999; Zhao et al, 2011). The core of Hippo signaling pathway is the kinase cascade of mammalian sterile 20-like kinase 1 (MST1)/MST2, Salvador1 (SAV1), large tumor suppressor kinase 1 (LATS1)/LATS2, and MOB kinase activator 1A (MOB1A)/MOB1B. Hippo pathway negatively regulates Yes-associated protein 1 (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ), which forms a complex with TEA domain family member (TEAD) transcription factors (TEAD1–4) to express target genes, resulting in cell proliferation and migration (Goulev et al, 2008; Huang et al, 2005).
Hippo signaling pathway is activated by cell–cell junctions or cell-density sensing via E-cadherin/α-catenin or mechanical signals via integrins to activate MST1/MST2 or LATS1/LATS2, resulting in phosphorylation of YAP/TAZ, followed by proteolytic degradation of YAP/TAZ to suppress expression of the target genes (Kim et al, 2011; Schlegelmilch et al, 2011). In addition, various scaffolding proteins, including Moesin-Ezrin-Radixin like (Merlin) tumor suppressor encoded by neurofibromin type 2 (NF2), are associated with the regulation by activation of some core complexes or suppression of nuclear localization of YAP/TAZ (Boopathy and Hong, 2019; Furukawa et al, 2017; Kim et al, 2011; Moya and Halder, 2019).
Because YAP/TAZ targets several promoters of ferroptosis-associated genes, including TfR1, ACSL4, and NOX2 via ANGPTL4 expression and NOX4 via EMP1 expression (Wu et al, 2019; Yang and Chi, 2020; Yang et al, 2020; Yang et al, 2019), susceptibility to ferroptosis would inversely depend on the activity of Hippo pathway (Fig. 4). Exceptionally, in hepatocellular carcinoma, YAP/TAZ upregulates SLC7A11, suppressor of ferroptosis, to induce ferroptosis resistance (Gao et al, 2021). Collectively, the interaction between ferroptosis and Hippo signaling pathway is component dependent and further studies are necessary on the relationship between Hippo signaling pathway components and ferroptosis sensitivity.
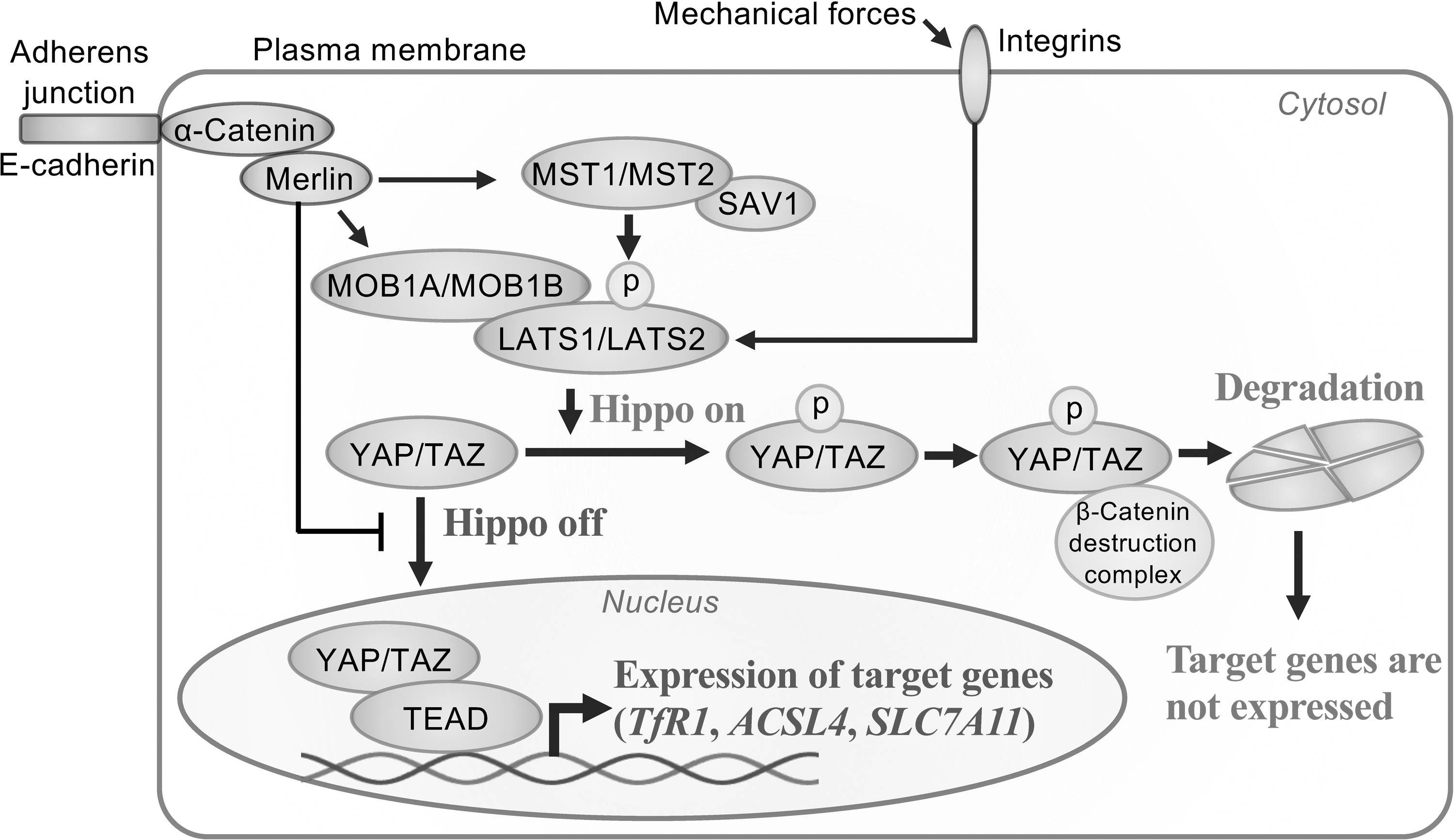
Immunological Interaction and Ferroptosis
In considering ferroptosis as a novel target of cancer therapy, we need to understand how ferroptosis of cancer cells eventually affects the tumor microenvironment. Whereas most studies about ferroptosis are performed in single-layer cultured cells and the role of ferroptosis in vivo remains largely elusive, some experimental progress indicates that ferroptosis can be an immunogenic cell death (ICD) (Efimova et al, 2020).
This is based on the facts that ferroptotic cells show common features of ICD, such as release of DAMPs, including high-mobility group box 1 (HMGB1) and ATP and translocation of calreticulin to the cell surface to activate immune responses, though the mechanisms for the releasing DAMPs are unknown and possibly differ from those of necroptotic or pyroptotic cells (Galluzzi et al, 2014; Kepp et al, 2019; Kolbrink et al, 2020; Medina and Ravichandran, 2016).
HMGB1 is released from ferroptotic cells in an autophagy-dependent manner (Wen et al, 2019). It causes inflammatory responses of bone marrow-derived macrophages by activating advanced glycosylation end products (AGE)/receptor for AGE (RAGE) pathway, which is a proinflammatory signaling cascade (Logan and Storey, 2018; Wen et al, 2019). In addition to HMGB1, ATP is also released from early ferroptotic cancer cells, which induce maturation of bone marrow-derived dendric cells (Efimova et al, 2020). Calreticulin (also known as calregulin, CRP55, or endoplasmic reticulum resident protein 60 [ERp60]) is a protein that binds to misfolded proteins to prevent export from ER to Golgi apparatus.
The translocation of calreticulin to cell membrane is known as “eat me signal” to cause engulfment of the cell by phagocytes or dendric cells. Though these processes have been observed during apoptosis, studies reported similar phenomena in ferroptotic tumor cells, inducing maturation of dendritic cells and infiltration of CTLs (Turubanova et al, 2019; Yu et al, 2020).
Whereas many studies suggest that ferroptosis is an ICD, ferroptotic pancreatic ductal adenocarcinoma cells harboring mutant KRAS G12D release KRASG12D protein via exosomes, which are taken up by macrophages, causing M2-like protumor polarization (Dai et al, 2020a). Prostaglandin E2 (PGE2), a major immunosuppressive factor, negatively affects anticancer immunity by blocking classical type-1 dendritic cell accumulation to tumor, thereby inhibiting CTLs (Wang and DuBois, 2015).
Ferroptotic tumor cells were reported to increase the release of PGE2 (Yang et al, 2014) though the effect is not elucidated yet. In contrast, under immune check point inhibitors such as anti-PD-L1, activated CD8+ T cells can secrete interferon-γ (IFN-γ), which induces downregulation of the expression of system Xc− (SLC7A11 and SLC3A2) and contributes to induce ferroptosis (Wang et al, 2019a) (Fig. 5).
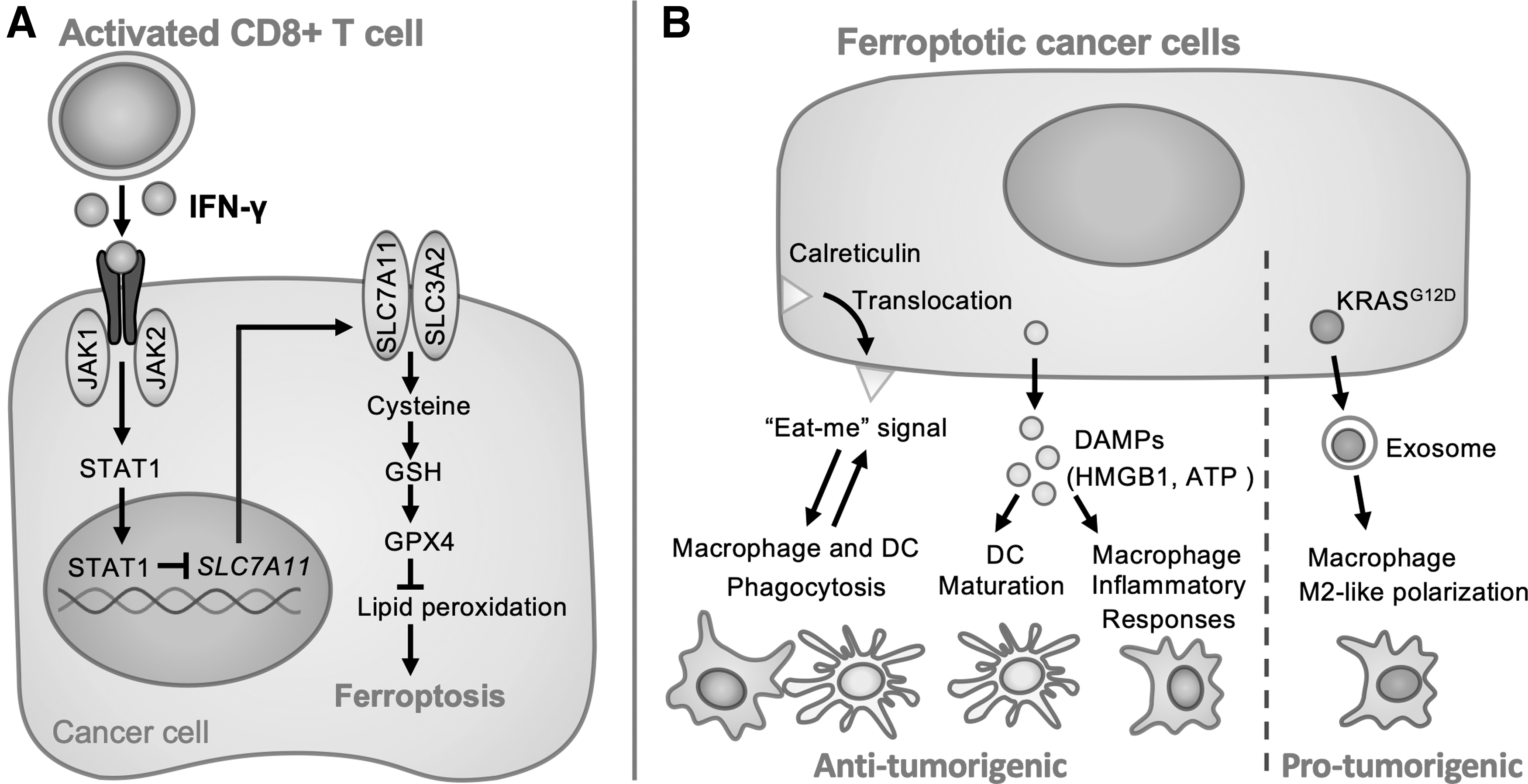
Taking together, ferroptosis can induce proinflammatory response by the release of DAMPs and the translocation of calreticulin. However, in some cases, ferroptosis may cause anti-inflammatory responses with KRASG12D and PGE2. Further studies on the association of immunological interaction with ferroptosis are necessary to develop precise therapy for each patient.
Ferroptosis Inducers
As already described, ferroptosis is a promising therapeutic endpoint for cancers. Various strategies were developed for ferroptosis induction. FINs are mainly classified into four types (Hassannia et al, 2019). Class I FINs act to reduce GSH, Class II FINs directly inactivate GPX4, Class III FINs deplete GPX4 and CoQ10 in association with squalene synthase (SQS)-mevalonate pathway, and Class IV FINs cause lipid peroxidation by an increase in labile iron pool via blocking iron metabolism or actively degrading heme. These drugs are expected to improve cancer treatment not only by increasing therapeutic choice but also by enhancing the effects of established anticancer drugs in combinational use (Liu and Wang, 2019) (Table 1).
Ferroptosis Inducers
CoQ10, coenzyme Q10; FPT1, ferroportin 1; FSP1, ferroptosis suppressor protein 1; GCL, glutamate-cysteine ligase; GSH, glutathione; HMGCR, HMG-CoA reductase; HO-1, heme oxygenase 1; GPX4, glutathione peroxidase 4; LIP, labile iron pool; NrF2, nuclear factor-erythroid 2-related factor 2; ROS, reactive oxygen species; Tf, transferrin; VDAC, voltage-dependent anion channel.
Among the Class I FINs, erastin is the first drug recognized as a FIN (Dixon et al, 2012). Erastin reduces GSH by inhibition of system Xc− and also inhibits mitochondrial voltage-dependent anion channel (VDAC) (Yagoda et al, 2007). Moreover, it was revealed that the RAF/MEK/ERK signaling pathway is important in erastin-induced ferroptosis in RAS-mutated carcinoma (Xie et al, 2016). Though erastin induces tumoricidal effects in preclinical studies in various cancer cells, actual use for living body is limited due to its poor solubility and instability. Therefore, more soluble and stable derivatives, piperazine erastin and imidazole ketone erastin, have been developed (Yang et al, 2014; Zhang et al, 2019).
Furthermore, among drugs approved by Food and Drug Administration (FDA), sulfasalazine, cisplatin, and sorafenib are recognized as Class I FINs. Sulfasalazine is currently used in the treatment of rheumatoid arthritis, ulcerative colitis, or Crohn's disease as an immunosuppressant, which inhibits System Xc− to induce ferroptosis. However, the effect of sulfasalazine is weaker than erastin (Dixon et al, 2012; Zhang et al, 2019). Cisplatin is a major antitumor drug, which is revealed to work partially as FIN by direct reduction of GSH (Guo et al, 2018; Ma et al, 2017). Sorafenib is a multitarget kinase inhibitor for the treatment of hepatocellular carcinoma, renal cell carcinoma, and thyroid cancer.
Sorafenib inhibits System Xc− as well to induce ferroptosis (Hou et al, 2019; Louandre et al, 2015; Louandre et al, 2013). Moreover, Olaparib, which is a poly (ADP-ribose) polymerase (PARP) inhibitor used for cancers exhibiting homologous recombination disorder, is also known to induce ferroptosis by p53-dependent downregulation of SLC7A11 (Hong et al, 2021).
Class II FINs directly inactivate GPX4 to induce ferroptosis. RSL3 is a well-studied direct inhibitor of GPX4 (Yang et al, 2014). Among the FDA-approved drugs, altretamine, which is an anticancer drug for ovarian cancer, is reported to inhibit GPX4 as Class II FINs (Woo et al, 2015).
Class III FINs are indirect inactivators of GPX4. FIN56 promotes autophagy-mediated degradation of GPX4 to induce ferroptosis (Shimada et al, 2016; Sun et al, 2021). Among the FDA-approved drugs, statins are reported as Class III FINs. Statins are inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase (HMGCR), which are commonly used to reduce serum cholesterol levels. Fluvastatin, lovastatin, and simvastatin inhibit HMGCR, leading to a decreased production of CoQ10 and selenoproteins, including GPX4, resulting in ferroptosis (Viswanathan et al, 2017).
Class IV FINs cause lipid peroxidation by iron overload or enhancement of catalytic reaction of iron. Withaferin A activates NRF2 to upregulate HMOX1, leading to degradation of heme and an increase in catalytic Fe(II) (Hassannia et al, 2018). BAY11-7085 also increases intracellular iron via upregulation of HMOX1 (Chang et al, 2018). Siramesine downregulates FPT1, and a combination of lapatinib and siramesine disrupts iron transport to induce ferroptosis by iron overload (Ma et al, 2016).
Nanomaterials-Based Cancer Therapy to Induce Ferroptosis
In addition to traditional drugs, nanoparticle-based drugs have been developed in the past two decades. Materials sized in nanoscales, generally in 1–100 nm at least in one dimension, possess many unique properties such as high surface/volume ratio and characteristic magnetic/electrical properties, which are distinct from micromaterials. For medical use, they are undertaken to design for efficient drug transportation and controlled release (Cheng et al, 2021). With a high surface/volume ratio, nanomaterials are expected to enhance the specificity of targeted therapy by assembling specified biomolecules on surface to specifically recognize target cells, leading to increase efficacy of the drugs with reduced cytotoxicity to untargeted normal cells.
Various forms of nanomaterials, such as nanoparticles, liposomes, solid lipid nanoparticles, nanostructured lipid, nanoemulsions, dendrimers, graphene, and metallic nanoparticles, have been intensively studied for cancer treatments. Such nanomaterials are already demonstrated to induce ferroptosis in cancer cells (Cheng et al, 2021; Zaffaroni and Beretta, 2021). Iron oxide nanoparticles (IONs) (Zanganeh et al, 2016), LOOH-treated IONs (Zhou et al, 2017), amorphous iron nanoparticles (AFeNPs) (Zhang et al, 2016), and FePt nanoparticles (Yue et al, 2017) can induce ferroptosis via iron overload. Polyethylene glycol-coated ultrasmall nanoparticles called Coined Cornel dots (C'dots) were approved by FDA, which induce iron overload by adsorption of extracellular iron and transporting them into the cells (Kim et al, 2016).
Arginine-capped manganese silicate nanobubbles (AMSNs) deplete GSH and inactivate GPX4 (Wang et al, 2018). Low-density lipoprotein nanoparticles, composed of natural ω-3 PUFA and docosahexaenoic acid, induce GSH depletion and accumulation of ROS to induce ferroptosis in liver cells (Ou et al, 2017). Furthermore, some photodynamic drugs have been designed to induce ferroptosis, such as a disulfide-bearing imidazole ligand coordinated with zinc to form an all-active metal organic framework nanocarrier to deplete GSH (Meng et al, 2019).
Nonthermal Plasma Therapy
Recently, nonthermal plasma (NTP; also as low-temperature plasma, LTP) therapy is one of the novel and promising options for cancer treatment at the preclinical stage. Plasma is the fourth state of physical matter, which is ionized gas, consisting basically of free electrons and positive ions (Rehman et al, 2016). Plasma is common across the space. However, states of solid, liquid, or gas are dominant on the Earth for the pressure and the temperature. Even on the Earth, plasma can be produced by supplying energy to gas for ionization, commonly with electrical discharge. Plasma is classified into two groups according to their temperature, “hot,” thermal plasma, and “cold,” NTP/LTP.
Thermal plasma can reach several thousand degrees Celsius, which has been used in manufacturing applications such as in metallurgy, casting, or chemical synthesis. In contrast, NTP (also known as LTP, cold atmospheric plasma, and nonequilibrium atmospheric pressure plasma) is on the temperature close to the ambient temperature, which can be irradiated to the live body. NTP is currently under intensive investigation in medicine, especially in the areas of wound healing, disinfection, endometriosis (Hayashi et al, 2020; Ishida et al, 2016), and cancer treatment (Kajiyama et al, 2017; Laroussi, 2020; Sato et al, 2019).
NTP is formed by ionized gas flow of Ar or He. Various reactive species, including •OH, H2O2, O2 −, and NO, are generated from the surrounding atmosphere. Thus, NTP works to load oxidative stress to the irradiated targets. In addition to direct NTP exposure, NTP can be handled in the forms of plasma-activated medium (PAM) or plasma-activated Ringer's lactate (PAL) as indirect strategies. Both direct and indirect NTP exposures induce ferroptosis specifically in cancer cells where abundant catalytic Fe(II) is responsible to induce Fenton reaction and ferroptosis (Furuta et al, 2018; Jiang et al, 2021b; Nakamura et al, 2021; Okazaki et al, 2014; Shi et al, 2017; Shi et al, 2016; Tanaka and Hori, 2017; Tanaka et al, 2015).
Whereas the responsible chemical reactions have not been completely identified yet, degradation of ferritin and simultaneous increase in catalytic Fe(II) and an increase in NO through inducible NO synthase through NF-κB activation and autophagy induction play important roles (Furuta et al, 2018; Jiang et al, 2021a). We would add here that NTP and PAM/PAL may cause apoptosis as well in certain cancers, such as glioblastoma (Tanaka et al, 2019; Utsumi et al, 2016).
This method is promising for the treatment of cancer with chemoresistance because the therapeutic mechanism is completely different from the other drugs and can target higher amounts of catalytic Fe(II) in cancer cells, which is fundamental for cancer proliferation. The limitation of NTP exposure is the short permeation, which can affect only a few millimeters in depth (Okazaki et al, 2014). Thus, it would be more appropriate for cancers on the epidermal or serosal surface, such as skin cancer, oral cancer, peritoneal disseminations, and the margins of operational resection (Fig. 6).
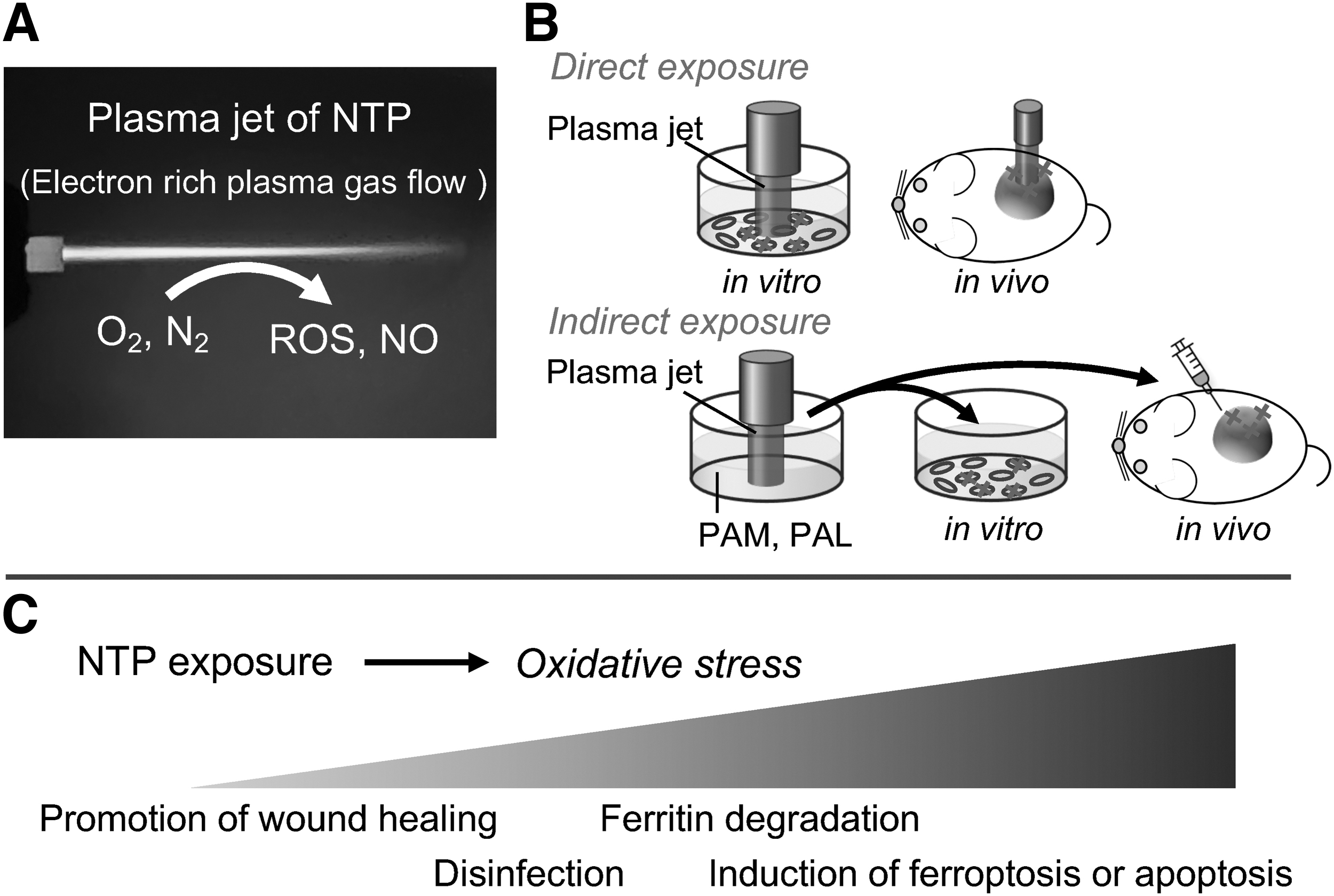
Conclusion
Ferroptosis is a novel form of nonapoptotic regulatory cell death, characterized by catalytic Fe(II)-dependent lipid peroxidation. Ferroptosis is driven by iron overload, incorporation of PUFAs onto membrane PLs via peroxisome-dependent and peroxisome-independent pathway, and is regulated by GPX4, CoQ10, or ESCRT-III.
Ferroptosis induction therapy is anticipated to innovate cancer therapy because ferroptosis induction is ideal to overcome problems to make cancers resistant to chemotherapy, such as EMT and possibly activated Hippo signaling pathway. Therefore, intensive research is in progress worldwide to establish ferroptosis inducing therapeutic methods of distinct modalities. Among the FINs, NTP therapy is one of the promising methods for the unique anticancer effect as a selective handy FIN for catalytic Fe(II)-rich cancer cells.
Footnotes
Authors' Contributions
Y.M. and S.T. conceived, wrote, and organized the article; prepared the figures; and contributed to the discussion.
Author Disclosure Statement
All authors declare no conflict of interest to present.
Funding Information
This study was supported, in part, by JST CREST (Grant Number JPMJCR19H4), JSPS KAKENHI (Grant Numbers JP16H06276 [AdAMS], JP19H05462, and JP20H05502), and Research Grant of the Princess Takamatsu Cancer Research Fund (19-25126).