
Abstract
Significance:
Diabetic peripheral neuropathy (DPN) associated with a diabetic foot ulcer (DFU) is likely to be complicated with critical factors such as biofilm infection and compromised skin barrier function of the diabetic skin. Repaired skin with a history of biofilm infection is known to be compromised in barrier function. Loss of barrier function is also observed in the oxidative stress affected diabetic and aged skin.
Recent Advances:
Loss of barrier function makes the skin prone to biofilm infection and cellulitis, which contributes to chronic inflammation and vasculopathy. Hyperglycemia favors biofilm formation as glucose lowering led to reduction in biofilm development. While vasculopathy limits oxygen supply, the O2 cost of inflammation is high increasing hypoxia severity.
Critical Issues:
The host nervous system can be inhabited by bacteria. Because electrical impulses are a part of microbial physiology, polymicrobial colonization of the host's neural circuit is likely to influence transmission of action potential. The identification of perineural apatite in diabetic patients with peripheral neuropathy suggests bacterial involvement. DPN starts in both feet at the same time.
Future Directions:
Pair-matched studies of DPN in the foot affected with DFU (i.e., DFU-DPN) compared with DPN in the without ulcer, and intact skin barrier function, are likely to provide critical insight that would help inform effective care strategies. This review characterizes DFU-DPN from a translational science point of view presenting a new paradigm that recognizes the current literature in the context of factors that are unique to DFU-DPN.
Introduction
A network of peripheral nerves connects the organs such as the skin, muscles, and internal organs to the spinal cord and the brain. Any damage to these nerves outside of the brain and spinal cord may cause weakness, numbness, and pain often felt in the hands and feet. When such damage occurs secondary to the incidence of diabetes, it is referred to as diabetic peripheral neuropathy (DPN). DPN is highly prevalent in both type 1 and type 2 diabetes mellitus (T1DM and T2DM). DPN is mostly discussed in the context of a neurological disorder. This article addresses DPN in the context of a diabetic ulcer, which is known to be complicated by infection (Bellomo et al, 2022; Sen, 2021; Sen et al, 2021).
Although the pathogenesis of DPN is typically linked to biochemical and molecular mechanisms induced by diabetes, infection of diabetic ulcers could also contribute to DPN. DPN encompasses clinical and subclinical conditions noted in a range of anatomical locations. Diabetes mellitus can cause damage, focal or diffuse, to peripheral somatic as well as autonomic fibers (Edwards et al, 2008). It is estimated that half of all patients with diabetes mellitus develop symptomatic peripheral neuropathy within 25 years of disease onset (Hicks and Selvin, 2019; Pirart, 1977). Patient age, disease duration, and quality of diabetes control are strong predictors, whereas etiology has no influence. C-fiber and autonomic nerve fiber dysfunction is common.
Distal symmetric sensorimotor neuropathy is reported in an estimated 30% of hospitalized patients with diabetes as either primary or secondary diagnosis, compared with 20% in the overall diabetic population (Volmer-Thole and Lobmann, 2016). Between 13% and 26% of patients with diabetes report painful chronic polyneuropathy (Volmer-Thole and Lobmann, 2016). The lower extremities are commonly affected by complications secondary to both type 1 as well type 2 diabetes. Foot ulceration and re-ulceration are the most frequently recognized complications (Armstrong et al, 2017). This review addresses DPN in the context of the diabetic foot (Carmichael et al, 2021; Chammas et al, 2016). Focus is directed toward molecular and cellular mechanisms underlying such DPN in the context of diabetic foot ulcer (DFU).
DFU and Complications Relevant to Neuropathy
At present, it is estimated that >450 million people are affected by diabetes worldwide. The current global prevalence is 9.3% (Saeedi et al, 2019). Diabetes causes peripheral artery disease (PAD) as well as DPN. DPN, in both T1DM and T2DM, accounts for more than 60% incidence of foot ulcers (Syafril, 2018). The 3-year mortality for people with diabetes increases from 13% to 28% with an ulcer (Murphy-Lavoie et al, 2021). Diabetic PAD and DPN, often studied separately, are mechanistically interrelated and responsible for downstream pathologies, such as the occurrence of DFUs. The six-point SINBAD (Site, Ischemia, Neuropathy, Bacterial infection, Area, and Depth) scale can be used to study DFU (Monteiro-Soares et al, 2020). Although effective glycemic control can reduce the incidence of diabetic neuropathy in type 1 diabetes, most patients with type 2 diabetes will develop this complication.
Greater than 60% of an estimated million nontraumatic limb amputations per year are performed in people suffering from diabetes. The presence of DFU doubles the risk of 10-year mortality in diabetics (Chammas et al, 2016; Rubio et al, 2020). For people with DFU, more than one third are expected to undergo amputation (Kim et al, 2018; Thewjitcharoen et al, 2020). Majority of the DFUs suffer from biofilm infection (Pouget et al, 2020). The impact of infection as a complicating factor is profound such that 85% of all wounds that led to amputation became chronic because of infection (Raghav et al, 2018).
Thus, DPN relevant to DFU is a topic that warrants dedicated attention. Unlike most other cases of diabetic neuropathies, the following factors act in concert to cause the pathogenesis of DFU-associated DPN (DFU-DPN): (1) deviations in glycemic status and its downstream complications, (2) vasculopathy and ischemia, (3) chronic inflammation, (4) diabetic skin, (5) biofilm infection, (6) metabolic syndrome (MetS), (7) hypertension, (8) genetic susceptibility, (9) psychosocial factors, (10) aging, (11) sex, and (12) lifestyle.
Glycemic Status Related Factors in Peripheral Neuropathy
Impaired glucose tolerance is highly prevalent (7.5%) worldwide and rapidly rising (Saeedi et al, 2019). The remarkable effects of glucose-lowering drugs to reduce cardiovascular risk in person with diabetes underscore the significance of glucose control and glycemic targets in patient care (Kalyani, 2021). In the context of the effect of glycemic status on the overall nervous system, the direct threat posed by hyperglycemia itself on an acute basis appears to be little to none. A far more serious threat is posed by hypoglycemia. In diabetic insulinoma patients, treatment with hypoglycemic agents may cause hypoglycemia (Volmer-Thole and Lobmann, 2016). In the nervous system, glucose deprivation causes neurotoxicity.
If glucose deprivation is further complicated by hypoxic insult as expected under ischemic conditions, the outcomes are far worse (Babu et al, 2022; Balch et al, 2020; Guo et al, 2018; Reichert et al, 2001). Clinically, hypoglycemia may cause neurocognitive disorders, including intoxicated behavior and mentation, seizures, and loss of consciousness. Under some conditions, hypoglycemia may induce hemiparesis. The brain itself is much more sensitive to hypoglycemia and is well equipped to tolerate hyperglycemia because of the presence of the blood–brain barrier (BBB), which reduces the brain exposure to two thirds of the peripheral blood level. Yet elevated HbA1c, a product of hyperglycemia, is associated with poor cognitive function (Anstey et al, 2015). This apparent disconnect calls for a critical look at the mechanistic underpinnings of diabetes-associated neuropathy.
Tight control of glucose levels using continuous subcutaneous insulin infusion (CSII) proved to be beneficial in providing symptomatic pain relief in diabetic patients and was accompanied by a significant improvement in pain scores. In contrast, sensory studies in the median nerve showed no significant changes during the study (Boulton et al, 1982). Short-term CSII had a beneficial effect on diabetic neuropathies by improving nerve conduction velocities (Gupta et al, 2020; Kronert et al, 1987). In young insulin-dependent diabetic subjects, CSII had beneficial effects on diabetic neuropathy as manifested by rescuing vibratory perception threshold of the great toe and the medial malleolus (Jakobsen et al, 1988).
In another study on young diabetics, the effects of CSII and multiple daily injections of insulin were studied on peripheral nerve function as measured by thermal and vibration threshold testing. In adolescents, the use of CSII lowered the rates of peripheral nerve abnormality (Zabeen et al, 2016). As addressed in a separate section below, life span is an important variable that predicts diabetic neuropathy. Although several studies have reported the beneficial effects of CSII in the young diabetics, that effect does not seem to hold in the elderly. In older people with type 1 diabetes, CSII lowered HbA1c, improved hypoglycemia and ketoacidosis, and reduced time spent in hospital but worsened the rates of diabetic neuropathy (Grammes et al, 2020). Such observations call for an understanding of the disease process of DPN at the molecular and cellular levels.
Insulin signaling is impaired in diabetes. Peripheral neuropathy is associated with insulin resistance independent of MetS (Han et al, 2015). Insulin resistance is closely associated with neurodegenerative disease comorbidity. T2DM patients, suffering from peripheral insulin resistance, are known to have higher risk for the development of Alzheimer's disease (AD), Parkinson's disease, or glaucoma (Qian and Zhang, 1993). Deficiencies in insulin signaling in animal models of type 1 and 2 diabetes cause AD pathology (Kimura, 2016). Insulin resistance induced by high-fat diet is associated with neurotoxic oxidative stress and dopamine depletion (Morris et al, 2010). Insulin resistance has been directly implicated in glaucomatous neurodegeneration.
Direct blockade of insulin receptor function by S961 resulted in loss of retinal ganglion cells (Faiq et al, 2019). These examples of neurodegenerative aspects of impairments in insulin signaling are directly relevant to DPN. In mice, a TACE inhibitor (TNFα converting enzyme, TACE) drug that prevented insulin resistance in T2DM also rescued DPN independent of any glucose-lowering effect (Maekawa et al, 2019). L-carnitine successfully managed peripheral neuropathy in diabetic mice via the induction of insulin-like growth factor 1 (IGF-1) (Wang et al, 2019). Direct intervention using IGF-1 was also beneficial in managing experimental peripheral neuropathy (Wang et al, 2018). In streptozotocin-induced diabetic rats, application of a pectin–insulin patch prevented the onset of symptoms indicative of peripheral neuropathy (Sibiya and Mabandla, 2018).
Both demyelination and axonal degeneration are implicated in diabetic neuropathy (Abuelwafaa et al, 2019; Malik, 2014; Roman-Pintos et al, 2016; Sullivan and Feldman, 2005). Demyelination and Schwann cell (SC) dysfunction are characteristic features of DPN. The myelin sheath around axons degenerates often because of depleted myelin sheath lipids (Mitro et al, 2014; Palavicini et al, 2020; Yuan et al, 2021). Replenishment of these lipids is achieved by hydrolysis of triglyceride-rich lipoproteins by lipoprotein lipase (LPL). LPL is rate limiting in the provision of triglyceride-rich lipoprotein-derived lipids into tissues. Deficiencies in neuronal LPL compromise neuronal lipid-sensing and systemic energy balance (Bruce et al, 2020). LPL also helps in uptake of lipids by SC, an important target to manage neuropathy (Eid et al, 2020).
SC-derived reactive oxygen species (ROS) cause cellular damage (Chang et al, 2021) and have been directly implicated in DPN (Eid et al, 2020). Insulin acts on its receptor in SCs to protect against DPN. Myelin proteins, known to be downregulated in diabetes, are rescued by insulin signaling (Rachana et al, 2016). The insulin signaling pathway is primarily conducted by tyrosine phosphorylation of receptor protein kinases, which in turn activates adaptor proteins. In SC as well as in the sciatic nerve, insulin signaling is impaired (Manu et al, 2017). Restoration of insulin supply and signaling in SC and nerves are important for the rescue of DPN. Impairment of insulin function may cause neuromuscular degeneration via defects in mitochondrial function (Lopez Del Amo et al, 2019). The next segment addresses the significance of mitochondrial dysfunction in DPN.
Molecular Mechanisms Associated with DFU-DPN
Mitochondrial dysfunction
Diabetic patients (T2DM) with lower extremity complications suffer from impaired mitochondrial oxidative phosphorylation (Tecilazich et al, 2013). Mitochondrial dysfunction causes distal axonopathy in diabetic neuropathy. Defects in the signaling of the adenosine monophosphate-activated protein kinase/PGC-1α pathway are responsible for such outcome (Roy Chowdhury et al, 2012). In DPN patients, Charcot foot is common. In this condition, the bones of the foot are weakened, and there is abnormal movement between bones. When the foot is weight bearing, small muscles of the foot contract to prevent this excessive bone movement. Muscle ATP is thus depleted under conditions where glucose uptake is blunted because of insulin resistance.
Magnetic resonance imaging of Charcot neuroarthropathy has revealed mitochondrial dysfunction based on the analyses of tissue ATP levels (Lymbouris et al, 2020). Chronic hyperglycemia causes functional dysregulation of mitochondrial bioenergetics via aberrant expression of advanced glycation end product/receptor for advanced glycation end products (AGE/RAGE) and hexosamine biosynthetic signaling pathways, which in turn causes oxidative stress and chronic inflammation (Stefano et al, 2016). Analysis of the molecular underpinnings of hyperglycemia-induced metabolic dysfunction recognizes these major mechanisms: (1) increased aldose reductase (AR) activity; (2) AGEs; () hexosamine signaling; (4) activation of protein kinase C (PKC); and (5) oxidative damage (Rolo and Palmeira, 2006).
Increased AR activity
AR or aldehyde reductase is a cytosolic NADPH-dependent oxidoreductase that catalyzes the reduction of glucose to sorbitol, the first step in the polyol pathway of glucose metabolism. Also known as glucitol, sorbitol is a sugar alcohol with poor cell permeability properties and the human body metabolizes it slowly. Sorbitol is metabolized to fructose by sorbitol-6-phosphate 2-dehydrogenase. Under conditions of hyperglycemia, sorbitol and fructose accumulate intracellularly causing osmotic tissue swelling and dysfunction. While AR inhibitors (ARI) have produced discouraging results in double-blind placebo-controlled ARI diabetic neuropathy trials, the scientific evidence supporting that AR is directly implicated in DPN is compelling and mounting (Oates, 2008; Rendell, 2021). Structure–function studies have elucidated epalrestat, the only antidiabetic ARI approved for use in humans, as a first-in-class phosphomannomutase 2 activator (Iyer et al, 2019). Slow progression of DPN may necessitate longer duration trials to prove efficacy.
Advanced glycation end products
In the diabetic skin, hyperglycemia induces nonenzymatic glycation of proteins, lipids, as well as nucleic acids. A heterogeneous set of molecules collectively referred to as AGEs are produced as a result. During such process, methylglyoxal (MGO) is produced as an intermediary. MGO reacts with free amino groups of lysine and arginine and with thiol groups of cysteine forming AGEs (Fig. 1). Hyperglycemia-induced nonenzymatic glycation involves nucleophilic addition reaction between a free amino group of a protein and a carbonyl group from a reducing sugar to form a freely reversible Schiff bases [R1R2C = NR′ (R′ ≠ H)]. Schiff base undergoes molecular rearrangements to generate Amadori products, which accumulate and produce AGE. MGO is a major precursor of nonenzymatic glycation of proteins and DNA, which goes on to form AGE.
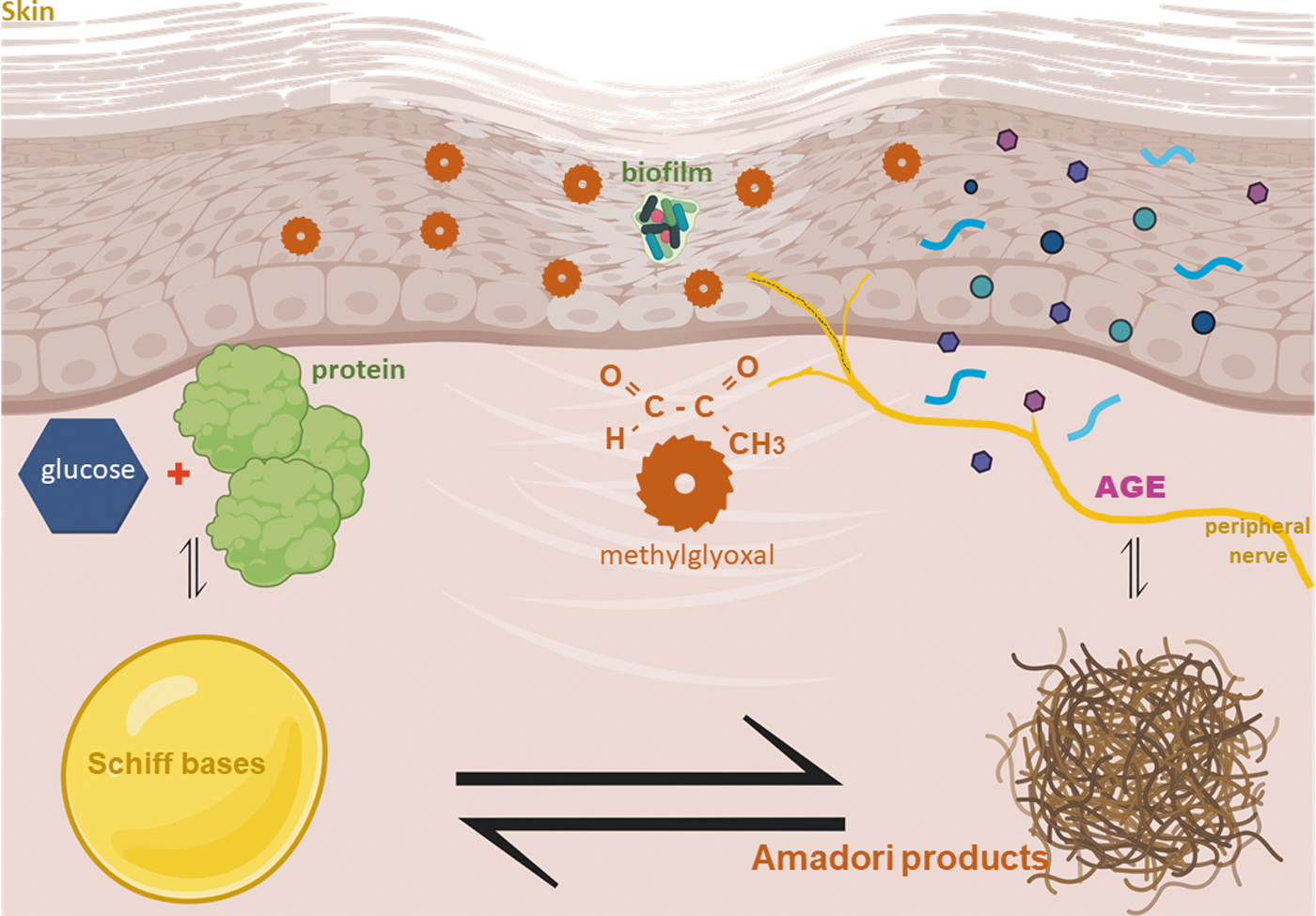
MGO cross-links with collagen contributing to ECMpathy. In the diabetic skin, MGO may directly bind to nerve endings causing DPN-related soreness (Bierhaus et al, 2012). MGO is also produced in the skin by oxidation of glycerin, abundantly present in moisturizers and skin lotions. Chronic presence of MGO in the skin causes oxidation of skin collagen protein as detected by carbonylation (Sugiura et al, 2021). Such skin protein carbonylation is also a hallmark of skin aging. Skin intrinsic fluorescent (also referred to as skin autofluorescence, SAF, in some studies) scores indirectly measure AGEs. Rising SAF score over 5 years was related to increased glycemic levels and decreased kidney function (Tomaszewski et al, 2021). The Helsinki Birth Cohort Study reports that SAF is associated with melancholic depressive symptoms (Eriksson et al, 2021).
SAF is an independent marker not only for DPN but also for diabetic retinopathy, diabetic kidney diseases, and cardiovascular disease. It is also a predictor of the complexity of T2DM complications (Wang et al, 2021). For T1DM patients, the SAF predicts ulcers and amputations (Rojubally et al, 2021). AGEs and the RAGE are elevated in the diabetic peripheral nerves (Zhang et al, 2018). One example of RAGE is high mobility group protein B1 (HMGB1), also known as AGER. HMGB1 levels are increased in diabetes and contribute to pain by modulating peripheral inflammatory responses (Bestall et al, 2018). Diaphanous-related formin 1 (DIAPH1), a cytoskeletal organizing molecule, interacts with the cytosolic domain of RAGE resulting in axonopathy/neuropathy (Zglejc-Waszak et al, 2021). RAGE deletion studies point toward its role in the development of DPN (de la Hoz et al, 2017; Juranek et al, 2013). Accumulation of AGEs in the peripheral nerves is an additional risk factor for the development of diabetic neuropathy (Papachristou et al, 2021).
Hexosamine signaling
In the domain of post-translational regulation of protein function, reversible addition of O-linked N-acetylglucosamine (O-GlcNAc) at serine and threonine residues has gained much attention. Several nuclear and cytoplasmic protein activities are subject to such regulation. The addition of O-GlcNAc to proteins is controlled by specific enzymes that are strategically localized. Furthermore, such addition is subject to sophisticated metabolic control. Transfer of O-GlcNAc to or from a protein may be viewed as the terminal step in a hexosamine signaling pathway (HSP). HSP has profound impacts on histone remodeling, transcription, proliferation, apoptosis, and proteasomal degradation.
Hyperglycemia is known to induce the hexosamine pathway and thereby impair wound healing (Kunkemoeller et al, 2019). As an alternative to glycolysis, upregulation of HSP is intended to metabolize excess glucose. Under conditions of hyperglycemia, fructose-6-phosphate, a glycolytic product, gets metabolized by HSP. During this metabolism, it is acted upon by glutamine:fructose-6 phosphate amidotransferase to produce UDP N-acetyl glucosamine (UDP-GlcNAc). This pathway leads to oxidative stress and related diabetic microvascular complications (Du et al, 2000; Nakamura et al, 2001; Pauer et al, 1976; Yang et al, 2001). Such complications contribute to DPN (Jalgaonkar et al, 2022; Yorek, 2022).
PKC activation
The PKC family of enzymes represents a major hub in cell signaling. PKC family members catalyze receptor-mediated hydrolysis of membrane phospholipids, thus unleashing a wide range of cellular signals (Callender and Newton, 2017; Eichberg, 2002). AGE induces PKC (Ido et al, 2001). In the diabetic skin, PKC is hyperactive and contributes to microangiopathy (Gutterman, 2002; Ngo et al, 2005). In this largest organ of the body, DPN starts with the retraction and loss of sensory terminals while the neuronal cell bodies remain preserved. Induced PKC activity is implicated in this process (Zochodne, 2014). PKC alpha, beta, and gamma subspecies are localized in the sensory axon terminals. There is ample evidence supporting that PKC regulates ion channel function.
Thus, it is suspected that PKC may control mechanoelectric transduction in sensory axon terminals (Masutani et al, 1994). Persistent activation of PKCδ has been reported in fibroblasts from diabetic patients. Insulin signaling and wound healing are thus impaired. PKCδ inhibition has produced favorable outcomes (Khamaisi et al, 2016). Ruboxistaurin, inhibitor of PKC βII activation, reverses the dysfunction of circulating endothelial progenitor cells and prevents exaggerated inflammatory response commonly associated with biofilm infection. Wound healing outcomes are thus improved (Das et al, 2018).
Oxidative damage
In patients, DPN is associated with oxidative stress and related thiol-disulfide imbalance (Abd El-Khalik et al, 2020; Buyukaydin et al, 2020; Ojalvo et al, 2017). Even in prediabetes, small C-fibers innervating the sweat glands could be affected by oxidative stress (Parfentyeva et al, 2014). In blood plasma of T2DM patients, high levels of lipid peroxidation and compromised antioxidant defenses are evident (Vairamon et al, 2009). The aged human skin is particularly affected by oxidative stress (Rinnerthaler et al, 2015). Oxidative stress is directly implicated in the development of DPN (Mallet et al, 2020). Here, we discuss some of the primary mechanisms contributing to oxidative stress in the diabetic skin. A major source of persistent ROS in the skin is chronic inflammation (Hu et al, 2015).
Often such oxidative stress disrupts skin barrier function making the organ more prone to infection-related complications (Ilves et al, 2021). Infection, in turn, increases the severity of oxidative stress, thus triggering a vicious cycle that impairs wound closure (Buvelot et al, 2017; Li et al, 2021). While moderate levels of ROS are necessary for wound repair, excessive levels cause oxidative stress and wound chronicity (Roy et al, 2006; Sen and Roy, 2008). NADPH oxidases represent a major source of superoxides and hydrogen peroxide in the skin under conditions of inflammation (Meyskens and Liu-Smith, 2017; Sen and Roy, 2008).
Thyroid stimulating hormone (TSH) is recognized as an independent risk factor for DPN progression. It aggravates SC apoptosis and demyelination via oxidative stress (Fan et al, 2021). Targeted inhibition of the NADPH oxidase-4 and liver X receptor pathway preserves SC integrity in diabetic mice (Eid et al, 2020). A plant-based drug used for the treatment of diabetic neuropathy, DA-9801, rescues against diabetic endothelial dysfunction by attenuating inflammation and related NADPH oxidase function (Hong et al, 2021). NADPH oxidases are also implicated in neutrophil extracellular traps (NETs)-related neuropathic complications as discussed in the Biofilm Infection section (Carmona-Rivera and Kaplan, 2016).
Nonenzymatic auto-oxidation of glucose is likely to be a significant contributor to skin oxidative stress under conditions of hyperglycemia. Under these conditions, excessive glucose can undergo auto-oxidation to enediol radical, which in turn can convert to the single electron reduction product of molecular oxygen, superoxide anion radical. Hydroxyl and hydroxyalkyl free radicals are involved in the auto-oxidation of monosaccharides (Thornalley and Stern, 1984). Superoxide dismutate forms hydrogen peroxide. Auto-oxidation of glucose may produce dicarbonyl ketoaldehyde that can produce ketamine upon reaction with the amino groups of protein. Protein oxidation markers are sharply elevated in the diabetic skin (Lee et al, 2015). Oxidative stress caused by auto-oxidation of glucose is directly implicated in diabetic neuropathy (Pop-Busui et al, 2006).
ROS are produced in the diabetic skin following modification of skin collagen by MGO, a highly reactive dicarbonyl. This causes endoplasmic reticulum (ER) stress of the diabetic skin (Nowotny et al, 2018). The ER is major site hosting the production and folding of approximately one third of cellular proteins. During ER stress, the capacity of ER to fold protein is limited and capped resulting in unfolded protein response (UPR). Unfolded or misfolded proteins thus accumulate in the ER lumen. ER stress is evident in the skin and can be pathogenic in this organ (Hassan et al, 2015; Jadeja et al, 2020; Park et al, 2019).
Both ER stress and UPR are known to be directly implicated in peripheral neuropathy and microvascular complications (O'Brien et al, 2014; Sankrityayan et al, 2019; Valenzuela et al, 2016). In DPN, pharmacological inhibitors of ER stress have produced favorable outcomes in experimental studies (O'Brien et al, 2014). Small-molecule ER stress inhibitors can be broadly categorized as natural chaperones, synthetic chaperones, or pharmacological chaperones that target UPR. They have been prescribed as therapeutic agents for various diseases (Prasad et al, 2022). Both hyperglycemia and hyperlipidemia contribute to diabetic ER stress relevant to DPN (Yang et al, 2022). Delineation of specific ER-stress dependent molecular pathways that contribute to the pathogenesis of DPN, in the context of a wide array of other causative factors, would provide critical insight.
Vasculopathy and Ischemia
Diabetic neuropathy often coexists with vasculopathy. Both are strong independent risk factors for the development of DFU (McNeely et al, 1995). Increased cross-linkage of type IV collagen may contribute to rigidity of the nerve as well as compromise blood vessel compliance and perfusion. During Wallerian degeneration, the basal laminal ensheathment around myelinated nerve fibers of SC is retained after the removal of myelin and axonal debris. A corrugated tube thus formed around SCs contributes to nerve rigidity (King et al, 1989). Interestingly, the strongest risk factor for the development of DFU was recognized as impaired cutaneous oxygenation, above neuropathy and vasculopathy (McNeely et al, 1995). Thus, in this segment, the mechanistic connection between these three major risk factors will be addressed in light of their mechanistic underpinnings.
Nerve ischemia, secondary to vasculopathy, is recognized as a common mechanism for the different patterns of diabetic neuropathies. Extensive work on the diabetic eye demonstrates that local small vessel disease causes neuropathy via ischemia (Biousse and Newman, 2014). Direct study of Diabetic Neuropathy in patients revealed that parts of the microvascular bed are occluded by thrombus. Electrophysiological studies showed a pattern of denervation that was distally predominant in some patients, suggesting that the neuropathy, at least in part, relates to the multiple small infarcts (Timperley et al, 1985). The metabolic consequences of ischemia directly contribute to the development of DPN (Nukada, 2014).
Limited peripheral nerve perfusion may directly contribute to the development of DPN by several mechanisms (Baumer et al, 2014). Mechanisms implicated in pathologies caused by peripheral nerve ischemia include prolonged inflammation, dysregulated Ca2+ transport, oxidative stress, and mitochondrial dysfunction (Bagdatoglu et al, 2006; Gholami et al, 2017; Rinker et al, 2013; Rinker et al, 2011). Peripheral nerve injury precipitates vascular dysfunction and endoneurial hypoxia worsening the disease condition (Lim et al, 2015).
Diabetes induces both macrovascular and microvascular endothelial dysfunction (Shi and Vanhoutte, 2017). Common occurrence of extracutaneous microvascular complications and DFU in patients with T2DM warrants a multidisciplinary approach of medical professionals that includes prevention of risk factors and good regulation of glycemia (Balaban et al, 2020). Technologies such as continuous glucose monitoring and flash glucose monitoring are providing insight into the root causes of glycemic variability. In public health studies, visit-to-visit glycemic variability is known to be associated with increased risk of vascular events (Hirakawa et al, 2014).
Such vascular complications cause diabetic neuropathy (Skrha et al, 2016). Higher HbA1c variability is associated with increased risks of all-cause mortality, cardiovascular events, and microvascular complications of diabetes independent of high HbA1c (Li et al, 2020). Microvascular endothelial dysfunction is typically characterized by blunted release of nitric oxide (NO), oxidative stress, elevated inflammatory mediators, abnormal angiogenesis, and faulty endothelial repair. PAD contributes to critical limb ischemia. Noninvasive measures, such as the ankle–brachial index, show that asymptomatic PAD is very common (Criqui and Aboyans, 2015). Independent of traditional risk factors, the presence of microvascular disease increases the risk of amputation alone and synergistically increases risk in patients with PAD (Beckman et al, 2019).
Diabetes mellitus is not only a strong risk factor for PAD, but it accelerates PAD making these patients more susceptible to ischemic events and impaired functional status compared with patients without diabetes (Thiruvoipati et al, 2015). Lesions of blood vessels supplying the nerve can cause peripheral neuropathy (Said, 1996). The pathogenesis of neuropathy and vasculopathy can be intertwined (Kolar et al, 2014). Subclinical diabetic small fiber peripheral neuropathy is significantly associated with PAD in patients with T2DM (Sheen et al, 2018). Classical vasculitic neuropathies, nerve large arteriole vasculitides and nerve microvasculitis, have been known for a long time (Naddaf and Dyck, 2015). Ischemic peripheral neuropathy is directly linked to livedoid vasculopathy (Soulages et al, 2022).
In the skin, biomarkers of nerve and vascular origin differentiate painful from painless DPN (Shillo et al, 2021). Mechanistically, high metalloproteinases account for both neuropathy and vasculopathy (Opdenakker and Abu El-Asrar, 2019). Diabetic patients with PAD have poorer lower extremity function than those with PAD alone. Diabetes-associated neuropathy is a factor that contributes to such compromised functionality (Dolan et al, 2002). Sudomotor dysfunction, common in T2DM patients, includes small fiber neuropathy, cardiovascular autonomic neuropathy, and peripheral sympathetic autonomic neuropathy. PAD is associated with sudomotor function (Chahal et al, 2017). PAD and neuropathy are causatively linked such that they influence each other and aggravate the condition of diabetic foot.
Chronic Inflammation
Nerves and ganglia outside the brain and the spinal cord make up the peripheral nervous system. Axons of this system run together in bundles called fibers. Multiple fibers form the nerve. The nerves, including connective tissue and blood vessels, reach out to the organs such as the skin, glands, and muscles. The nerves of the peripheral nervous system do not effectively regenerate in response to severe neural injury. Peripheral nerve tissues are primarily made up of SCs, nerve axons, and the blood–nerve barrier (BNB). The BNB, a physiological boundary between the peripheral nerve axons and the blood stream, prevents influx of harmful substances circulating in the blood into peripheral nerves.
It is mainly localized in the microvessels of peripheral nerves within the peripheral neural parenchyma (endoneurium) and consists of vascular endothelial cells bound by tight junction proteins, pericytes attached to the outer side of the vascular endothelial cells, and basal laminae covering these two types of cells (Takeshita et al, 2020). The BNB forms a tight and highly regulated interface in the human body similar to the BBB. Structurally, BNB contains a unique dynamic but selective blood–tissue interphase within the peripheral nerve endoneurium. This is made up of endoneurial microvessels within the nerve fascicle and the investing perineurium.
To reach the endoneurial extracellular space, blood-borne molecules need to cross the endoneurial vascular endothelium or the perineurium that surrounds the nerve fascicle (Richner et al, 2018). Inflammation and nerve edema disrupt the BNB, thus facilitating DPN (Takeshita et al, 2020). Chronic inflammation is a hallmark of DFU (Singh et al, 2016). DPN is associated with increased abundance of inflammatory cells within the peripheral nerve (Hagen and Ousman, 2021). Functional evaluation of BNB at the site of DFU and its significance in DPN as well as wound chronicity is thus warranted.
Diabetic ulcers are often presented with associated cutaneous vasculitis (Shavit et al, 2018). Vasculitis is an inflammatory process affecting the vessel wall and leading to its compromise or destruction and subsequent hemorrhagic and ischemic events. In peripheral nerves, vasculitis compromises nerve vessels causing vascular ischemia, axonal degeneration, and neuronal demyelination (Maiuolo et al, 2021). Diabetic inflammatory neuropathies are often associated with vasculitis. Vasculitis, typically involving inflamed blood vessels supplying the peripheral nerve, is causatively linked to neuropathy of the affected nerve (Said, 1996). Vasculitic neuropathies can be characterized by acute-to-subacute onset of painful sensory and motor deficits that result from inflammatory destruction of nerve blood vessels and subsequent ischemic injury (Gwathmey et al, 2014).
The Diabetic Skin
Chronic diabetes markedly compromises the structure and function of the skin. Breach of skin barrier function may be viewed as an open wound. Diabetes compromises barrier function of the intact skin, opening an invisible wound and thus making the skin vulnerable to infection. Functional wound closure relies on restoration of skin barrier function at the site of wound repair (Barki et al, 2019; Ghatak et al, 2015; Roy et al, 2014; Sen and Roy, 2021 ). In the diabetic skin, restoration of epidermal barrier function after wound healing is delayed increasing the risk of infection (Gonzalez et al, 2021; Koivukangas et al, 1999; Percival et al, 2012).
The diabetic skin shows elevated markers of oxidative stress because of weakened antioxidant defense system (Dimaki et al, 2019; Kim et al, 2021; Wu et al, 2016; Yang et al, 2020). Oxidative stress of the skin is known to compromise barrier function (Hwang et al, 2018; Katsuyama et al, 2017; Rojo de la Vega et al, 2017; Schafer et al, 2012; Yokoyama et al, 2019). Compromised tight junctions of the skin epithelium, as evidenced by depleted tight junction scaffold proteins zona occludens 1 and 2, account for such functional defect of the skin (Roy et al, 2014).
NO physiologically generated by skin cells plays a critical role on ensuring cutaneous vascular health. Diabetic skin suffers from limited NO production and enhanced NO consumption resulting in compromised skin perfusion (Yang et al, 2016). In the ischemic skin, upregulation of matrix metalloproteinases and downregulation of tissue inhibitor of metalloproteinase 1 in the diabetic dermal fibroblasts threaten homeostasis of skin extracellular matrix (ECM) (Wu et al, 2016). Dysregulated ECM accounts for weaknesses in skin integrity, and biomechanical properties making the organ more vulnerable to breach of barrier function and therefore wound occurrence (Bermudez et al, 2011). The diabetic human skin is vulnerable to oxidative damage and loss of barrier function (Cho et al, 2014). Such defect in skin function breaches a major host defense mechanism as the skin loses its ability to function as barrier against microbial entry into the body. Furthermore, lack of nutritional immunity in diabetic skin infections promotes Staphylococcus aureus virulence (Thurlow et al, 2020).
The diabetic skin is characterized by lower cutaneous nerve fiber density, which subsequently leads to impaired sensory nerve functions (Reichert et al, 2017). Cells from the diabetic skin showed a reduced capacity to induce neurite outgrowth. This limitation is caused by deficiency in neurotrophic factors, such as nerve growth factor (NGF). Keratinocytes from the diabetic skin show insulin resistance and increased expression of proinflammatory cytokines demonstrating the persistent effect of diabetes mellitus on human skin cells. Lowered activity of glyoxalase enzyme activity in diabetic keratinocytes is responsible for the induction in proinflammatory cytokines and limitations in NGF.
MGO is a toxic by-product of hyperglycemia. The diabetic skin is limited in its ability to detoxify MGO. Overall, the diabetic skin environment is not well suited to support nerve function (Reichert et al, 2017). The epidermis of the diabetic skin suffers from depletion of nerves and neuropeptides. In particular, calcitonin gene-related peptide is low in the diabetic epidermis. DPN was associated with lowered abundance of epidermal protein gene product 9.5 (PGP 9.5) as well as calcitonin gene-related peptide nerve counts. Such epidermal depletion of neurotrophic factors was noted at a time when vascular disease had not yet been established (Levy et al, 1992). In the skin, NGF and its precursor (pre-proNGF and proNGF) regulate neuronal function via the tyrosine kinase receptor TrkA and the pan-neurotrophin receptor p75NTR signaling pathway.
NGF binding with TrkA triggers tyrosine kinase-initiated signaling. Prepro-NGF seems to be preferentially taken up and transported by the pan-neurotrophin receptor p75NTR in the diabetic skin (Yiangou et al, 2002). Such p75NTR signaling lead to neuronal apoptosis via interaction with sortilin, a third receptor. This pathway, evident in the diabetic skin, achieves outcomes that are diametrically opposite in end result compared with the survival/differentiation functions of NGF signaling. Importantly, the cytotoxic effects of the p75NTR pathway are not limited to neural cells and may affect other cells of the skin as well. Overall, the diabetic skin is characterized by biomechanical weakness and a depleted neurotrophic environment. Recent developments in neurotrophic skin reprogramming in vivo offer hope.
The topical electrophoretic tissue nanotransfection (TNT) offers a non-viral approach to topically reprogram tissues through a nanochanneled device validated with well-established and newly developed reprogramming models (Gallego-Perez et al, 2017). It is reported that neurotrophic TNT may directly convert skin fibroblasts into electrophysiologically active induced neuronal cells in vivo. Recent work demonstrates that neurotrophic TNT is effective in causing neurotrophic enrichment of the diabetic skin stroma.
Neurotrophic TNT, friendly for point-of-care applications outside of laboratory settings, induced endogenous skin NGF and other co-regulated neurotrophic factors such as Nt3. Such intervention spared the loss of cutaneous PGP 9.5+ mature nerve fibers in the diabetic skin. This work establishes feasibility that in vivo reprogramming can be effective in inducing changes in the diabetic skin microenvironment in a way that can be leveraged for therapeutic purposes such as the rescue of pre-existing nerve fibers from its predictable path of loss under conditions of diabetes (Roy et al, 2020b).
Biofilm Infection
As discussed above (see The Diabetic Skin section), the diabetic skin is at a high risk of compromised barrier function making it vulnerable to infection. Wound biofilm is polymicrobial three-dimensional aggregates that are encased in microbial extracellular polymeric substances, which provide protection against host defense systems as well as antimicrobial drugs (Deng et al, 2020; Sen et al, 2020). Importantly, wound biofilm are shaped by an iterative process involving microbial offense and host defense at the wound site, which fortify the surviving pathogens with adaptive molecular processes that pose severe threat to the host (Ganesh et al, 2015). Wound biofilm represents a major threat to the already weakened ECM of the diabetic skin setting the stage for wound recurrence (Roy et al, 2020a).
Wound biofilms have been directly linked to wound chronicity (Sen, 2019). Standard hospital tests measuring colony forming units are likely to underestimate biofilm infection as biofilm bacteria can be hypometabolic and low on colony formation. Thus, the search for fast and accurate bacterial sensing and biofilm monitoring is currently on (Parlak and Richter-Dahlfors, 2020; Pugia et al, 2021). Hyperglycemia favors biofilm formation as approaches to lower glucose are effective in reducing such infection (Houot and Watnick, 2008). Under conditions of abundance, glucose is transported into microbial cells followed by phosphorylation to glucose-6-phosphate at the expense of EIIAGluc protein dephosphorylation. Dephosphorylated EIIAGluc protein thus accumulates and favors biofilm formation. In contrast, under glucose lowered conditions, components of the phosphoenolpyruvate phosphotransferase system PTS are phosphorylated, thus diminishing biofilm formation (Nagar and Schwarz, 2015).
Vast majority of all DFUs are infected by polymicrobial biofilm. DFU biofilm contributes to disease progression and chronicity of the lesion, the development of antibiotic resistance, and makes wound healing difficult to treat (Pouget et al, 2020). Metataxonomic characterization of the polymicrobial burden in DFU and related biofilm has helped understand the composition of chronic wound pathogenic biofilm (Suryaletha et al, 2018). The host nervous system can also be inhabited by bacteria. Because electrical impulses are an integral component of microbial physiology and signaling (Sen et al, 2020), microbial colonization of the host's neural circuit is likely to influence transmission of action potential (Abashina and Vainshtein, 2021).
Bacteria have been directly implicated in optic neuropathy (Vaphiades and Golnik, 2007). Small-bowel bacterial overgrowth in diabetic subjects is associated with cardiovascular autonomic neuropathy (Zietz et al, 2000). Thus, characterization of mechanisms underlying DFU-associated neuropathy would be incomplete without factoring in the threat posed by biofilm infection. The identification of perineural apatite in diabetic patients with peripheral neuropathy suggested bacterial involvement. Additional evidence supporting a role of infection in DPN lays the foundation for a new paradigm wherein microbial processes deserve attention in the context of DPN pathogenesis and management (Mat Saad et al, 2013; Richard et al, 2012; Sommer and Lee, 2001).
The molecular mechanisms by which wound polymicrobial biofilm or its products may directly contribute to DFU associated neuropathy remains to be studied. There is ample support for the notion that biofilm products may contribute to microvascular diseases. For example, at high concentrations, redox-active bacterial pigment pyocyanin inactivates NO, which is in limited supply in the diabetic skin (Hempenstall et al, 2015; Warren et al, 1990). Pyocyanin can cause endothelial dysfunction by causing oxidative stress and inhibiting prostacyclin production (Kamath et al, 1995; Muller, 2002). Biofilm is known to contribute to several pathologies that are likely to contribute to local neuropathy. The presence of biofilm induces oxidative stress (Fig. 2) and chronic inflammation (Fig. 3) (Cappelli et al, 2005; Hochstim et al, 2010; Rowinska et al, 2021; Saini et al, 2017).
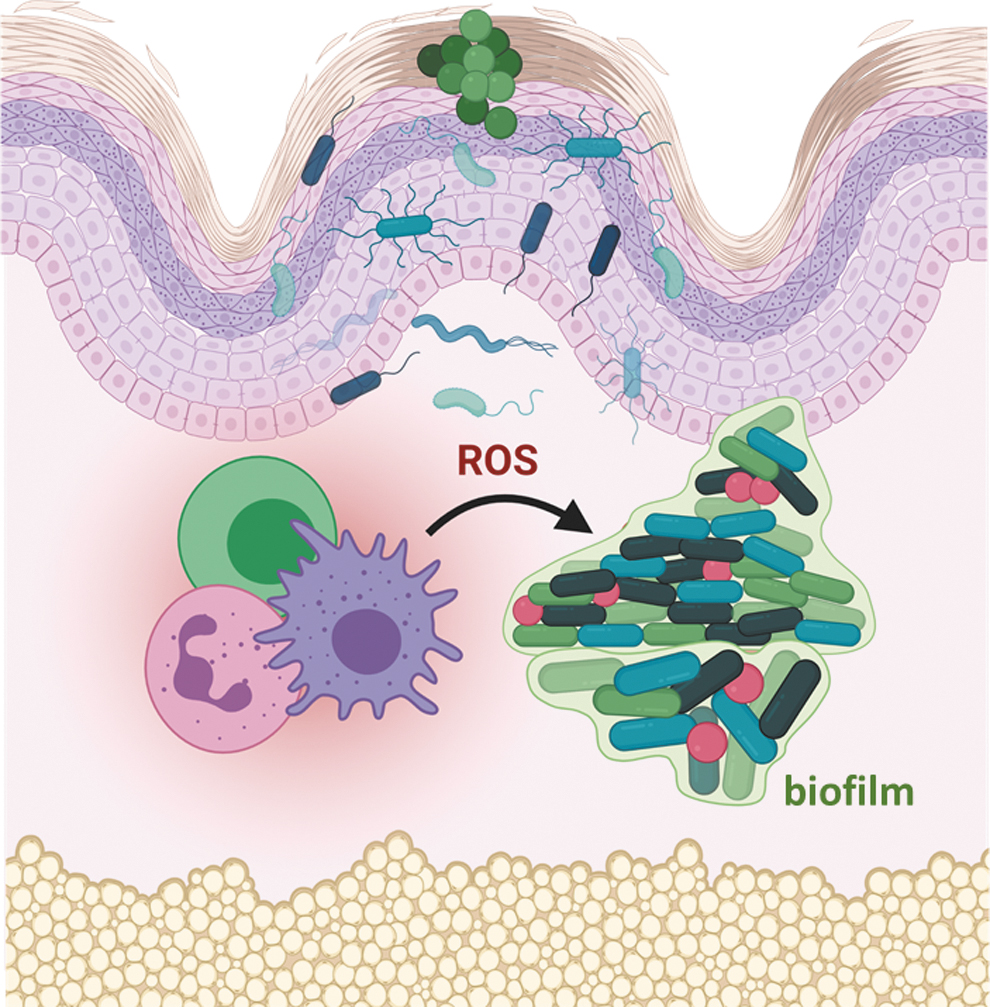
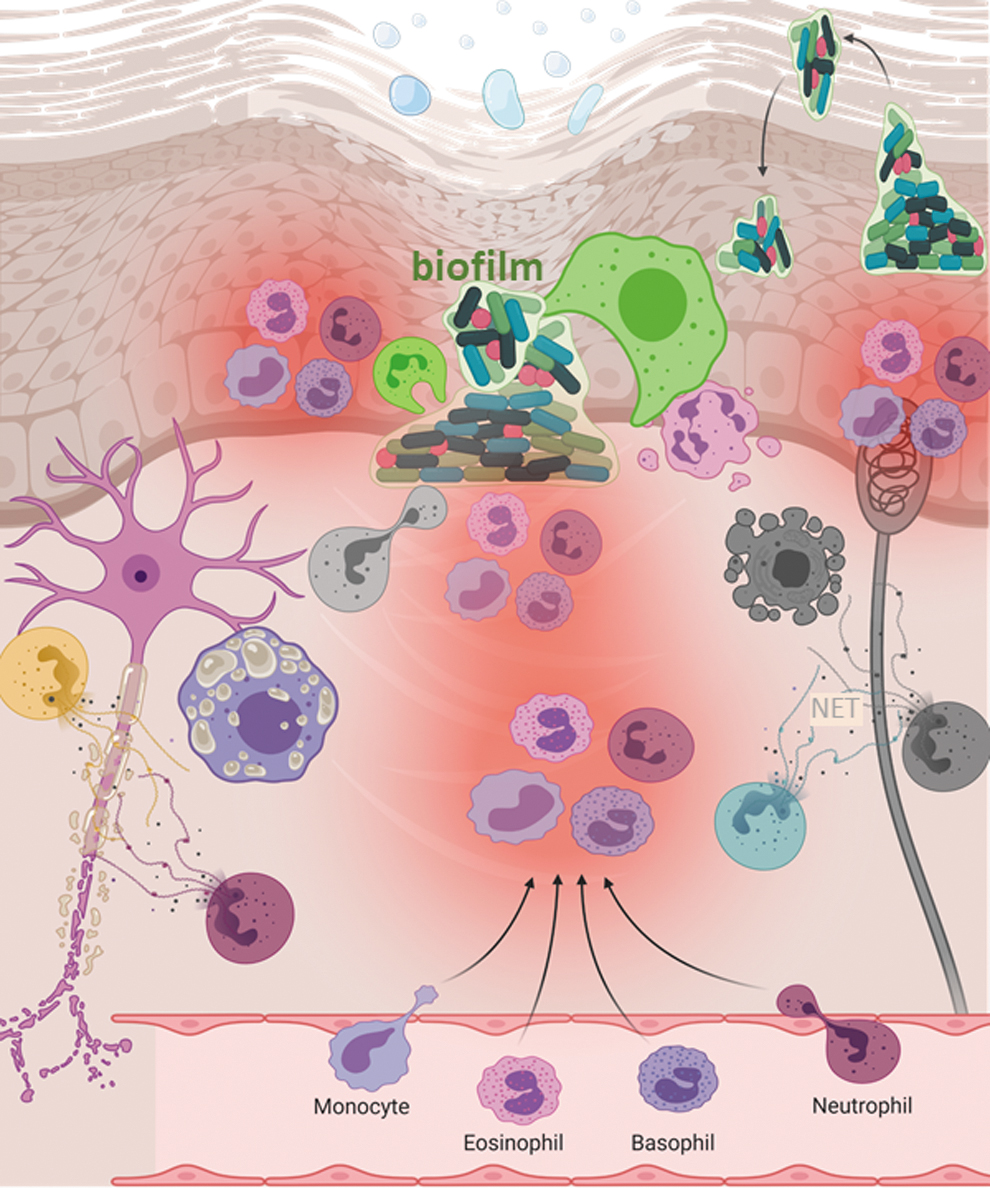
Direct evidence of bacterial biofilm causing neuroinflammation is also evident (Snowden et al, 2012). A typical host defense response to biofilm infection is neutrophil hyperactivity and the formation of NETs. NETs include granule-derived proteins with microbicidal activity. Suicidal NETosis release a lattice, composed of DNA associated with proteins citrullinated by protein-arginine deiminase 4, from neutrophils. Such biofilm-sensitive neutrophil hyperactivity may cause vasculitis (Arneth and Arneth, 2021; Kimura et al, 2021; Lee et al, 2017; Soderberg and Segelmark, 2018). Cutaneous vasculitis may contribute to DPN (Collins and Periquet, 2008; Finsterer, 2009; Said, 1996; Younger, 2004).
Skin with compromised barrier function is also likely to be affected with cellulitis (Abolhasani et al, 2021; Rossolini and Stefani, 2009). Cellulitis is skin infection presented as a poorly demarcated, warm erythematous area with associated edema and tenderness to palpation with no drainage, discharge, or abscess (Brown and Hood Watson, 2021). Diabetes, obesity, and old age are major predisposing factors for cellulitis (Bristow, 2008; Cranendonk et al, 2017; Koutkia et al, 1999). Cellulitis is reported in Charcot foot (Sommer and Lee, 2001). Charcot foot is a rare foot deformity noted in those with DPN that is clinically viewed as a serious complication. Cellulitis can cause tissue damage and death (gangrene). The infection can affect the peripheral nervous system. The cellulitis foot with no tissue loss can be divided into the neuropathic or neuroischemic foot with cellulitis alone (Edmonds et al, 2020).
Metabolic Syndrome
DPN and MetS neuropathies often co-exist. MetS neuropathy preferentially affects small unmyelinated axons early in its course, and it may also affect autonomic and large fibers (Kazamel et al, 2021). MetS exacerbates neuropathy risk in diabetic patients. In T1DM, neuropathy is closely linked to glycemic control. However, in T2DM, neuropathy is linked to dyslipidemia, central obesity, hypertension, insulin resistance, and glucose control. Of note, several clinical studies failed to improve the prevalence of neuropathy after intensive glucose control in T2DM patients (Lee et al, 2020). The search for factors, beyond hyperglycemia, that cause DPN is therefore of interest. Indeed, exercise, dietary restriction, and bariatric surgery show some early favorable effects in managing DPN (Stino et al, 2020).
Patients with diabetes and MetS, compared with those with T2DM alone, show poorer peripheral nerve structure and function outcomes (Issar et al, 2021). MetS increases the risk for cryptogenic sensory peripheral neuropathy. MetS neuropathy preferentially affects small unmyelinated axons early in its course, and it may also affect autonomic and large fibers. Inflammation and dyslipidemia play major roles in the pathogenesis of MetS neuropathy (Kazamel et al, 2021). It is widely acknowledged that in T2DM patients, diabetes and obesity drive peripheral neuropathy (Callaghan et al, 2018). Interventions targeting MetS are likely to be effective in preventing or treating DPN (Callaghan et al, 2018).
Hypertension
It is estimated that roughly 75% of adults with diabetes also have hypertension (Long and Dagogo-Jack, 2011). Half of all patients suffering from hypertension alone exhibit insulin resistance (Reaven, 2003). It is therefore expected that diabetes and hypertension may share underlying risk factors, such as ethnicity, familial polygenic factors, dyslipidemia, and lifestyle determinants. Peripheral vascular disease is commonly noted in patients with long-standing diabetes or hypertension (Long and Dagogo-Jack, 2011). Hypertension in the diabetic is implicated in nephropathy. Plasma levels of tissue inhibitor of metalloproteinase-1 in patients with T1DM associate with early diabetic neuropathy as well as nephropathy (Papadopoulou-Marketou et al, 2021).
Hypertension causes endothelial dysfunction (Virdis and Taddei, 2011) and compromises skin perfusion via its effect on the skin microvasculature (Concistre et al, 2020). Hypertensive patients show reduced responsiveness of cutaneous microcirculation (Farkas et al, 2005; Lindstedt et al, 2006). Patients with familial predisposition to hypertension show erratic dynamics of microvascular perfusion with diminished chaotic ischemic flow (Gryglewska et al, 2011). A close inverse association between skin microvascular function and salt sensitivity has been evident. Such observation suggests a role for generalized microvascular defects as a link between salt sensitivity, insulin resistance, and hypertension (de Jongh et al, 2007). As discussed above, these effects of hypertension are likely to have causative impact on DPN.
Genetic Susceptibility
Peripheral neuropathy can be caused by mutations in genes expressed by neural and SCs (Ghosh and Tourtellotte, 2021). Several heterogeneous groups of susceptible genetic loci are known to contribute to the development of DPN (Prabodha et al, 2018). Interestingly, neuropathy is evident in many patients who are prediabetic. Yet it is absent in others with long-standing diabetes pointing toward a genetic predisposition (Imamura and Maeda, 2011; Kirthi et al, 2021; Papanas et al, 2011; Prabodha et al, 2018; Witzel et al, 2015). Depending on the case, such neuropathies may be manifested during development, childhood or adulthood. Axonal degeneration, demyelination, and loss of motor and sensory neurons leading to functional deficits are expected. Wider use of targeted gene sequencing as well as exome sequencing is likely to provide more insight into the genetic basis of DPN.
Recent work has identified a genetic locus on chromosome 2q24 predicting peripheral neuropathy risk in patients with T2DM. The minor allele of the lead single-nucleotide polymorphism (SNP) decreased DPN odds by 36%. This locus was successfully validated. The minor protective allele at this locus was associated with higher tibial nerve expression of an adjacent gene (sodium voltage-gated channel alpha subunit 2 [SCN2A]) coding for human voltage-gated sodium channel NaV1.2 (Tang et al, 2019). Neurotrophic fibroblast growth factor 2 (FGF2) is effective in neuroprotection, neurogenesis, and angiogenesis. SNPs at the 3′UTR of the FGF2 gene and their interaction with environmental factors have been associated with risk for DPN.
Subjects carrying the T allele at the rs1048201 locus in the FGF2 gene had a significantly lower risk of developing DPN compared with subjects carrying the C allele. Subjects with the G genotype at the rs6854081 locus had an exceptionally higher risk of developing DPN than subjects with the T allele. Individuals harboring the G allele at the rs7683093 locus had a markedly higher risk of DPN than patients with the C allele. Individuals having the A genotype at the rs1476215 locus had a significantly higher risk of DPN than individuals carrying the T allele. An interaction between age and alcohol consumption and the SNP rs7683093 was evident (Jiang et al, 2021). Genetic susceptibility to alcohol-induced DPN is discussed below (see the Lifestyle section).
Psychosocial Factors
Diabetes distress, a psychological barrier to diabetes self-management, represents a psychosocial burden in patients with DFU and DPN. T2DM patients reporting high self-stigma may have lower social support (Botchway et al, 2021). Social stressors are known to impede wound healing (Sen, 2019). Individuals with DPN show depressive symptoms (Vileikyte et al, 2005). Depression and especially DFU-specific emotions are likely to contribute to DFU chronicity (Vileikyte and Gonzalez, 2014). DPN can be painful.
Depression, anxiety, low quality of life, and poor sleep are associated with pain in painful diabetic neuropathy (Kioskli et al, 2019). Mindfulness-based stress reduction, a psychosocial intervention, was effective in improving DPN outcomes under conditions of optimized pharmacotherapy (Nathan et al, 2017). Psychosocial stress, via the central hypothalamic–pituitary–adrenal (HPA) axis and the peripheral intracutaneous HPA axis, may modify the skin immune system, barrier function, and wound healing (Hunter et al, 2015). A closer look at psychosocial factors and their significance in DFU-associated DPN is warranted.
Aging
Functional testing of patients with diabetic neuropathy shows lower axonal diameters in smaller fibers of the elderly (Shibuta et al, 2010). Skin epidermal innervation is compromised with aging and in sensory neuropathy (Lauria et al, 1999). Premature aging-related peripheral neuropathy has been evident in a mouse model of progeria (Goss et al, 2011). Age-related deterioration of the immune system is often referred to as immunosenescence. In the elderly, immunosenescence drives neuroinflammation such that larger numbers of inflammatory cells are found within the peripheral nerve.
Age-related activation of NFκB-light-chain-enhancer of activated B cells related aberrant cytokine expression, and proinflammatory changes in the bone marrow are responsible for such neuroinflammatory condition. Furthermore, the aged skin has higher abundance of AGE and elevated levels of AGE receptors (Hagen and Ousman, 2021). The observation that macrophage depletion ameliorates peripheral neuropathy in aging mice supports a causative role of neuroinflammation in age-related DPN (Yuan et al, 2018).
Sex
Women with long-standing T1DM have shown a trend toward higher neuropathic pain than men (Cardinez et al, 2019). For those with T2DM, the presence of an ACE D allele is associated with increased risk of peripheral neuropathy in women. An important role of sex in the development of DPN is proposed (de la Hoz et al, 2017). Sex-dependent differences in the renin–angiotensin system are thought to be implicated in the development of peripheral neuropathy. Postmenopausal women are at an even greater risk of peripheral neuropathy. The decline in the serum estrogen levels as a function of age is critical in the development of peripheral neuropathy (Singh et al, 2016). In women at midlife, peripheral neuropathy limits physical functioning and increases the risk of future disability (Ylitalo et al, 2013).
Women with T2DM are at higher risk for macrovascular complications than men. Diabetic neuropathy was more prevalent in these women. Interestingly, the interval from T2DM diagnosis and onset of neuropathy was shorter in men than in women. This may be related to testosterone and neurosteroid deficiency that is common in men with diabetes (Kamenov et al, 2010). Experimental studies have revealed sex-specific decrease in neuroactive steroid levels in male diabetic animals. This alters mitochondrial function and axonal transport contributing to the sex difference observed in diabetic neuropathy (Pesaresi et al, 2011).
Gonadectomy in female, but not in male, diabetic animals protects the peripheral nervous system via elevating the levels of dehydroepiandrosterone (DHEA) in the female sciatic nerve (Pesaresi et al, 2011). DHEA restored nerve conduction velocity only in females. Furthermore, it was only in females that DHEA exerted neuroprotective actions on thermal sensitivity or molecular parameters, such as gene expression of myelin proteins. Intra-epidermal nerve fiber density, lowered by diabetes, was corrected by DHEA only in females. Sex-specific mechanisms underlying DFU-associated DPN are therefore of interest.
Lifestyle
Poor lifestyle choices such as comfort eating, poor diet, smoking, inactivity, and alcoholism are implicated in up to 40% of premature deaths in the United States and contribute to persistent disparities in health. Socioeconomic inequalities are recognized to be a major determinant of lifestyle (Dieteren and Bonfrer, 2021; Foster et al, 2021). Lifestyle interventions are effective in reducing DPN severity (Ghavami et al, 2018). Long-term intensive lifestyle interventions significantly decreased questionnaire-based evaluation of DPN (Look, 2017). Smoking has been identified as a risk factor for the development of microvascular complications such as DPN (Clair et al, 2015). A systematic review and meta-analysis addressed the association between smoking and DPN. The odds of developing DPN were higher in patients who smoked (Clair et al, 2015).
Alcoholism may directly contribute to DPN (Zahr et al, 2019). The prognosis of alcoholic peripheral neuropathy is good and independent of age provided that intake of alcohol is discontinued and other causes of neuropathy, such as diabetes, are carefully excluded (Hillbom and Wennberg, 1984). Alcohol dependence syndrome is a chronic lifestyle problem. It is known to cause neurological complication, including DPN. It is associated with ethanol neurotoxicity and nutritional deficiencies such as thiamine/vitamin B1 malabsorption. Charcot osteoneuropathy is typically seen secondary to diabetes mellitus (0.1%–5%). Charcot foot suffers from loss of sensitivity and DPN.
Interestingly, chronic alcoholism has been associated with Charcot foot outcomes in a nondiabetic patient (Qanneta and Bove Aleu, 2020; Shibuya et al, 2008). Comparison of diabetic patients with or without foot ulcer showed history of increased alcohol consumption in those with ulcers (Altenburg et al, 2011). In Asians, the differences of alcohol tolerance are mainly determined by aldehyde dehydrogenase 2 (ALDH2) polymorphism. The study of T2DM patients divided by ALDH2 phenotype and alcohol consumption revealed that alcohol consumption is associated with DPN, especially in drinkers who are genetically alcohol intolerant (Suzuki et al, 2003).
Conclusion
Specific risk factors for DFU and post-DFU complications such as biofilm infection of the wounded diabetic skin make DFU-DPN unique (Fig. 4). Mechanistic understanding of the interplay of the factors that cause and complicate DFU-DPN is necessary to better define this substantial health care problem. Under current conditions of clinical care, DFU-DPN is diagnosed too late. It is important to recognize that only moderate-to-severe DPN is detected by the standard 10 g monofilament test and foot examination. Point-of-care tests aimed at detecting small fiber damage are valuable for the timely detection of DPN.
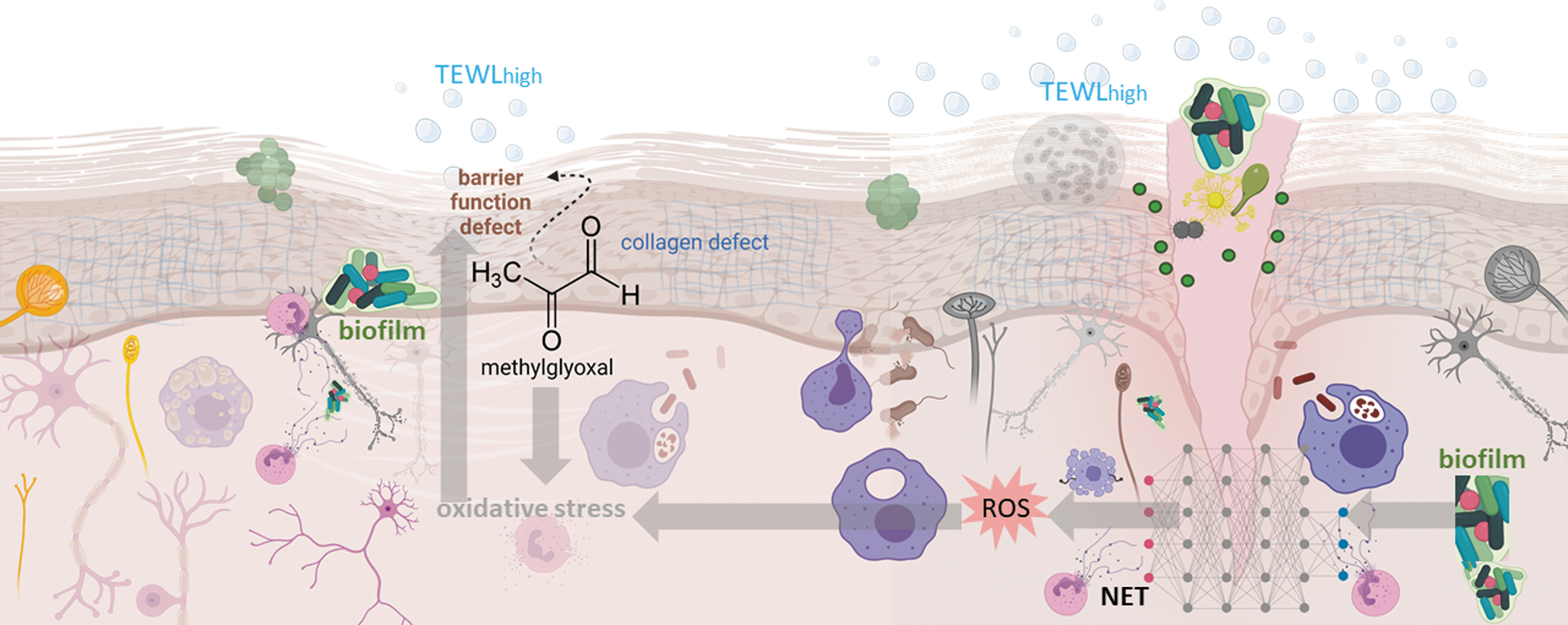
At present, thermal thresholds, microneurography, evoked potentials, sudomotor function, laser Doppler flare, and skin biopsy tests meet those needs. DFU-DPN is nested in the diabetic skin, which is likely to suffer from compromised barrier function. The presence of biofilm infection is highly likely and expected to complicate DPN in ways not usually evident elsewhere in the body. The DFU-DPN is unlike any other neuropathy and warrants dedicated attention.
Footnotes
Authors' Contributions
Conceptualization: C.K.S., S.R., and S.K. Writing—original draft preparation: C.K.S., S.R., and S.K. Writing—review and editing, C.K.S., S.R., and S.K.
Acknowledgment
We thank Dr. Shomita Mathew-Steiner for proofreading the article.
Author Disclosure Statement
All authors have no competing financial interests.
Funding Information
Work on diabetic wounds and neurodegeneration in the laboratory is supported by the following National Institutes of Health grants: U01DK119099, R01DK128845, R01NS042617, R01DK125835, and R01DK114718.