
Abstract
Aims:
Cystathionine β-synthase (CBS) is essential for homocysteine (Hcy) transsulfuration, yielding cysteine as a common precursor of hydrogen sulfide (H2S), glutathione (GSH), and other sulfur molecules, which produce neuroprotective effects in neurological conditions. We previously reported a disruption of microglial CBS/H2S signaling in a Parkinson's disease (PD) mouse model. Yet, it remains unclear whether CBS affects nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 3 (NLRP3) inflammasome activity and other pathologies in PD.
Results:
Microglial CBS expression decreased after lipopolysaccharide (LPS) stimulation. Elevated GSSG (the oxidized GSH) content and decreased H2S generation were found in the brains of microglial cbs conditional-knockout (cbs cKO) mice, whereas serum and brain Hcy levels remained unaltered. Moreover, microglial cbs cKO mice were susceptible to NLRP3 inflammasome activation and dopaminergic neuron losses caused by LPS injection into the substantia nigra, whereas cbs overexpression or activation produced opposite effects. In vitro studies showed that cbs overexpression or activation suppressed microglial NLRP3 inflammasome activation and interleukin (IL)-1β secretion by reducing mitochondrial reactive oxygen species (mitoROS) level. Conversely, ablation of cbs enhanced NLRP3 expression and mitoROS generation and augmented microglial NLRP3 inflammasome activity in response to adenosine triphosphate challenge, which was blocked by the mitoROS scavenger.
Innovation and Conclusion:
The study demonstrated an elevated GSSG level and reduced H2S generation, which correlated with a susceptible status of microglia in the brain of cbs cKO mice. Our findings reveal a critical role of CBS in restraining the microglial NLRP3 inflammasome by controlling redox homeostasis and highlight that activation or upregulation of CBS may become a potential strategy for PD treatment.
Introduction
Cystathionine β-synthase (CBS) is the rate-limiting enzyme of the transsulfuration pathway, playing an essential role in the metabolic conversion of homocysteine (Hcy) to cysteine (Hu et al., 2011). Hence, CBS deficiency is closely related to Hcy buildup, referred to as hyperhomocysteinemia (hHcy) when it exceeds 15 μM in the human plasma (Jhee and Kruger, 2005). More than 150 cbs gene mutations have been identified. However, the role of CBS is largely underestimated. Studies reveal that CBS is able to synthesize hydrogen sulfide (H2S), which is a gaseous signaling molecule and serves as a neuromodulator in the brain. H2S donors suppressed neuroinflammation by modulating nuclear factor kappa-B (NF-κB) and AMP-activated protein kinase activity (Hu et al., 2007; Jia et al., 2020) and exerted neuroprotective effects in rodent and cellular models of Parkinson's disease (PD) (Hu et al., 2010; Kida et al., 2011; Vandiver et al., 2013).
Innovation
Our study showed an elevated GSSG (the oxidized glutathione) level and reduced hydrogen sulfide production in the brains of microglial cbs conditional-knockout (cbs cKO) mice compared with cbs fl/fl littermates, with no altered serum or brain homocysteine levels. This correlated with a preactivated status of cbs-deficient microglia, more susceptible to proinflammatory stimulation. By reconstituting redox homeostasis, cystathionine β-synthase (CBS) activation or overexpression suppressed microglial nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 3 (NLRP3) inflammasome activation and alleviated dopaminergic neuron losses in the midbrain of a neuroinflammation-related Parkinson's disease (PD) mouse model. The findings advance our understanding of CBS function in the brain and reveal that modulating CBS may be of potential for PD treatment (Fig. 1).
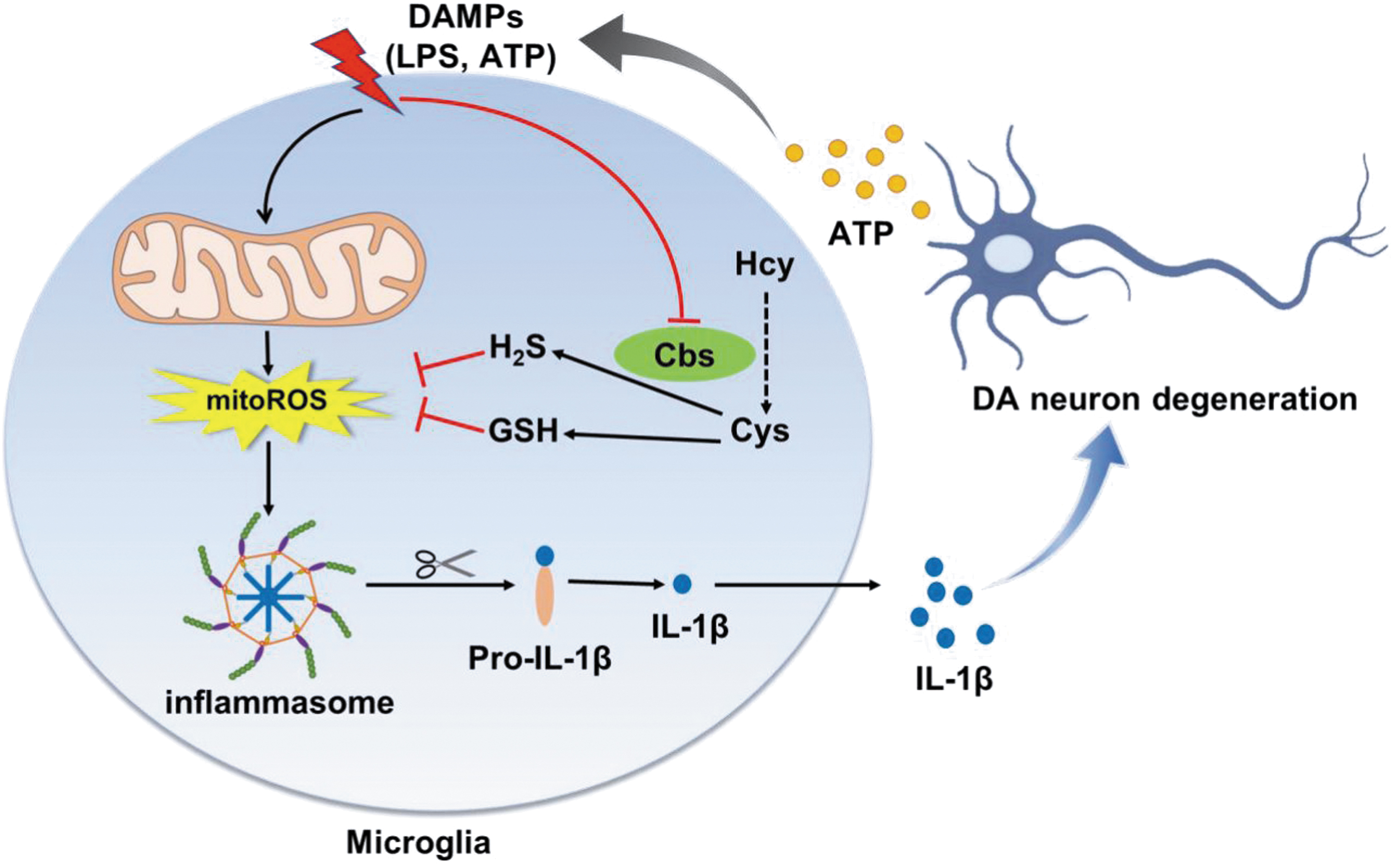
These implicate an important role of CBS in the brain. Indeed, CBS is highly expressed in the brain, in addition to the liver and kidney (Enokido et al., 2005; Panagaki et al., 2022; Shan et al., 2019). Our previous study identified an abundance of CBS protein in microglia and revealed the decrease of microglial CBS expression in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced PD mouse model (Yuan et al., 2018). Moreover, cbs overexpression attenuated glial activation and locomotor deficits in these PD mice. These prompted us to intensively explore the function of microglial CBS in the brain, particularly in PD.
PD is a neurodegenerative disorder characterized by motor and nonmotor symptoms, mainly due to the loss of dopaminergic (DA) neurons in the substantia nigra (SN) and the formation of α-synuclein inclusion body. Neuroinflammation and oxidative stress are closely related and greatly contribute to PD pathogenesis (He et al., 2020; Teleanu et al., 2022). Inflammasome is a multiprotein complex composed of nucleotide binding and oligomerization domain-like receptor (NLR) family member, apoptosis-associated speck-like protein containing a CARD domain (ASC), and pro-caspase-1, serving as intracellular sensors to a variety of stresses, including reactive oxygen species (ROS) accumulation and mitochondria or lysosome disruption. This assembled complex triggers the cleavage of pro-caspase-1 into its active form, which subsequently induces pro-interleukin (IL)-1β/IL-18 maturation and secretion, leading to pronounced inflammation (Zhou et al., 2011).
Inflammasome activation acts as a cascade linking oxidative stress to neuroinflammation. Several subtypes of inflammasomes have been reported. Among these, NLR pyrin-domain-containing protein 3 (NLRP3) inflammasome is greatly implicated in PD progression (Qiao et al., 2018). NLRP3 expression was reported to be increased in the brains of patients with PD and various murine models (Gordon et al., 2018; von Herrmann et al., 2018). Exome sequencing identified a rare NLRP3 polymorphism associated with a lower risk of developing PD (von Herrmann et al., 2018). The mice with genetic knockout of caspase-1 or nlrp3 was resistant to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced PD-like phenotypes (Qiao et al., 2017; Yan et al., 2015). Moreover, fibrillar α-synuclein triggered microglial NLRP3 inflammasome activation and IL-1β release, driving sustained neuroinflammation in PD (Gordon et al., 2018). Therefore, NLRP3 inflammasome is a potential target for PD treatment.
In this study, we reported that microglial CBS expression declined in response to lipopolysaccharide (LPS) stimulation. In vivo studies demonstrated that microglia-conditional cbs depletion (cbs cKO) augmented the NLRP3 inflammasome activation, IL-1β generation, and DA neuron losses caused by LPS injection into the SN, whereas cbs overexpression or activation produced opposite effects. In vitro studies further revealed the involvement of mitochondrial reactive oxygen species (mitoROS) in the regulation of CBS on microglial NLRP3 inflammasome activity. Consistently, elevated GSSG (the oxidized glutathione [GSH]) content and reduced H2S generations were found in the brains of cbs cKO mice compared with cbs fl/fl littermates, whereas the serum and brain Hcy levels remained unaltered. Together, our study identified an essential role of CBS in restraining the NLRP3 inflammasome by controlling redox homeostasis in microglia and suggested that activation or upregulation of CBS may become a potential strategy for the treatment of PD and other neuroinflammation-related diseases.
Results
Microglial CBS expression was downregulated in response to LPS stimulation
To study whether CBS expression was altered in response to inflammatory insults in vivo, we bilaterally injected 7.5 μg LPS into the mouse SN via a stereotaxic apparatus (Fig. 2A). This procedure was reported to trigger NLRP3 inflammasome activation and DA neuron losses in the SN (Chen et al., 2019; Mao et al., 2017), recapitulating the key pathological features of PD. The protein and mRNA levels of toll-like receptor 4 (TLR4), CBS, and NLRP3 inflammasome components in the ventral midbrain (containing SN) were examined. As shown in Figure 2B–K, the protein and mRNA levels of TLR4, which recognizes and binds to LPS, rapidly increased at day 3, and persisted till day 7 and 14 after LPS injection. The mRNA levels of myeloid differentiation primary response protein 88 (myd88), the adaptor protein downstream to TLR4 activation, dramatically enhanced at day 3 but rapidly declined at day 7 and 14 after LPS treatment.
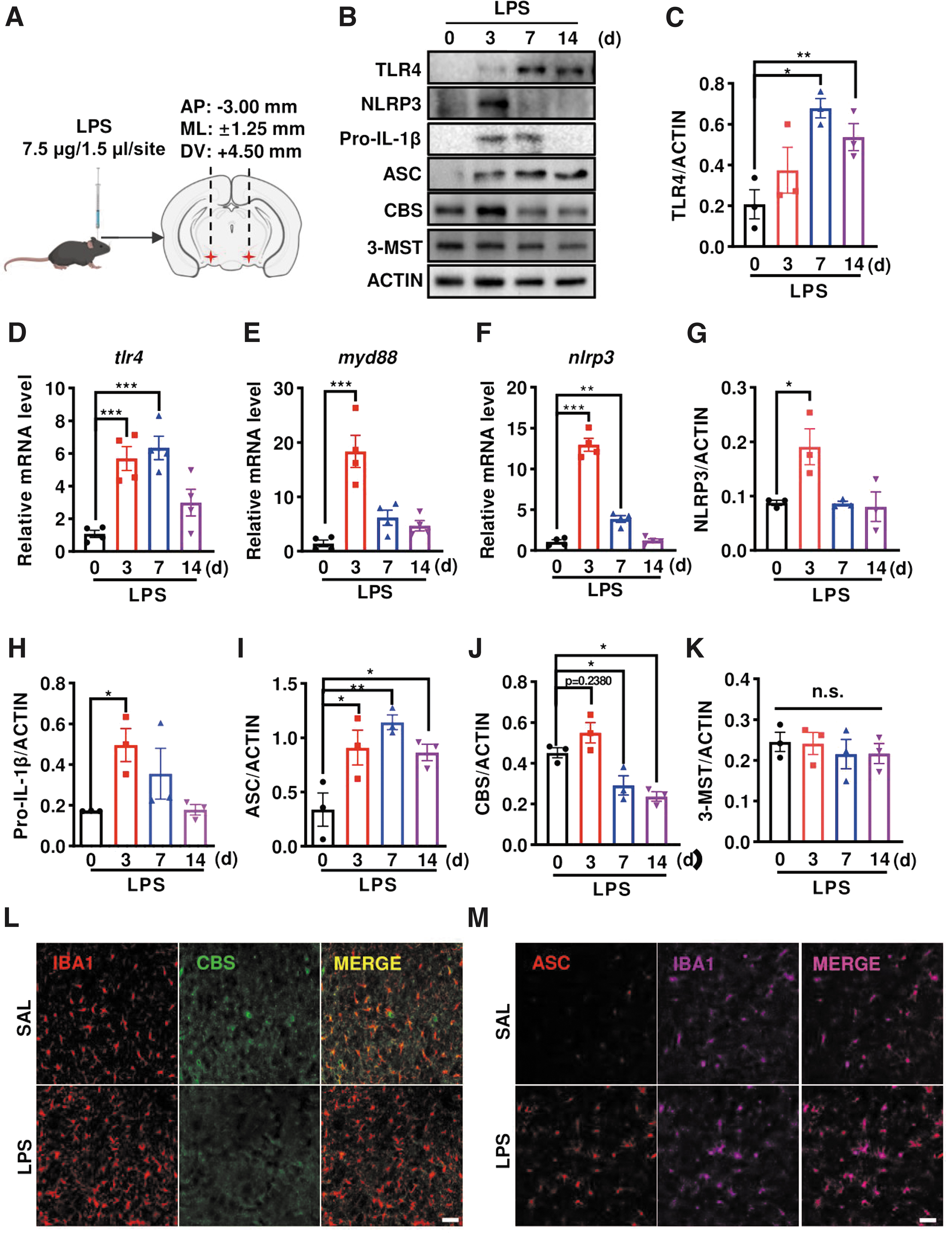
Similarly, the levels of NLRP3 mRNA and protein, as well as pro-IL-1β protein, robustly increased at day 3 post-treatment, followed by the decreases and almost returning to the basal level at day 0. These indicated that LPS injection triggered an immediate response of TLR4/MyD88 and priming in NLRP3 inflammasome, consistent with previous reports (He et al., 2013; Kim et al., 2019). Moreover, the adaptor protein ASC, which is essential for NLRP3 inflammasome assembly, also increased early at day 3 and persisted to be elevated even at day 7 and 14 after LPS injection. Interestingly, CBS protein level declined gradually at day 7 and 14, with a mild but not significant increase at day 3 following LPS injection.
Yet, the protein expressions of 3-mercaptopyruvate sulfurtransferase (3-MST), another reported H2S-producing enzyme in the brain, remained unaltered. As expected, immunostaining identified an increased number of ionized calcium binding adapter molecule 1 (IBA1+) microglia in the substantia nigra reticular (SNr) of LPS-injected mice compared with the saline (SAL) group. CBS predominantly colocalized to IBA1+ microglia. And the CBS intensity decreased in the LPS-injected group (Fig. 2L). In addition, we found an enhanced intensity of ASC, which predominantly colocalized to IBA1+ cells, in the SNr of LPS-injected mice (Fig. 2M). These were consistent with the Western blot data mentioned above.
LPS was well established to trigger an immediate response and NLRP3 transcription in microglia. We then studied the changes of these proteins in LPS-stimulated microglia (Fig. 3A–E) and observed a rapid increase of NLRP3 protein expression at 3 h, followed by the decreases at 12 and 24 h in LPS-stimulated murine BV2 microglia cell line. CBS protein level gradually decreased, consistent with the in vivo results. 3-MST or cystathionine-γ-lyase (CSE, another H2S synthetase and the enzyme also involved in the transsulfuration pathway) did not change in LPS-stimulated BV2 cells. We also repeated the experiments in human-derived microglia cell line HMC3.
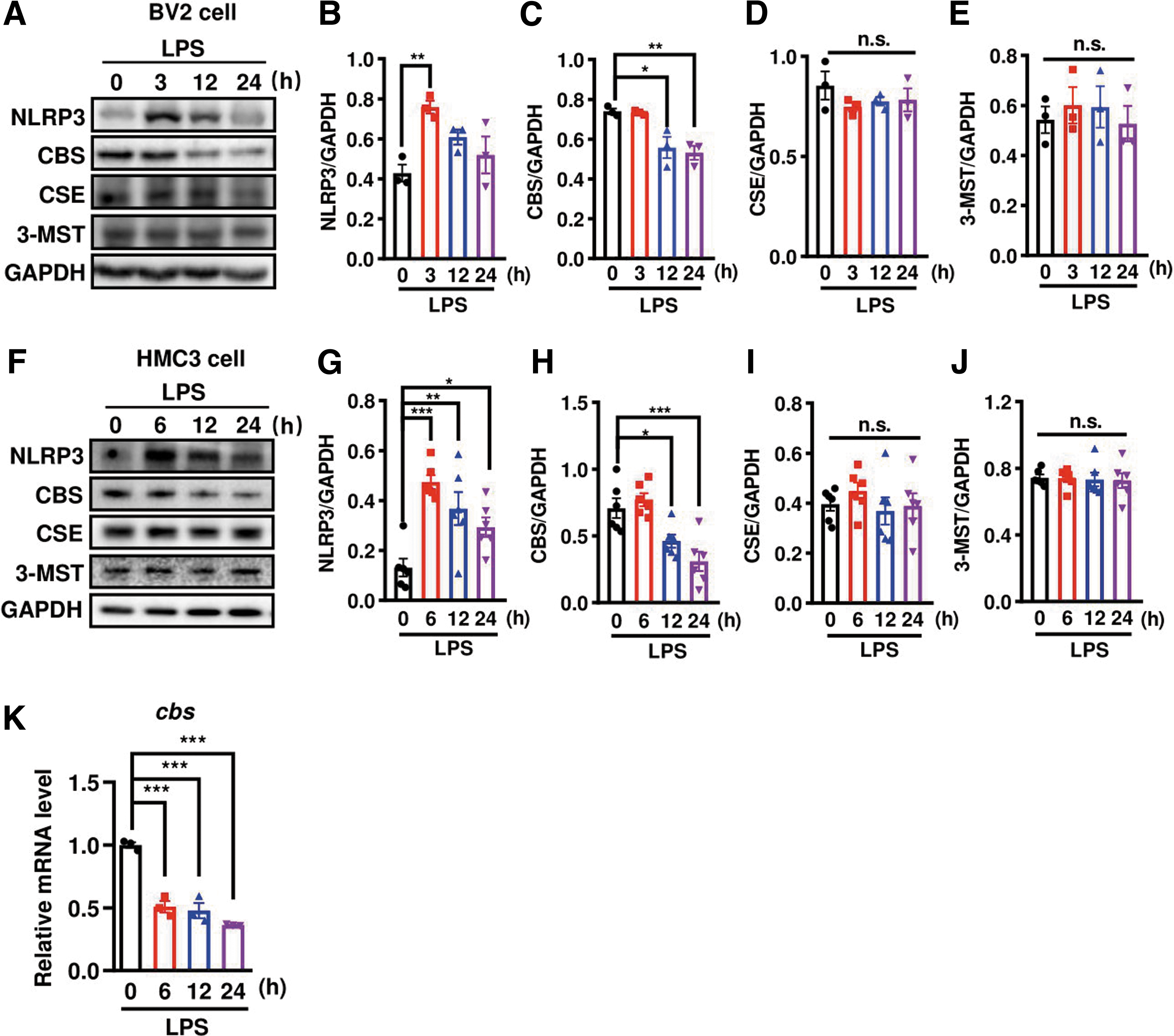
The temporal changes of NLRP3 and CBS protein expressions were similarly observed in LPS-stimulated HMC3 cells (Fig. 3F–J). A transient increase of NLRP3 protein level was found at 6 h after stimulation, whereas CBS protein level declined time dependently with 24 h after LPS stimulation. Neither CSE nor 3-MST protein level changed. In LPS-treated primary microglia culture, cbs mRNA level also decreased (Fig. 3K), in line with its protein expression change. This indicated that microglial cbs transcription was suppressed in response to LPS. These in vivo and in vitro observations imply that CBS may play a role in regulating the neuroinflammatory responses in microglia.
Microglial cbs depletion disrupted H2S generation and GSH redox homeostasis in the mouse brain
To understand the in vivo relevance of microglial CBS downregulation, we generated tamoxifen-inducible microglial cbs cKO mice by crossbreeding cbs fl/fl with Cx3cr1 CreERT2 mice. The offspring cbs fl/fl littermates served as controls. These cbs cKO mice were normal in appearance and fertility even after tamoxifen injection. No difference was observed in the body weight (Fig. 4A). Immunostaining, in combination with confocal scanning, confirmed the deficiency of CBS in IBA1+ cells in the brains of tamoxifen-injected cbs cKO mice (Fig. 4B).
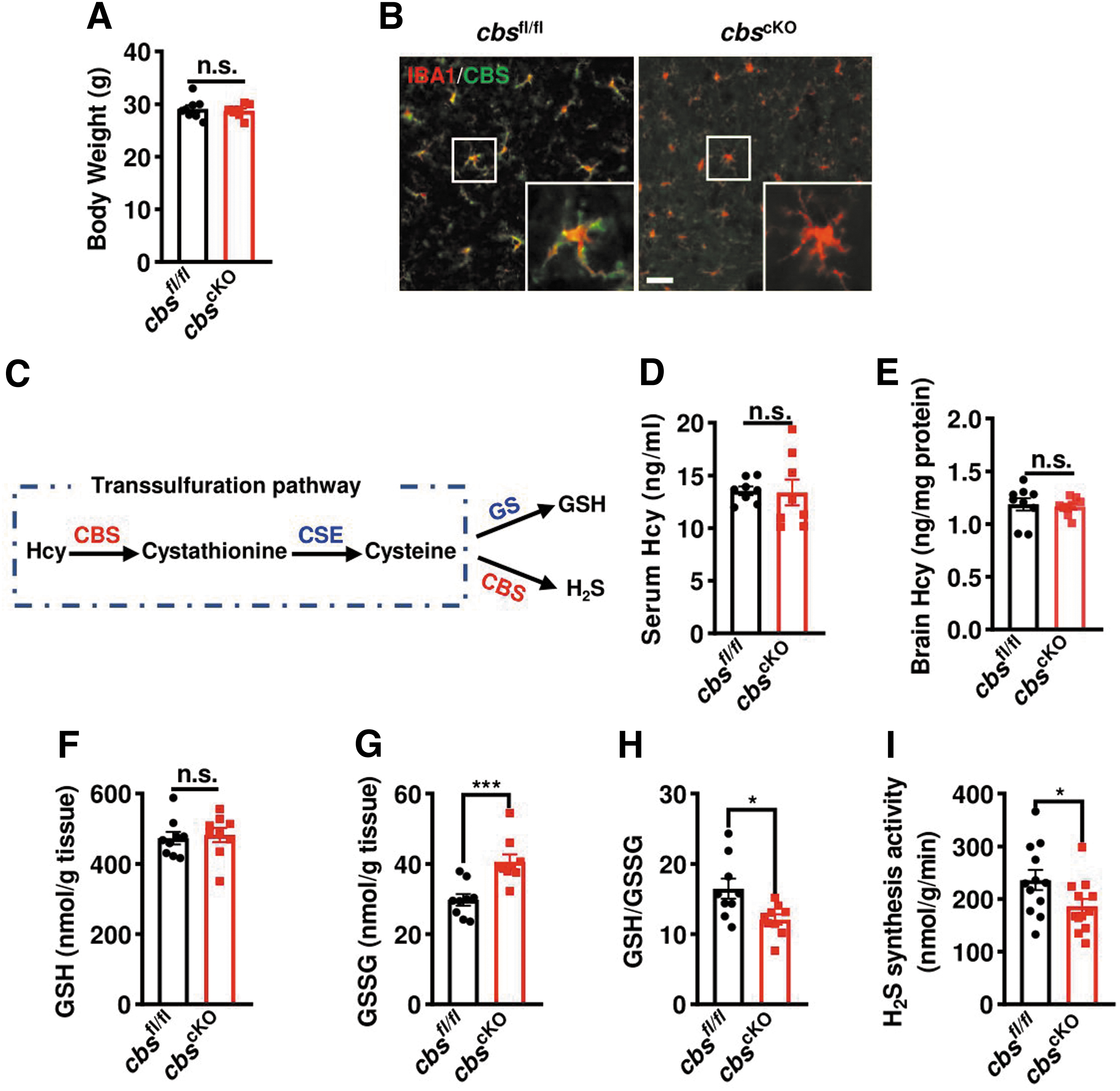
CBS is responsible for the metabolic conversion of Hcy to cysteine (Fig. 4C), which is the precursor of all organic sulfur molecules, such as H2S and GSH (Sbodio et al., 2019). cbs mutation or deficiency caused hHcy and liver injury in humans and mice (Robert et al., 2005). Therefore, we tested if Hcy level was changed, but no significant difference was found in the serum or brain Hcy levels between cbs cKO mice and cbs fl/fl littermates (Fig. 4D, E). Intriguingly, the GSSG level, the oxidized form of GSH, in the brains of microglial cbs cKO mice was higher than that in cbs fl/fl littermates while GSH level remained unchanged (Fig. 4F, G), with a lower ratio of GSH over GSSG in cbs cKO mice (Fig. 4H). And the H2S synthesis activity in cbs cKO mice also decreased (Fig. 4I). These suggest that microglia-specific depletion of cbs mainly impaired the generation of its downstream sulfur molecules, which may disrupt the redox homeostasis in the brain.
Microglial cbs-deficient mice were susceptible to inflammasome activation and DA neuron losses in the SN caused by LPS injection
Oxidative stress and chronic neuroinflammation are implicated in the pathogenesis of PD. Redox homeostasis and H2S are essential in controlling microglial phenotype and function (Hu et al., 2007; Rojo et al., 2014). Therefore, tamoxifen-injected cbs cKO mice were used to investigate the relevance of microglial cbs deficiency in the neuroinflammation-triggered mouse model of PD. The experimental design is shown in Figure 5A. More robust increases of pro-IL-1β and ASC protein levels, as well as IL-1β content, were found in the SN of LPS-injected cbs cKO mice compared with LPS-injected cbs fl/fl mice (Fig. 5B–E). We observed no significant difference either in pro-IL-1β protein level or IL-1β contents between SAL-treated cbs cKO mice and their littermates. However, a four-fold elevation of il-1β mRNA level was found in microglia acutely isolated from adult cbs cKO mice compared with controls (Fig. 5F).
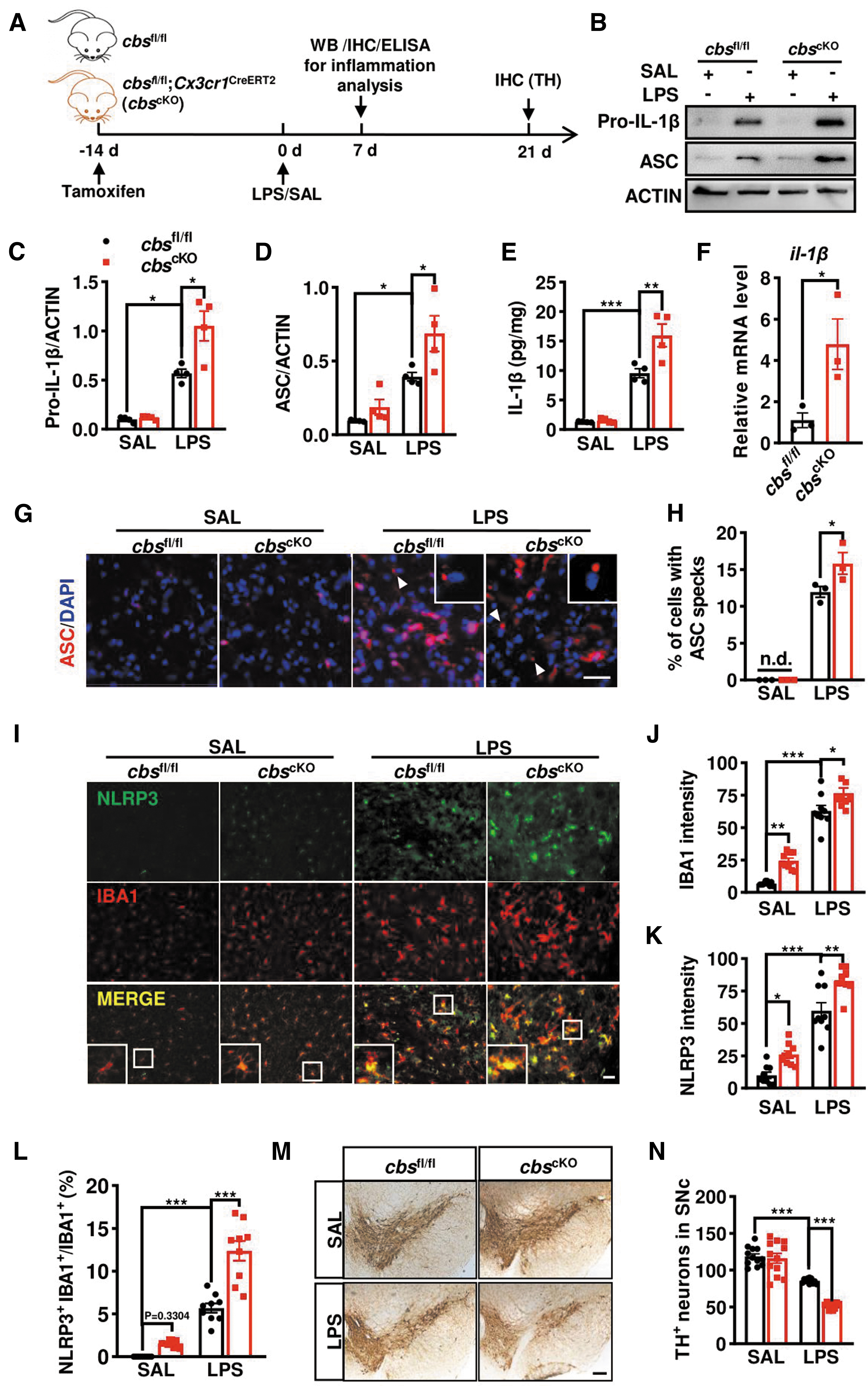
In addition, a higher percentage of cells containing the ASC speck, a singular, perinuclear and punctate “speck” structure with ∼1 μm in diameter, was identified in the SNr of LPS-treated cbs cKO mice than that in cbs fl/fl littermates (Fig. 5G, H). The speck was almost undetected in SAL-treated groups. IBA1 and NLRP3 double staining showed that NLRP3 fluorescence was almost undetectable in SAL-treated mice but dramatically enhanced following LPS injection, with higher NLRP3 intensities in microglial cbs cKO mice than in controls. Similar changes were observed in IBA1 intensities. Consistent with the elevated il-1β mRNA level in isolated microglia from adult cbs cKO mice, we found mild increases of IBA1 and NLRP3 intensity in SAL-treated cbs cKO mice compared with cbs fl/fl littermates. Moreover, NLRP3 predominantly localized to IBA1+ cells, and the ratio of NLRP3+IBA1+ over IBA1+ microglia in LPS-injected cbs cKO mice was higher than that in cbs fl/fl littermates (Fig. 5I–L). These findings indicated that CBS depletion already affected microglial status and augmented the NLRP3 inflammasome activation and IL-1β production in mice.
Microglia-mediated neuroinflammation causes damages to DA neurons, which is implicated in PD. Therefore, the potential impact of microglia-specific CBS depletion on DA neurons in the SN was also studied. We observed no difference in the number of tyrosine hydroxylase positive (TH+) neurons between SAL-injected cbs cKO and cbs fl/fl mice. However, the number of TH+ neurons decreased about 28% in LPS-injected cbs fl/fl mice compared with SAL-treated cbs fl/fl mice, and the decrease reached 55% in LPS-injected cbs cKO mice compared with SAL treatment (Fig. 5M, N), indicating that ablation of microglial cbs gene augmented the LPS-induced DA neuron damage in the SN.
The motor performance was also evaluated as patients with PD suffered from motor dysfunction. All tests including open field, rotarod, and pole climbing did not show any difference between cbs cKO and cbs fl/fl mice after SAL treatment. LPS caused a moderate locomotor impairment in cbs fl/fl mice, as indicated by a decrease of total travel distance during open field test. The decreases of total traveled distance and the duration in the central area were more severe in cbs cKO mice relative to cbs fl/fl controls on day 21 after LPS injection (Supplementary Fig. S1A–C). However, either rotarod or pole climbing test did not reveal any difference among the four tested groups (Supplementary Fig. S1D–E), indicating that microglial CBS depletion exacerbated the LPS-induced locomotor deficits, without obvious effect on motor coordination.
cbs overexpression and activation mitigated NLRP3 inflammasome activation and DA neuron losses in the SN caused by LPS injection
We then asked whether cbs overexpression could alleviate NLRP3 inflammasome activation and IL-1β production induced by LPS in vivo. To achieve this, recombinant AAV-cbs or AAV-vec was delivered into the SN at 2 weeks before LPS or SAL stereotaxic injection (Fig. 6A). Immunostaining showed the decreases of IBA1+ cells number and its fluorescence intensity in the SN of AAV-cbs mice compared with AAV-vec counterparts at day 7 after LPS injection (Fig. 6B–D). LPS-induced increases of NLRP3, ASC, mature caspase-1, and IL-1β were attenuated in AAV-cbs mice compared with AAV-vec mice (Fig. 6E–K). The efficiency of cbs overexpression was verified by Western blot in parallel.
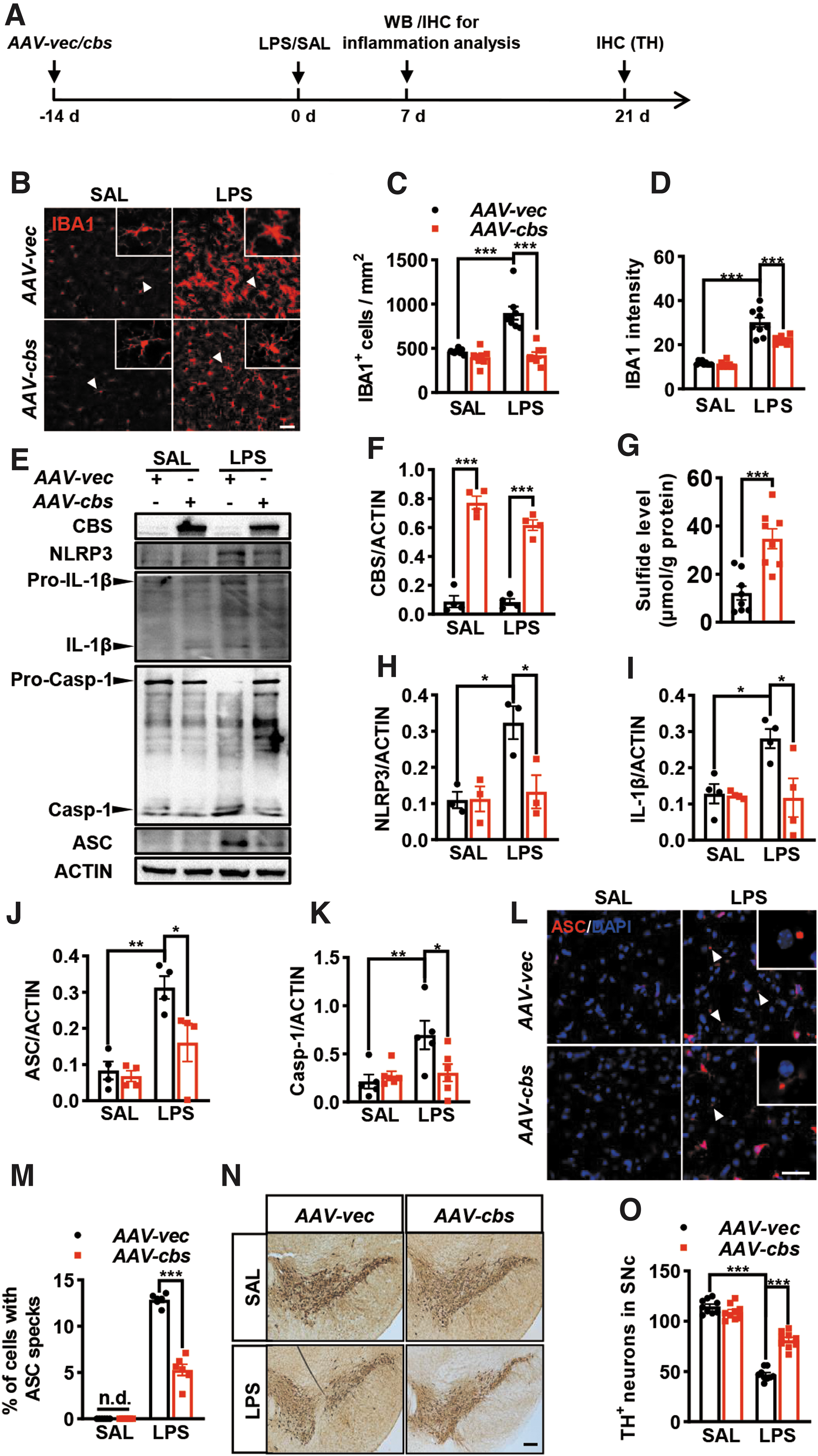
The sulfide level assay also supported the successful establishment of cbs overexpression in the midbrain delivered with AAV-cbs compared with AAV-vec (Fig. 6G). Remarkably, neither IBA1+ cells number or IBA1 intensity, nor NLRP3 inflammasome protein expression was altered in SAL-injected two groups of mice. Immunostaining identified a higher percentage of cells containing the ASC speck in the SN of both AAV-vec and AAV-cbs mice following LPS injection (Fig. 6L, M). However, the percentage of cells with ASC speck in LPS-injected AAV-cbs mice was much lower than that in LPS-injected AAV-vec mice. An obvious loss of TH+ neurons was found in the SN pars compacta of LPS-treated AAV-vec mice, which was attenuated in AAV-cbs-injected mice (Fig. 6N, O).
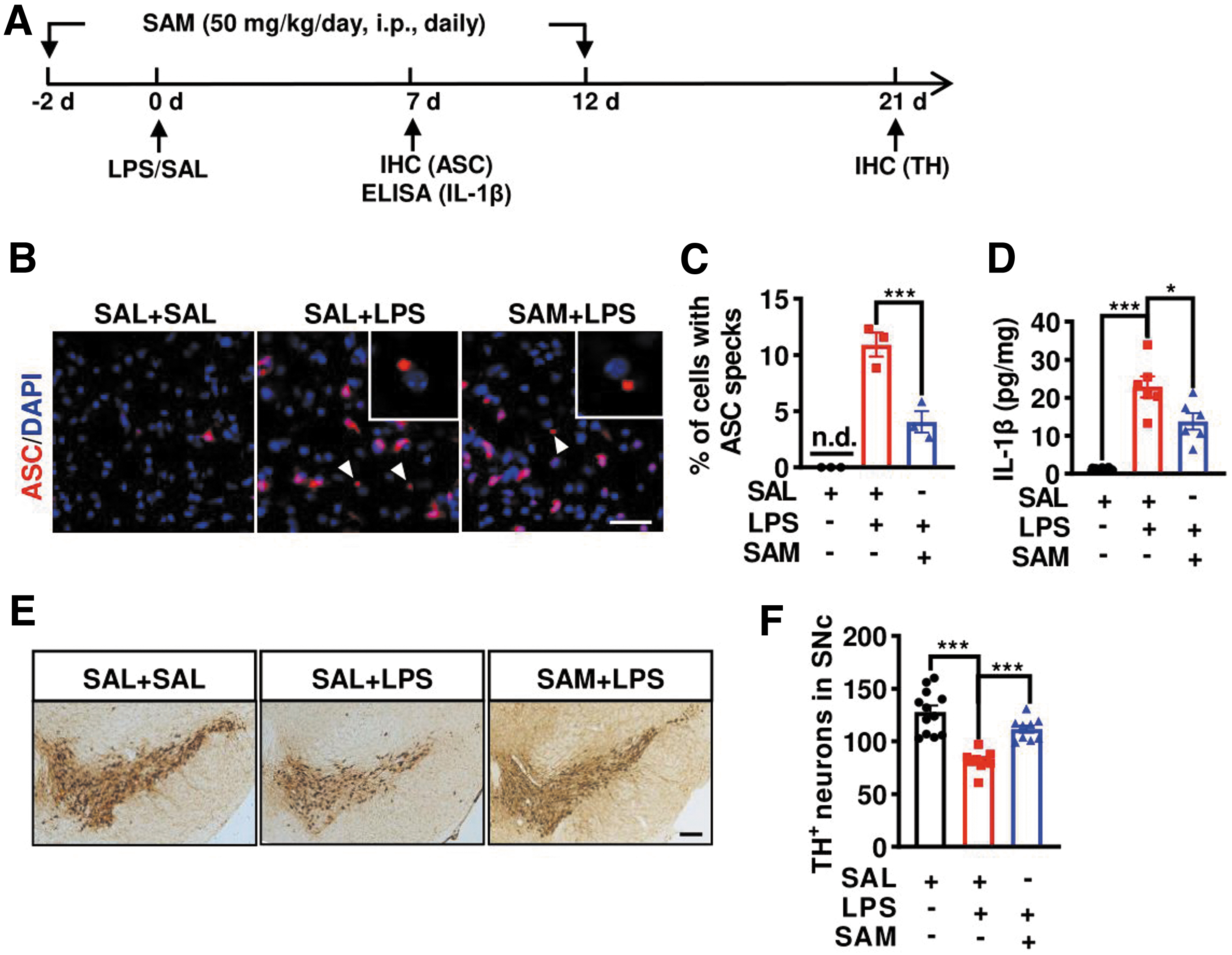
Apart from genetic approach, we used the pharmacological technique to modulate the CBS activity. CBS activator S-adenosyl methionine (SAM, 50 mg/kg) was intraperitoneally (i.p.) administered once daily for 2 weeks, and SAL or LPS was injected into the SN on the third day after SAM injection, as illustrated in Figure 7A. Similar to cbs overexpression, SAM administration not only reduced the quantity of cells containing the ASC speck but also alleviated IL-1β level in the SN of LPS-injected mice, as revealed by immunostaining and enzyme-linked immunosorbent assay (ELISA) results (Fig. 7B–D). LPS-caused TH+ neurons loss in the SN was also alleviated in the SAM+LPS group compared with the LPS treatment-alone group (Fig. 7E, F). These data suggest that cbs overexpression or activation can suppress NLRP3 inflammasome assembly and indirectly protect DA neurons against LPS-triggered injury in the SN.
cbs activation and overexpression suppressed NLRP3 inflammasome activation and IL-1β secretion in microglia
We then introduced a commonly used in vitro model to verify the in vivo findings and explore the underlying mechanism. To achieve this, BV2 or primary microglial culture cells were briefly primed with 100 ng/mL LPS for 3 h and then subjected to the classic NLRP3 inflammasome activator adenosine triphosphate (ATP) for 0.5 h. The CBS activator SAM or H2S-releasing compounds GYY and sodium hydrosulfide (NaHS) were added into LPS-primed microglia before ATP. Western blotting showed that ATP-induced elevations of IL-1β and caspase-1 levels in the supernatant fractions were significantly reduced in SAM-pretreated cells compared with the vehicle group, although both pro-caspase-1 and cleaved caspase-1, as well as IL-1β levels in the lysates, were similar between the two groups (Fig. 8A). The effect of SAM was mimicked by exogenously applied H2S donors.
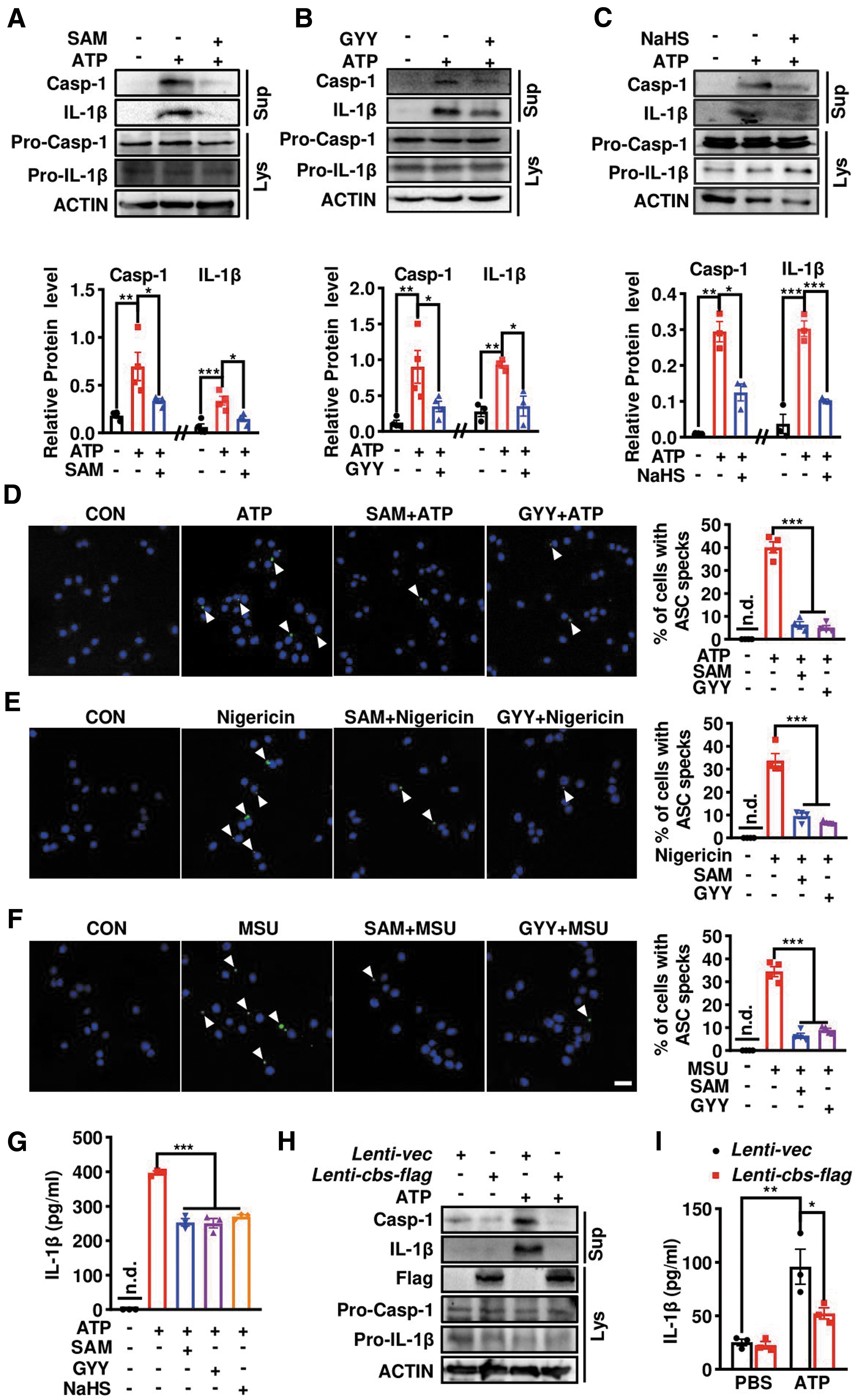
As shown in Figure 8B, C, preincubation with GYY or NaHS consistently prevented the accumulations of caspase-1 and IL-1β in the supernatant triggered by ATP, with fewer effects on these proteins in the lysate fraction. Immunostaining with anti-ASC displayed that ATP caused ∼40% of microglial cells forming a perinuclear ASC speck, which was almost undetected in ATP-untreated cells. Microglial cells pretreated with SAM or GYY before ATP challenge showed less ASC speck formation (Fig. 8D). The inhibition of SAM and GYY on the ASC speck was also observed in microglia stimulated by two other NLRP3 inflammasome activators, nigericin and monosodium urate (MSU) (Fig. 8E, F), implying a common suppression by CBS-H2S axis on NLRP3 inflammasome assembly triggered by different insults. Supportively, ELISA also showed that SAM and two H2S-releasing compounds reduced ATP-induced IL-1β secretion into the supernatant of LPS-primed microglia (Fig. 8G).
Similar to SAM, lentivirus-mediated flag-tagged cbs overexpression (Lenti-cbs-flag) also reduced the mature IL-1β and caspase-1 protein levels in the supernatant of ATP-challenged LPS-primed microglia compared with Lenti-vector-infected microglia, as indicated by Western blotting (Fig. 8H) and ELISA (Fig. 8I). H2S measurement using the fluorescent probe HSip-1 DA revealed an enhanced fluorescence intensity in Lenti-cbs-flag-infected BV2 cells compared with the vector group, which also confirmed the successful cbs overexpression (Supplementary Fig. 2A, B). These data suggest that CBS-H2S signaling can inhibit NLRP3 inflammasome and IL-1β generation in microglia.
cbs knockdown triggered microglia into a susceptible status to inflammasome activation
The effect of microglial cbs deficiency on NLRP3 inflammasome and IL-1β release was also verified in vitro. First, we used si-cbs to knockdown cbs in BV2 microglial cell line. A significant increase of supernatant IL-1β level was observed in si-cbs-transfected cells compared with scramble small interfering RNA (siRNA)-transfected cells following ATP stimulation, as consistently revealed by Western blot (Fig. 9A) and ELISA (Fig. 9B). Remarkably, cbs knockdown caused elevations in both pro- and mature IL-1β protein levels in the lysate fraction, although the mature form was undetected in the supernatant of ATP-unstimulated group.
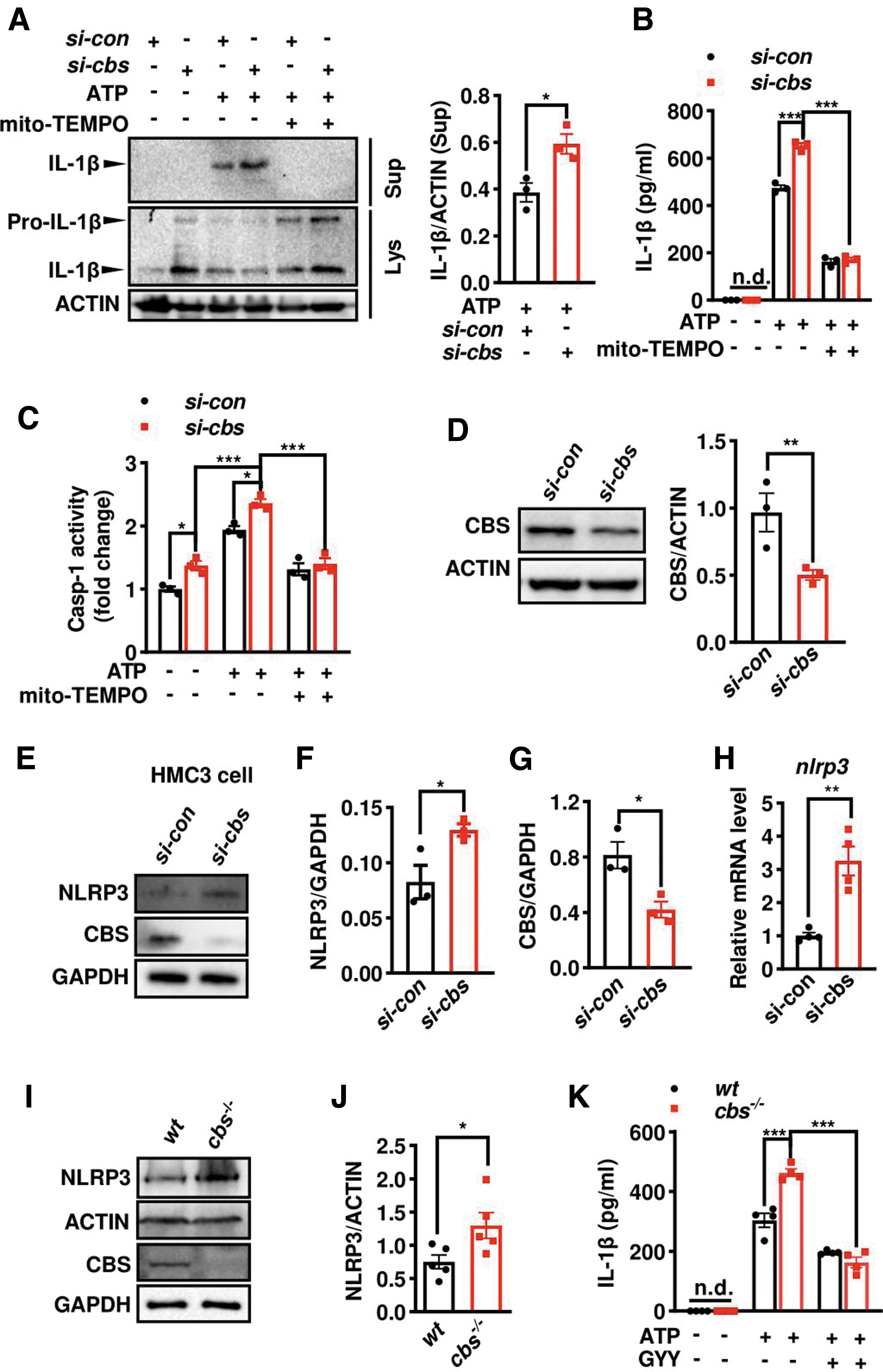
Also, a higher caspase-1 activity was found in cbs-knockdown cells than that in controls, regardless of ATP stimulation or not (Fig. 9C). Moreover, cbs silencing-induced elevations in IL-1β secretion and caspase-1 activity were completely blocked by mito-TEMPO, a mitoROS scavenger, consistent with previous reports that mitoROS was critical for inflammasome activation. The efficiency of cbs silencing was verified by Western blot, which reached ∼50% relative to scramble siRNA transfection (Fig. 9D). The findings were confirmed in human-derived microglial cell line. NLRP3 protein and mRNA levels elevated in cbs-knockdown HMC3 cells compared with controls (Fig. 9E–H).
We also verified the findings in wild-type (wt) and cbs-deficient (cbs− /− ) primary microglia cultures prepared from neonatal cbs fl/fl and Cx3cr1 Cre; cbs fl/fl pups, respectively. Western blotting confirmed the absence of CBS band in cbs− /− microglia. An elevated NLRP3 protein level was observed in cbs− /− microglia (Fig. 9I, J), consistent with a mild increase of NLRP3 in SAL-treated cbs cKO mice compared with cbs fl/fl controls (Fig. 5K). We did not detect IL-1β in cbs− /− microglia in the absence of ATP via ELISA. However, a higher IL-1β level was found in cbs− /− microglia compared with wt microglia after ATP stimulation, which was alleviated by GYY treatment (Fig. 9K).
CBS inhibited NLRP3 inflammasome via lowering mitoROS levels
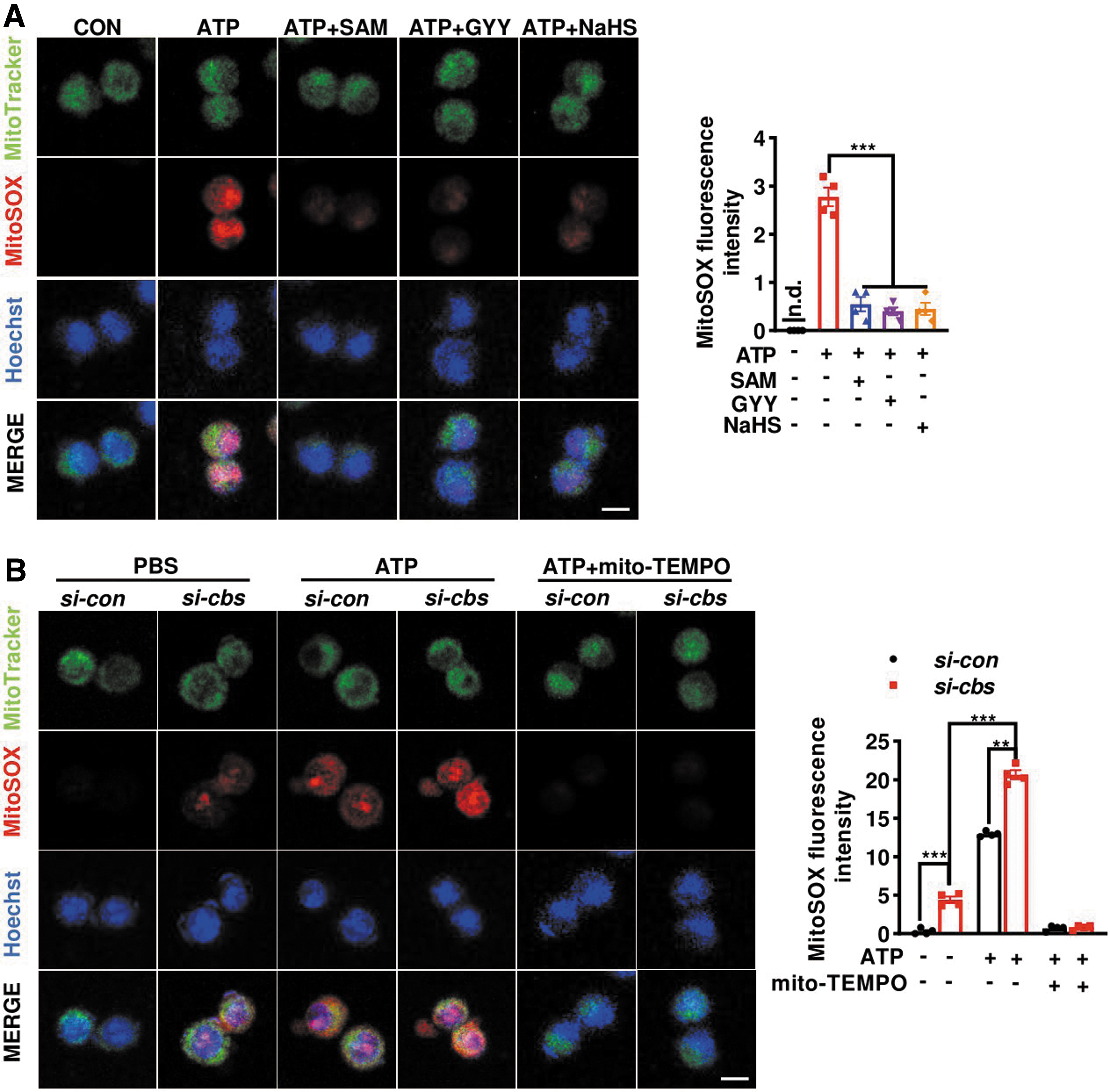
Finally, we explored the mechanism by which CBS-H2S axis suppressed NLRP3 inflammasome assembly. Previous studies reported a critical role of mitoROS in triggering inflammasome activation (Banoth and Cassel, 2018; Zhou et al., 2011). The above-mentioned data in Figure 4 pointed to a disruption of redox homeostasis in microglia due to cbs depletion. Therefore, mitoROS level was monitored by measuring the fluorescence of mitoSOX, a mitochondrial superoxide indicator. As reported, the microglial mitoSOX intensities dramatically increased at 0.5 h after ATP stimulation.
The increase was attenuated not only by SAM but also by GYY and NaHS pretreatment (Fig. 10A). In contrast, cbs knockdown resulted in higher mitoROS levels in both phosphate-buffered saline (PBS)- and ATP-stimulated microglia. Moreover, ATP- and cbs silencing-induced increases in mitoSOX intensity were blocked by mito-TEMPO, confirming the involvement of mitoROS (Fig. 10B). Collectively, the data suggest that CBS-mediated transsulfuration pathway may be critical in maintaining the redox homeostasis in microglia and that dysregulation of CBS expression may result in mitoROS generation and promote inflammasome activation in response to inflammatory challenges.
Discussion
H2S is a signaling molecule and its releasing compounds have been reported to exert multiple functions in various tissues. However, its physiological role is yet to be determined. The exact quantity of H2S in real conditions is unknown. Thus, the enzymes responsible for endogenous H2S production have received attention in recent years. H2S can be produced from cysteine by CBS, CSE, and 3-MST. Notably, the enzymes distribute in a tissue- and cell-specific manner. CSE is predominant in the cardiovascular system, whereas CBS and 3-MST are abundant in the brain. CBS expression changes temporally in the brain, where it declines in the late embryonic stage but increases after birth (Robert et al., 2003). In adult mouse brain, CBS is ubiquitously expressed but enriched in the Purkinje cell layer and hippocampus. We and other groups previously reported an enriched CBS expression in microglia (Du et al., 2014; Zhao et al., 2017).
Yet, its role in microglia is poorly defined. In this study, we provided the in vivo and in vitro evidence that microglial CBS expression declined in response to LPS stimulation. 3-MST was unaffected. Using microglial cbs cKO mice and cbs-deficient microglia, we demonstrated that cbs depletion promoted NLRP3 inflammasome activation and IL-1β secretion by enhancing mitoROS generation, whereas cbs overexpression or activation, as well as H2S donors, inhibited NLRP3 inflammasome activity. Moreover, cbs depletion augmented microglial activation and DA neuron losses in the SN of LPS-treated mice, which mimicked the neuroinflammation-related pathological changes in PD. Our study suggests that microglial CBS may serve as a target to suppress the NLRP3 inflammasome and that upregulation of CBS can be beneficial for PD (summarized in Fig. 1).
An impact of H2S on inflammasome was reported previously. For instance, H2S inhibited the NLRP3 inflammasome in bone marrow-derived macrophages, and higher NLRP3 inflammasome activity and IL-1β yield were observed in cse-deficient bone marrow-derived macrophages in response to stimuli (Castelblanco et al., 2018). However, little is known regarding the effect of CBS on inflammasome. The CBS activator SAM inhibited the NLRP3 inflammasome cascade and restored neurological deficits at 1 day postcerebral hemorrhage by suppressing microglial purinergic P2X7 receptor expression (Zhao et al., 2017). The mechanism may not be limited to this. In this study, we showed that SAM and H2S donors also attenuated microglial NLRP3 inflammation triggered by MSU and nigericin, which caused inflammasome activation independent of P2X7.
Instead, mitoROS, a critical regulator of the inflammasome, may be involved. We found that mitoROS level was enhanced in cbs-deficient cells but reduced in SAM- or H2S donor-treated cells. The elevations in IL-1β secretion and caspase-1 activity in ATP-stimulated cbs-deficient microglia were abrogated by mito-TEMPO. We constructed the tamoxifen-induced microglial cbs cKO mice and identified the role of microglial CBS in regulating redox homeostasis and inflammasome activity in the brain. We provided the evidence that cbs ablation may trigger microglia into a preactivated status as pro-IL-1β and NLRP3 expressions already increased in cbs-depleted murine and human-derived microglia even without stimulation. CBS may act as a brake-like molecule, orchestrating the redox homeostasis and inflammatory status in microglia.
NLRP3 inflammasome activation involves two steps, priming and complex assembly. Priming is generally triggered by pattern recognition receptor such as TLR4 signaling in response to LPS stimulation, which results in the transcription of NLRP3 and pro-IL-1β/18 via NF-κB activation. The inflammasome assembly is activated by numerous molecular or cellular events, including mitochondrial dysfunction, ion homeostasis disruption, and lysosomal damage. This process is accompanied by rapid relocations of NLRP3 and ASC into a singular, perinuclear, punctate “speck” structure of ∼1 μm in diameter. Interestingly, a recent study demonstrated that the ASC speck and NLRP3 inflammasome were spatially and temporally distinct (Nagar et al., 2021).
The speck was a dynamic structure that amplified the NLRP3 response to weak stimuli by facilitating the formation and release of small NLRP3: ASC complexes, which in turn activated caspase-1. ASC plays an essential role in small NLRP3 clusters assembly into the large speck (Chen and Chen, 2018). We found that NLRP3 transcription and translation consistently enhanced on day 3 after LPS treatment, implying that the transcriptional priming of NLRP3 is mainly involved. Moreover, NLRP3 expression rapidly declined on day 7 and 14. This was consistent with the immediate responses of TLR4/MyD88 signaling triggered by LPS.
Likely, the LPS-induced rapidly increased transcription and expression of NLRP3 may be sufficient for the “threshold” to recruit and associate with ASC for inflammasome assembly. Supportively, we observed that ASC protein level also increased early at day 3, but it persisted to be elevated even at day 7 and 14 after LPS injection. As an adaptor protein, ASC brings together NLRP3 and caspase-1 and facilitates the perinuclear “speck” assembly via its oligomerization. The damaged DA neurons caused by LPS-induced neuroinflammation may release a few molecules such as ATP to facilitate the inflammasome assembly and IL-1β release in the brain. Microglial NLRP3 inflammasome may be a sustained source of neuroinflammation that caused progressive DA neuron losses, which was initially triggered by a single injection of LPS into the SN.
Elevated IL-1β level was reported in the biofluids of PD patients compared with normal controls (Zhang et al., 2016; Zhou et al., 2016). Direct injection with tumor necrosis factor-α and IL-1β into the midbrain caused DA neuron losses in young adult rats (Carvey et al., 2005). Moreover, pharmacological inhibition of NLRP3 inflammasome or genetic depletion of NLRP3 or caspase-1 was shown to attenuate the PD-related pathologies in various murine models of PD (Gordon et al., 2018; Han et al., 2021; Lee et al., 2019). Excessive generation and release of IL-1β and other cytokines from activated microglia may produce damages to DA neurons, which can release ATP into extracellular space and then induce NLRP3 inflammasome assembly in the primed or activated microglia. This may form a vicious cycle. Hence, any strategy that inhibits inflammasome or blocks the IL-1β activity may be of potential to retard PD progression.
In this study, we showed that CBS, but not CSE or 3-MST, was downregulated in LPS-stimulated microglia, consistent with the previous report (Yuan et al., 2018). Indeed, CSE predominantly expresses in the cardiovascular system. Although 3-MST was reported to be abundant in the brain and localize to neurons in several brain regions (Shibuya et al., 2009), its distribution in glial cells is yet to be determined. We identified the expression of 3-MST in both murine- and human-derived microglial cell lines via Western blot. Yet, 3-MST protein level did not change in LPS-stimulated microglia. Altered microglial CBS was also demonstrated in cerebral ischemia (Zhang et al., 2017), hemorrhage, and traumatic brain injury (Zhang et al., 2013). This strongly suggests a unique role of CBS in microglia. But the transcription factors that regulate cbs expression are poorly defined. In this study, we found cbs mRNA levels decreased at 6 h, and this decline persisted till 24 h in LPS-stimulated microglia. However, a transient increase of NLRP3 transcription and expression followed by gradual declines was observed in LPS-stimulated BV2 and HMC3 microglia. It is well known that LPS-induced TLR4 signaling triggers nlrp3 transcription via NF-κB. Therefore, we speculated that NF-κB or other transcription factors triggered by various cytokines may negatively regulate cbs transcription. This warrants to be studied further.
H2S was demonstrated to exert neuroprotection in PD via the antioxidant and anti-inflammatory effects (Hu et al., 2010; Kida et al., 2011; Yuan et al., 2018). In 2009, Snyder and colleagues revealed for the first time that H2S signals through protein S-sulfhydration (Mustafa et al., 2009). They further reported the modification of parkin (a PD-related gene encoding protein) by H2S, which enhanced its catalytic activity as a ubiquitin E3 ligase. Parkin sulfhydration was shown to be markedly reduced in the brains of patients with PD, which may account for its contribution in sporadic PD (Vandiver et al., 2013).
Intriguingly, CBS expression and H2S production were reported to be reduced in the astrocytes and brains of DJ-1 (another important PD-related gene)-knockout mice, although the mechanism was unknown (Bae et al., 2013). All these link CBS-H2S signaling to PD. Disrupted CBS-H2S axis in microglia, which led to redox imbalance and inflammatory responses, may at least in part contribute to the pathogenesis of PD. Hence, CBS activators may have the potential to produce benefits for PD. Currently, there is a lack of specific agonists for CBS. So, more efforts are warranted to be put into this field.
In humans, cbs deficiency or mutation is one of the most common causes of hHcy, a risk factor for stroke and neurodegeneration. The mice with cbs homogeneous depletion showed a high mortality at 1 month after birth (Robert et al., 2005; Vitvitsky et al., 2004). Hence, conditional knockout or knock-in of cbs in specific cells is powerful for a deeper understanding of CBS function in health and disease. In the present study, we did not find alterations in the serum or brain Hcy levels of tamoxifen-treated microglial cbs cKO mice. However, an increase in GSSG, along with a lower ratio of GSH to GSSG, was observed in the brains of cbs cKO mice, indicating the occurrence of oxidative stress due to microglia-specific cbs depletion. Consistently, we found elevated mitoROS levels in cbs-deficient microglia. ATP caused more robust IL-1β secretion in these microglia, and this was partially ameliorated by the H2S donor.
Intriguingly, CBS not only contributes to H2S synthesis but also participates in the transsulfuration pathway of Hcy conversion to cysteine, which is the common precursor of sulfur molecules, including H2S, GSH, and taurine (Sbodio et al., 2019). H2S inhibits oxidative stress, and GSH serves as a major antioxidant in the brain. These two molecules may play a role in reducing mitoROS and thus suppressing NLRP3 inflammasome in microglia responding to various stimuli. In addition to H2S and GSH, taurine is indeed an enigmatic amino acid, which has been shown to modulate endoplasmic reticulum stress, Ca2+ homeostasis, and neuronal activity. It displayed the potential in ameliorating the changes against several neurological disorders (Jakaria et al., 2019). Moreover, a lower taurine level was reported in the cerebrospinal fluid of PD patients compared with age-matched controls (Engelborghs et al., 2003). Therefore, the exact role of CBS in the brain, which is closely related to the functions of these sulfur molecules, remains to be intensively explored.
In summary, this study identifies a role of CBS in suppressing NLRP3 inflammasome and IL-1β generation in microglia via the regulation of redox homeostasis using in vitro and in vivo studies with tamoxifen-induced cbs cKO mice. The findings provide the evidence as to how CBS controls the redox state in microglia and modulates neuroinflammation in a PD mouse model, and suggest that CBS may be a promising target for the treatment of neuroinflammation-related diseases such as PD.
Materials and Methods
Animals and stereotaxic surgery
To generate mice with microglia-specific cbs depletion, Cx3cr1 Cre mice (#025524) or Cx3cr1 CreERT2 mice (#020940), both kindly gifted by Prof. Jian Cheng (Soochow University), were crossbred with cbs fl/fl mice (GemPharmatech, Nanjing, China) followed by genotyping. The F1 offspring (Cx3cr1 Cre; cbs fl/+ or Cx3cr1 CreERT2; cbs fl/+) were crossbred with cbs fl/fl mice to obtain microglia-specific cbs-knockout mice (Cx3cr1 Cre; cbs fl/fl, cKO) or tamoxifen-inducible cKO (Cx3cr1 CreERT2; cbs fl/fl), respectively, and their littermates cbs fl/fl. Cx3cr1 CreERT2; cbs fl/fl mice (3-month-old) were i.p. administered tamoxifen (75 mg/kg/day; Sigma) for 5 days and used for studies 2 weeks later. C57BL/6 mice (6–8 weeks, male) were purchased from Shanghai Laboratory Animal Center and housed in a specific pathogen free-grade animal room with food and water ad libitum. Experimental procedures were performed according to the guidelines of the Institutional Animal Care and Use Committee of Soochow University.
For surgery, all mice were anesthetized with ketamine (100 mg/kg)/xylazine (10 mg/kg), placed in a stereotaxic apparatus, and bilaterally injected with LPS (7.5 μg/1.5 μL/site) or SAL into the SN with coordinates relative to bregma: anterior–posterior, −3.00 mm; medial–lateral, ±1.25 mm; dorsal–ventral, +4.50 mm at a rate of 0.2 μL/min. To explore the potential effect of cbs overexpression on PD-related neuroinflammation, recombinant adeno-associated virus encoding cbs (AAV-cbs; OBIO, Shanghai, China) or its vector (AAV-vec) was delivered into the SN as previously described (Yuan et al., 2018) 2 weeks before LPS or SAL injection.
Western blotting
Cell and tissue lysates were prepared using RIPA buffer with protease inhibitor cocktail, as previously described (Yuan et al., 2018). Lysates were separated by 10% sodium dodecyl sulfate–polyacrylamide gels, transferred onto polyvinylidene fluoride membranes, and then blocked in 5% nonfat powdered milk in 0.1% Tris-buffered SAL/Tween 20 for 1 h. Next, membranes were individually incubated with the following primary antibodies: anti-CBS (1:1500, 14787-1-AP; Proteintech, China), anti-CSE (1:500, SAB1405673; Santa Cruz), anti-3-MST (1:1000, A11587; Abclonal, China), anti-TLR4 (1:1000, SC-293072; Santa Cruz), anti-NLRP3 (1:1000, 15101S; Cell Signaling), anti-Pro/cleaved-IL-1β (1:200, I3767; Sigma), anti-ASC (1:1000, #67824; Cell Signaling), anti-Pro/cleaved-Caspase-1 (1:500, SC-56036; Santa Cruz), anti-Flag (1:1000, YM3001; Immunoway), anti-ACTIN (1:5000, SC-47778; Sigma), and anti-GAPDH (1:10,000, 60004-1-IG; Proteintech) at 4°C overnight.
After that, the membranes were washed and incubated with the corresponding secondary antibody (Jackson ImmunoResearch Lab) for 1 h at room temperature. Protein bands were detected using a chemiluminescence kit (P10300; NCM Biotech, China), and densitometric analysis was performed using the ImageJ software.
For supernatant IL-1β blotting, culture supernatants were harvested and centrifuged at 200 g for 10 min to remove floating cells. The resulting supernatant (∼600 μL) was then added to an equal volume of ice-cold methanol and 1/4 volume of chloroform, followed by vigorous vortex and spinning at 15,000 rpm for 5 min in a precooled centrifuge. Next, methanol was added to the pellet and vortexed again, followed by centrifugation at 15,000 rpm for 5 min at 4°C. The sediment was incubated in a 37°C water bath for 5 min to remove methanol by volatilization. Finally, lysis buffer and 5 × loading buffer were added to the pellet for Western blotting, as described previously.
Quantitative polymerase chain reaction
Total RNAs were isolated from the SN and cells using the TRIzol method. The first-strand cDNA was synthesized using the cDNA synthesis kit (R323; Vazyme, China). Mouse and human quantitative polymerase chain reaction (qPCR) primers were provided by Genewiz (Suzhou, China).
qPCR was performed using the specific primers listed as follows: Mouse tlr4 (F5′-ATGGCATGGCTTACACCACC-3′, R5′-GAGGCCAATTTTGTCTCCACA-3′), mouse myd88 (F5′-TCATGTTCTCCATACCCTTGGT-3′, R5′-AAACTGCGAGTGGGGTCAG-3′), mouse nlrp3 (F5′-ATTACCCGCCCGAGAAAGG-3′, R5′-TCGCAGCAAAGATCCACACAG-3′), mouse gapdh (F5′-GAAGGTCGGTGTGAACGGAT-3′, R5′-AATCTCCACTTTGCCACTGC-3′), mouse cbs (F5′-CCTATGAGGTGGAAGGGATT-3′, R5′-TGTAGTTCCGCACAGAGTCA-3′), mouse il-1β (F5′-TACCAGTTGGGGAACTCTGC-3′, R5′-TGGAAAAGCGGTTTGTCTTC-3′), human NLRP3 (F5′-GATCTTCGCTGCGATCAACAG-3′, R5′-CGTGCATTATCTGAACCCCAC-3′), and human GAPDH (F5′-GGAGCGAGATCCCTCCAAAAT-3′, R5′-GGCTGTTGTCATACTTCTCATGG-3′). Relative levels of mRNA were assessed with Hieff® qPCR SYBR Green Master Mix Kit (11202ES08; Yeasen, China) and quantified using the 2−ΔΔCt method by normalization to mouse gapdh or human GAPDH.
Immunohistochemistry
For histological study, mice were euthanized and transcardially perfused with SAL, followed by 4% paraformaldehyde for fixation. The brains were then dissected, postfixed overnight, and dehydrated in an increasing gradient of 10%–30% sucrose solution. Coronal sections (18 μm) were cut with a cryostat (Leica, Wetzlar, Germany). For immunostaining and bright field visualization, sections were treated with 3% hydrogen peroxide for 30 min to quench endogenous peroxidase activity and then blocked in 5% bovine serum albumin (BSA) in PBS with 0.25% Triton X-100 for 1 h at room temperature.
Next, sections were incubated with anti-TH (1:1000, HPA061003; Sigma) at 4°C overnight. Following brief washes, sections were incubated with HRP-conjugated secondary antibody for 1 h, stained using a 3,3-diaminobenzidine detection kit (GK500710; Gene Tech, China), and photographed under a microscope (Carl Zeiss, Germany). The number of TH+ neurons in the SN pars compacta was counted using the “cell counter” plug-in in Fiji software and corrected by a researcher blinded to the testing groups. Every fifth section throughout the SN was counted, and at least three sections per animal were included for analysis.
For immunofluorescent staining, sections were directly blocked in 5% BSA/PBS containing 0.25% Triton X-100 and incubated with the following primary antibodies: anti-IBA1 (1:1000, 019-19741; Wako, Japan), anti-CBS (1:100, SC-67154; Santa Cruz), and anti-NLRP3 (1:200, 15101S; Cell Signaling). Next, sections were washed in PBS for 5 min three times and incubated with the appropriate Alexa Fluor 488- or Alexa Fluor 555-conjugated secondary antibody (Invitrogen) for 1 h at room temperature. After that, sections were washed again and mounted onto slides using Fluoroshield mounting medium with DAPI. All sections were observed and photographed under a fluorescence microscope or scanned by confocal microscopy (Carl Zeiss). Fluorescence intensities were quantified using the ImageJ software.
Cell culture and treatment
Primary microglial cultures were prepared from the brains of neonatal (P1–P3) pups, as previously described (Yuan et al., 2018). In brief, cortical tissues were dissected, and meninges were removed. Next, tissues were digested and filtered through a 70-μm strainer, followed by centrifugation at 1000 rpm for 10 min. The pellets were suspended and cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin–streptomycin. The medium was replaced every 3 days. Once they reached confluence, microglial cells were harvested by orbital shaking at 180 rpm for 2 h and seeded into culture dishes for further experimentation. The murine BV2 microglial cell line was maintained in DMEM with 10% FBS and 1% penicillin–streptomycin in a CO2 incubator at 37°C.
To induce NLRP3 inflammasome activation, cells were primed with 500 ng/mL LPS (Escherichia coli O55:B5, L2880; Sigma) for 3 h, washed and cultured for 12 h, followed by stimulation with ATP (2.5 mM, 30 min), nigericin (5 μM, 1 h), or MSU (250 μg/mL, 3 h). ATP and MSU were purchased from Sigma (A7699; U2875), whereas nigericin was purchased from Millipore (N7143). The CBS activator SAM (A4377; Sigma), the H2S slow-releasing donor morpholin-4-ium 4-methoxyphenyl (morpholino) phosphinodithioate (GYY) (SML2470; Cayman Chemical), and the H2S fast-releasing salt NaHS (161527; Sigma) were added 12 h before NLRP3 inflammasome stimulation.
Serum and brain Hcy level assay
For determination of the serum Hcy level, blood was collected from the eyeballs of mice under anesthesia and placed at 4°C overnight, followed by centrifugation to obtain serum samples. To quantify the brain Hcy level, whole brain tissues were isolated and added with PBS for full homogenization using a sonicator (JY92-IIDN; NingboXinYi, China), followed by centrifugation. Hcy levels in the resulting supernatant and serum were measured with the ELISA kit following the manufacturer's protocol (LV30834; Animal Union Biotechnology, China). The protein concentration of the brain lysate was determined using a bicinchoninic acid assay kit (23225; Thermo). The Hcy content in the serum and brain is presented as ng/mL and ng/mg protein, respectively. The latter was normalized by the protein concentration of the corresponding lysate.
Measurement of reduced and oxidized GSH
GSH content in the brain was measured according to the protocol provided by the GSH and GSSG Assay Kit (S0053; Beyotime, China). In brief, the hippocampus was homogenized with protein removal reagent at a ratio of 1:10 (w/v) and centrifuged at 10,000 g for 10 min at 4°C. For total GSH detection, 10 μL supernatant sample was added to 150 μL total GSH detection working solution and incubated at room temperature for 5 min. Then, 50 μL of 0.5 mg/mL NADPH solution was added and mixed. The absorbance at 412 nm was immediately measured with a microplate reader (TECAN Infinite M200 Pro, Switzerland). For the GSSG assay, the supernatant was first added to the GSH scavenger and incubated for 60 min, followed by the same procedures as described above for GSH detection. Total GSH or GSSG level was calculated from the standard curve, and the ratio of GSH/GSSG was shown as [GSH]/[GSSG].
H2S-generating activity assay
Brain H2S synthesis activity was determined using the protocol, as previously described (Yan et al., 2022). The brain tissues were isolated from cbs fl/fl and cbs cKO mice and added with 12% (w/v) PBS for full homogenization using a sonicator. The tissue homogenates were transferred into a 50-mL tube and cooled down on ice for 10 min. Then, 10 mM l-cysteine and 2 mM pyridoxal 5-phosphate were added into the tube. Next, 1.5-mL centrifuge tube with a piece of filter paper was soaked in 1% zinc acetate solution and placed in the 50-mL tube.
The tube was filled with nitrogen gas for 20 s and then immediately capped and shaken on a 37°C shaker for 90 min. Next, trichloroacetic acid solution (50%) was added to each sample and reacted with zinc acetate. After incubation for 1 h, N,N-dimethyl-p-phenylendiamine sulfate (20 mM) in HCl (7.2 M) and FeCl3 (30 mM) in HCl (1.2 M) were added, and the absorbance of the solutions was measured at a wavelength of 670 nm (TECAN Infinite M200 Pro). H2S production was calculated against a standard curve of NaHS solution (3.9–250 μM). An aliquot of the homogenate was used for protein concentration measurement using a bicinchoninic acid assay kit (23225; Thermo). Data were normalized by the protein concentration and presented as nmol/g/min.
IL-1β measurement
The IL-1β levels in the culture supernatants and tissue lysates were determined using ELISA kits (88-7013-88; eBioscience) according to the manufacturer's instructions.
Adult microglial isolation and il-1β mRNA quantification
The adult microglial population was acutely isolated from cbs fl/fl and cbs cKO mice as we recently reported (Tu et al., 2021). In brief, mice were anesthetized and perfused with 50 mL PBS. Whole brains were immediately dissected and homogenized in 5 mL Hank's balanced salt solution in a glass homogenizer. The microglial population was isolated from the brain homogenate using a 30%–70% Percoll gradient after myelin debris removal. Total RNA was then extracted, reverse transcribed into cDNA, and subjected to a qPCR assay.
ASC speck detection by immunostaining
For ASC speck visualization in vitro, cells on coverslips were briefly washed in PBS and fixed with 4% paraformaldehyde. Next, cells or brain sections were incubated with 5% BSA with 0.25% Triton X-100 in PBS for 1 h and then probed with anti-ASC (1:500, #67824; Cell Signaling) at 4°C overnight. Thereafter, cells or sections were washed in PBS three times and incubated with Alexa Fluor 488- or Alexa Fluor 555-conjugated secondary antibody for 1 h at room temperature. Finally, the coverslips were mounted onto slides with Fluoroshield mounting medium containing DAPI (H-1200; Vector Laboratories). Images were taken under a confocal microscope (LSM700; Carl Zeiss), and quantification was performed by calculating the percentage of cells with singular and speck-like (1–10 μm in diameter) ASC-positive signals using the ImageJ software.
Open field test
Open field test was performed in a square arena (40 × 40 × 40 cm) with black walls and the white floors, divided into 16 equal parts. The four squares in the center were defined as the central region, whereas the other squares were defined as the peripheral regions. Mice were habituated for at least 1 h before the experiment. For testing, mice were placed in the center facing the same direction and allowed to explore freely for 10 min in a quiet and undisturbed room. Mouse activities were tracked and recorded by ANY-Maze system (Stoelting). The open field arena was thoroughly cleaned with 70% ethanol between each trial.
Rotarod test
Rotarod test was used to assess motor coordination and balance in mice. The rotarod apparatus consisted of a rotating rod (3 cm in diameter) with a nonslip surface. Mice were habituated to the rotarod apparatus at 3 days before the experiment. Each mouse was placed on the rotating rod, which rotated at a constant speed of 20 rpm over a period of 5 min. On the testing day, each mouse was placed on the rotating rod, and the rod was then accelerated from 0 to 40 rpm within 5 min. The latency to fall off the rod was recorded. Each mouse was tested for three trials with a 30-min interval. The average latency to fall of each mouse was then calculated for further analyses.
Pole climbing test
The pole apparatus consisted of a vertical pole (50 cm in height and 1 cm in diameter) with a rough surface. The base of the pole was covered with bedding to prevent the injury due to accidental drop. Mice were gently placed on the top of the pole, and the time taken to descend to the floor was recorded. Each mouse was tested for three trials, with a 30-min interval. The average time was calculated for further analyses.
Genetic manipulation of the cbs gene in vitro
To induce cbs overexpression, BV2 cells were infected with lentivirus encoding cbs (Lenti-cbs-flag) or its vector (OBIO) at a multiplicity of infection of 20. For cbs knockdown in primary microglia and human-derived microglia cell line HMC3, siRNAs targeting cbs (5′-CAGAUAUUCUGAGGAAAAUTT-3′ and 5′AUUUUCCUAGAAUACUCUGTT-3′) and scramble siRNA (GenePharma, Shanghai, China) were transfected using HiPerFect Transfection Reagent (301704; Qiagen, Germany) for 72 h.
Detection of H2S content in BV2 cells using HSip DA probe
BV2 cells were seeded into a 24-well plate and infected with lentivirus-carried cbs gene or vector on the second day. H2S content was detected according to previous report (Sasakura et al., 2011), using HSip-1 DA probe (SB22; Dogindo, China) at 60 h postinfection. Briefly, cells were washed twice with serum-free DMEM. Then, the HSip-1 DA stock solution (1 mM) was diluted with serum-free DMEM medium, and 200 μL of HSip-1 DA working solution (5 μM) was added to the wells and incubated at 37°C for 30 min. After that, the supernatant was removed, and cells were washed twice with 1 × PBS before observed and imaged using confocal fluorescence microscopy (Zeiss, Germany). The intracellular H2S content was indicated by HSip-1 DA fluorescence intensity.
Caspase-1 activity assay
Caspase-1 activity in the cell lysates was evaluated by measuring the cleavage of the caspase-1 substrate YVAD-AFC using a commercial caspase-1 assay kit from BioVision (K110-100) according to the manufacturer's instructions. The substrate cleavage was monitored by measuring the emission signal at a wavelength of 505 nm with a 400 nm fluorescence excitation on a fluorescence multi-well plate reader (TECAN Infinite M200 Pro).
MitoROS measurement
MitoROS were assessed using the fluorescent probe mitoSOX (M36008; Invitrogen). Five micromolars mitoSOX, along with 0.5 μM MitoTracker Green (M7514; Invitrogen), was added to the cells and incubated at 37°C for 15 min. Next, cells were washed with PBS twice and stained with 10 μM Hoechst 33342 (H1399; Sigma) for 15 min. Images were taken under an LSM700 confocal microscope (Zeiss), and the fluorescence intensity was calculated using the ImageJ software.
Statistical analysis
Data are presented as the mean ± standard error of the mean of at least three independent experiments. The statistical significance was evaluated using GraphPad Prism 8. The two-group differences were compared using Student's unpaired t-test and one-way analysis of variance (ANOVA) for multiple groups under single variance or two-way ANOVA for multiple groups under two variances followed by Tukey's post hoc analysis. The significance level was defined as p < 0.05. Electronic laboratory notebook was not used.
Author's Contributions
L.-F.H. and X.-O.H. conceived and designed the study. Y.-J.M., X.Y., and Y.-T.M. performed the experiments. M.W. and Y.L. involved in primary microglia culture. C.-S.P. isolated the adult microglia. C.-F.L. contributed to the revision of the article. L.-F.H wrote the article. L.-F.H. and X.-O.H. provided the funding support. All authors read and approved the submitted version.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the National Natural Science Foundation of China (81870997, 82104190, 82171251), the Key Project of Natural Science Foundation of Jiangsu Provincial Higher Education Institutions (No. 21KJA180003), and the Six Major Talents Peak in Jiangsu Province (YY-053). This study was also partly supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Jiangsu Province's Young Medical Talents Program (QNRC2016872), and the Discipline Construction Program of the Second Affiliated Hospital of Soochow University (XKTJTD202004).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.