
Abstract
Aims:
Lipid peroxidation occurring in lung adenocarcinoma (LUAD) cells leads to ferroptosis. Lysophosphatidylcholine acyl-transferase 3 (LPCAT3) plays a key role in providing raw materials for lipid peroxidation by promoting esterification of polyunsaturated fatty acids to phospholipids. Whether LPCAT3 determines ferroptosis sensitivity and the mechanism by which its expression is regulated in LUAD has not been reported.
Results:
LPCAT3 and acyl-coenzyme A (CoA) synthetase long-chain family member (ACSL)4 levels were positively associated with ferroptosis sensitivity in LUAD cell lines. Overexpression of LPCAT3 and ACSL4 sensitized LUAD cells to ferroptosis, while LPCAT3 and ACSL4 knockout showed the opposite effect. Zinc-finger E-box-binding (ZEB) was shown to directly bind the LPCAT3 promoter to stimulate its transcription in a Yes-associated protein (YAP)-dependent manner. An interaction between YAP and ZEB was also observed. E1A-binding protein p300 (EP300) simultaneously bound with YAP and ZEB, and induced H3K27Ac for LPCAT3 transcription. This mechanism was verified in primary LUAD cell and xenograft models. The ACSL4, LPCAT3, and YAP combination can jointly determine LUAD ferroptosis sensitivity.
Innovation:
The binding site of ZEB exists in the −1600 to −1401 nt region of LPCAT3 promoter, which promotes LPCAT3 transcription after ZEB binding. ZEB and YAP bind, and the ZEB zinc-finger cluster domain and YAP WW domain are crucial for their binding. EP300 may bind with YAP via its Bromo domain and with ZEB via its CBP/p300-HAT domain. In addition, the combination of ACSL4, LPCAT3, and YAP to determine ferroptosis sensitivity of LUAD cells is better than prostaglandin-endoperoxide synthase 2 (PTGS2), transferrin receptor (TFRC), or NADPH oxidase 1 (NOX1).
Conclusion:
LPCAT3 transcription is regulated by YAP, ZEB, and EP300. LUAD ferroptosis sensitivity can be determined by the combination of ACSL4, LPCAT3, and YAP. Antioxid. Redox Signal. 39, 491–511.
Introduction
Ferroptosis, an iron-dependent form of regulated cell death that was first identified in 2012, is induced by intracellular lipid peroxidation (Dixon et al., 2012). Acyl-CoA synthetase long-chain family member (ACSL)4 and lysophosphatidylcholine acyl-transferase 3 (LPCAT3) play an irreplaceable role in the formation of lipid peroxides. Arachidonic acid (AA) and adenosine deaminase (AdA) are the two most critical polyunsaturated fatty acids (PUFAs) that serve as raw materials for lipid peroxidation (Kagan et al., 2017).
ACSL4 catalyzes the addition of coenzyme A (CoA) to AA and AdA (Kuwata and Hara, 2019), and LPCAT3, a member of the lysophospholipid acyl-transferase family, promotes the esterification of AA and AdA to saturated lysophospholipids including lysophosphatidylcholine, lysophosphatidylethanolamine, and lysophosphatidylserine by participating the Lands cycle (Ichu et al., 2020; Reed et al., 2022; Thomas et al., 2018). Coupled with an additional step of peroxidation by LOXs, lipid peroxides can be generated to induce ferroptosis (Sun et al., 2021).
Innovation
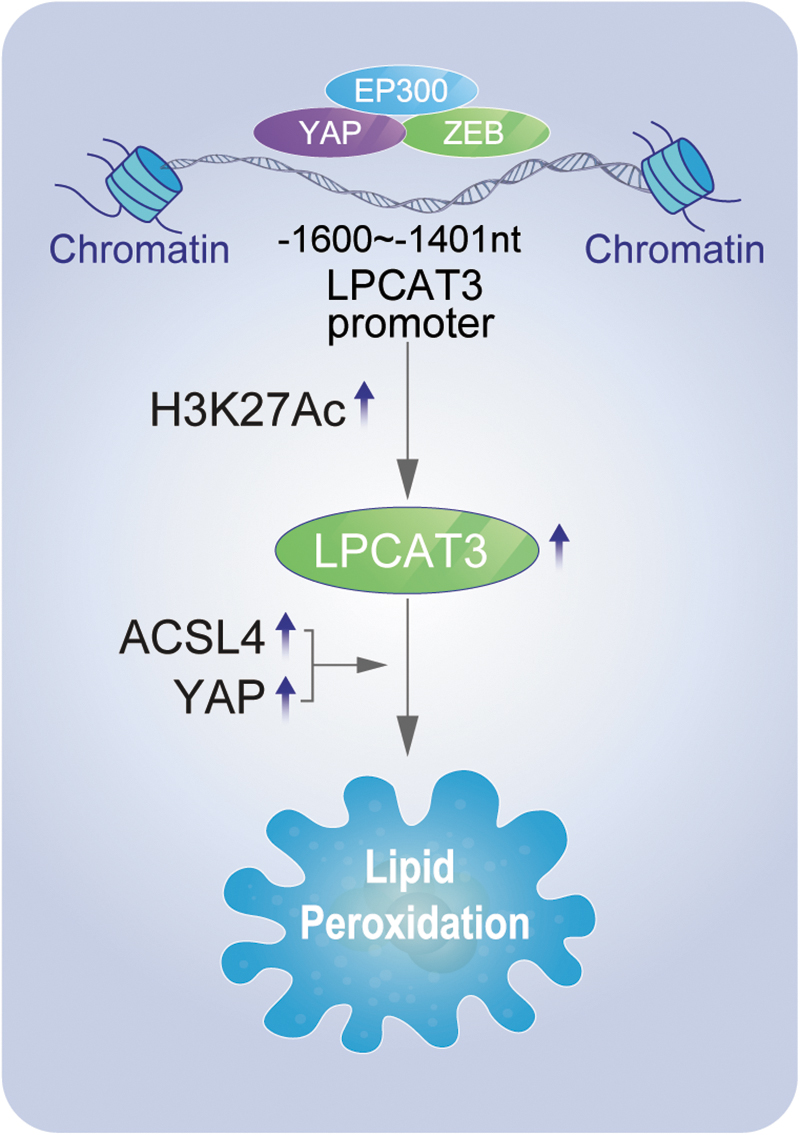
We dissected in detail the molecular mechanism by which zinc-finger E-box-binding (ZEB) and Yes-associated protein (YAP) promoted lysophosphatidylcholine acyl-transferase 3 (LPCAT3) transcription. Our results showed that the LPCAT3 promoter contained a ZEB-binding site in the −1600 to −1401 nt region. ZEB bound at this site and initiated transcription of the LPCAT3 gene. YAP acted as a cotranscriptional factor to promote ZEB-mediated transcription of LPCAT3. ZEB and YAP interacted via the zinc-finger cluster domain of ZEB and the WW domain of YAP.
The E1A-binding protein p300 (EP300) histone acetyltransferase bound to YAP and ZEB simultaneously through its Bromo and CBP/p300-HAT domains; it induced H3K27Ac to promote the transcription of LPCAT3. Both LPCAT3 and acyl-coenzyme A (CoA) synthetase long-chain family member (ACSL)4 were positively correlated with ferroptosis sensitivity of lung adenocarcinoma (LUAD) cells. Finally, the combination of ACSL4, LPCAT3, and YAP can better judge the ferroptosis sensitivity of LUAD cells than other markers. Therefore, this study has clinical application potential.
In addition, Fe2+ promotes the formation of reactive oxygen species (ROS) through the Fenton reaction, and is indispensable for lipid peroxidation. Sufficient iron is thus necessary for ferroptosis (Hassannia et al., 2019; Yoshida et al., 2019). Yes-associated protein (YAP) promotes the transcription of both ACSL4 and transferrin receptor (TFRC), and is therefore an important promoter of ferroptosis (Wu et al., 2019).
Researchers have explored the application of ferroptosis in the clinical treatment of tumors. Sorafenib, a multikinase inhibitor, has shown efficacy against a wide variety of tumors (Pollard et al., 2022). One of the main mechanisms for Sorafenib to eliminate tumor cells is to cause ROS production and ferroptosis (Coriat et al., 2012; Galmiche et al., 2014). Other drugs that promote ferroptosis include erastin, RSL3, piperazine erastin, imidazole ketone erastin (IKE), sulfasalazine, FIN56, and ferroptocide, but they are rarely used for clinical cancer treatment (Liang et al., 2019).
One reason ferroptosis-related therapies may be a strategy for clinical tumor treatment is that ferroptosis is more likely to occur in tumor cells with a high degree of malignancy. In tumor cells with a high metastatic tendency, E-cadherin and downstream NF2 are inhibited and YAP activity increases; ACSL4 and TFRC transcription are then activated, resulting in increased ferroptosis sensitivity (Wu et al., 2019). Moreover, O-GlcNAcylation of YAP further enhances the stimulating effect of YAP on TFRC (Zhu et al., 2021).
Lung cancer is the most common cancer in the world and has a high mortality rate (Sung et al., 2021). Lung adenocarcinoma (LUAD) is the main type of lung cancer, accounting for ∼40% of lung cancer cases (Kim et al., 2020). The 5-year overall survival of patients with LUAD remains poor. Therefore, the identification of new treatment methods or adjuvant therapy is necessary to improve the treatment of LUAD patients (Liang et al., 2022; Wang et al., 2022a). Notably, promoting ferroptosis of LUAD cells has been confirmed to eliminate LUAD cells resistant to cisplatin, AZD9291, and icotinib (Zhang et al., 2021).
Interestingly, the association between YAP and ferroptosis is also confirmed in LUAD. When ferroptosis occurs, adenylate cyclase 10 (ADCY10) functions as an upstream regulator of YAP. Under the stimulation of glutamate, ADCY10 can generate cAMP through ATP, resulting in glutamine-fructose-6-phosphate transaminase 1 (GFPT1) being phosphorylated and inhibited by protein kinase A (PKA), inhibiting the O-GlcNAcylation of YAP.
In turn, it inhibits the transcription of ferritin, which is conducive to the conversion of intracellular bound iron into labile iron through ferritinophagy. The expression of ADCY10 and ferroptosis sensitivity are both increased with the increase of the malignancy of LUAD, and there is a significant positive correlation between them. Therefore, it is clear that ADCY10 may be used as a sensitive marker of ferroptosis (Zhang et al., 2021). The above findings indicated that the induction of ferroptosis in LUAD cells is a promising new therapeutic strategy or may be used as an adjuvant strategy to the existing treatments.
In addition, in LUAD, 15 ferroptosis-related genes including voltage-dependent anion channel 2 (VDAC2), glutaminase 2 (GLS2), fms-related receptor tyrosine kinase 3 (FLT3), toll-like receptor 4 (TLR4), phosphorylase kinase catalytic subunit gamma 2 (PHKG2), phosphogluconate dehydrogenase (PGD), pannexin 1 (PANX1), KRAS proto-oncogene (KRAS), phosphatidylethanolamine-binding protein 1 (PEBP1), arachidonate 15-lipoxygenase (ALOX15), arachidonate 12-lipoxygenase, 12R type (ALOX12B), ACSL3, CDGSH iron sulfur domain 1 (CISD1), FA complementation group D2 (FANCD2), and solute carrier family 3 member 2 (SLC3A2) can be combinedly used to determine the prognosis of LUAD, and the effect is better than that of any single indicator (Ren et al., 2021). Therefore, the identification of effective markers for ferroptosis in LUAD may help identify tumors that may be suitable for ferroptosis-related or targeted treatments, but existing indicators are still very lacking.
The Hippo signaling pathway, through a cascade of kinases, controls tissue growth and organ size in animals by regulating cell proliferation, division, and death (Pan, 2022). The YAP transcription regulator is the main effector molecule of this pathway in humans.
Large tumor suppressor kinase 1/2 (LATS1/2), activated by mammalian STE20-like kinases 1/2 (MST1/2), suppresses the activity of YAP by phosphorylation (Moya and Halder, 2019). However, YAP can be activated by the O-GlcNAcylation to promote the transcriptional enhancer factor domain family member (TEAD)-, transcription factor CP2 (TFCP2)-, and cyclic adenosine monophosphate response element-binding protein (CREB)–transcriptional systems, leading to overexpression of growth-related genes and even tumor development (Li et al., 2021; Wang et al., 2013; Zhang et al., 2017a; Zhang et al., 2017b).
Recent studies have shown that the Hippo/YAP pathway plays an important and complex role in tumor cell ferroptosis. YAP promotes the transcription of both ACSL4 and TFRC, and is therefore an important promoter of ferroptosis (Wu et al., 2019); and this finding provided mechanistic insights into the observations that cancer cells with mesenchymal or metastatic properties are highly sensitive to ferroptosis (Hangauer et al., 2017; Wu et al., 2019).
YAP also activates S-phase kinase-associated protein 2 (SKP2) and arachidonate lipoxygenase 3 (ALOXE3) transcription, and makes cells more prone to ferroptosis (Qin et al., 2021; Yang et al., 2021). With the increase of tumor malignancy, YAP can further enhance the expression of ferritin and enhance the iron reserve. In the presence of a ferroptosis stimulator, a large amount of labile iron is released, which benefits lipid peroxidation and ferroptosis (Wang et al., 2021b; Zhang et al., 2021). However, other studies have shown that YAP upregulates solute carrier family 7 member 11 (SLC7A11) expression, leading to inhibition of ferroptosis (Gao et al., 2021). YAP may thus regulate ferroptosis in both positive and negative ways. Better understanding of the mechanism by which YAP regulates ferroptosis may provide a more effective method for ferroptosis-related tumor treatments.
The objectives of this study were as follows: to (i) determine whether LPCAT3 can be used as a marker of ferroptosis sensitivity; (ii) explore the molecular mechanism of LPCAT3 regulation in LUAD; (iii) further analyze the relationship between Hippo/YAP signaling pathway and ferroptosis; and (iv) identify a combination of markers that effectively indicates ferroptosis sensitivity. In this study, we found that ACSL4 and LPCAT3 were significantly positively correlated with ferroptosis sensitivity.
The transcription of LPCAT3 and ACSL4 was activated by YAP and zinc-finger E-box-binding (ZEB), and ZEB directly interacted with the LPCAT3 promoter. ZEB directly bound with YAP, and the binding was mediated by their zinc-finger cluster (ZF) and WW domains. YAP- and ZEB-induced LPCAT3 transcription relied on E1A-binding protein p300 (EP300). EP300 may bind with YAP via its Bromo domain and bind with ZEB via its CBP/p300-HAT domain. This mechanism was verified in LUAD cell lines, primary cells, and nude mouse xenograft models.
We used the primary cell models to show that the combination of three markers, YAP, ACSL4, and LPCAT3, could assess the ferroptosis sensitivity of LUAD. Therefore, YAP, ACSL4, and LPCAT3 might be helpful for the ferroptosis-related diagnosis and treatment of LUAD.
Results
LPCAT3 and ACSL4 are positively related to ferroptosis sensitivity in LUAD
Ferroptosis sensitivity varies across various LUAD cell lines (Zhang et al., 2021). Here, we selected six LUAD cell lines, including A549, H358, H1299, H1650, PC9, and H1975, and treated them with erastin (a ferroptosis inducer that binds and inhibits VDACs [VDAC2/VDAC3], and directly inhibits the cystine/glutamate antiporter system XC − activity to reduce glutathione [GSH] level) (Dixon et al., 2012; Xie et al., 2016) to detect cell death and other ferroptosis-related indicators, including lipid ROS, labile iron, malondialdehyde (MDA), GSH, and 4-hydroxynonenal (HNE). The change of these indicators was the most significant in the H1975 cell line and the least significant in A549 cells after erastin treatment, indicating that among the six cell lines, H1975 cells had the greatest ferroptosis sensitivity, while A549 cells had the lowest ferroptosis sensitivity (Fig. 2A–F and Supplementary Fig S1A).
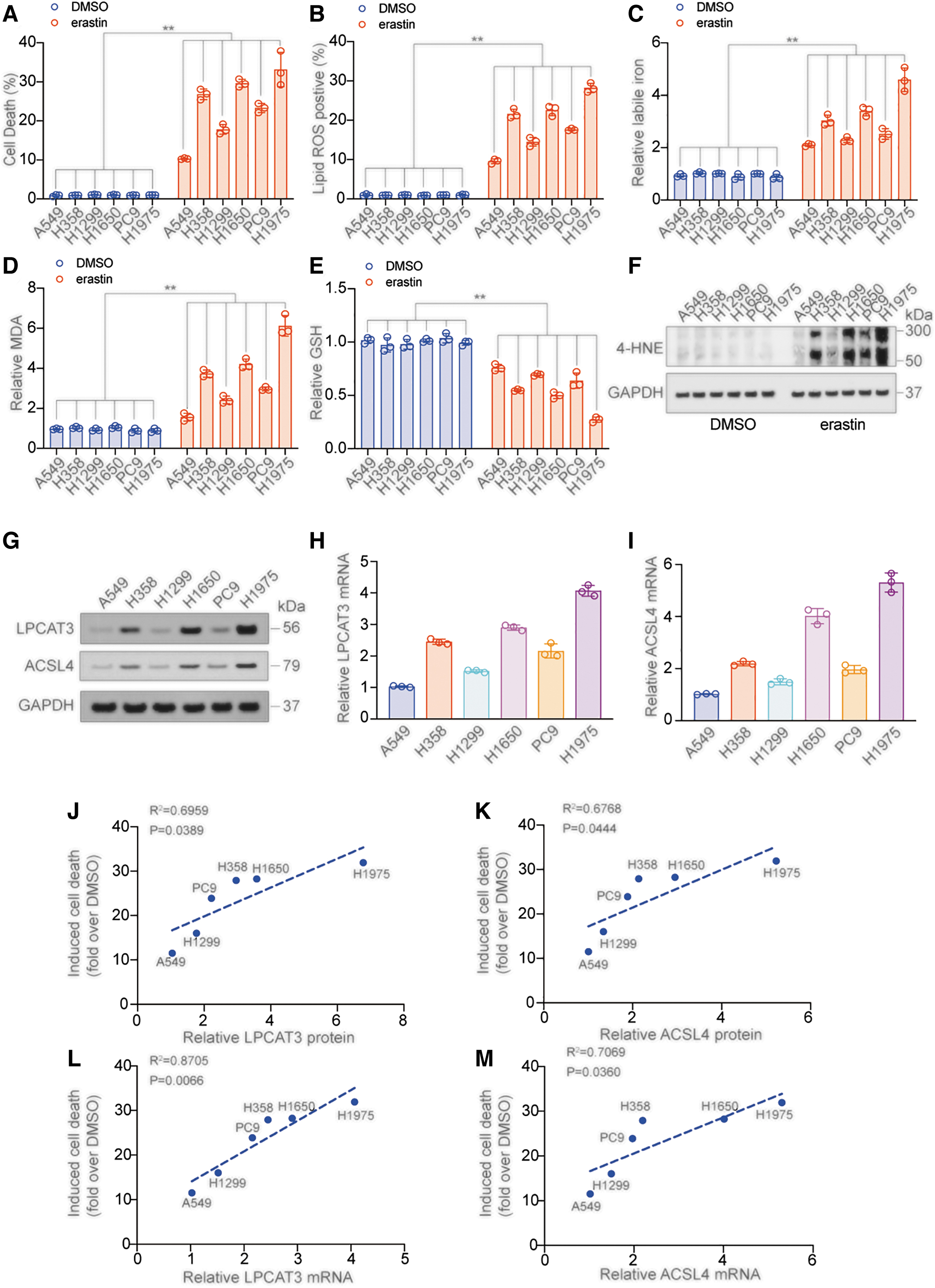
Previous studies have confirmed a positive correlation between the expression of ACSL4 and the ferroptosis sensitivity in cells (Kagan et al., 2017; Yuan et al., 2016). As LPCAT3 is a key enzyme in the next step of the ACSL4-catalyzed lipid peroxide reaction, we explored whether it also functions as a ferroptosis sensitivity marker. The mRNA and protein expressions of LPCAT3 and ACSL4 were the highest in H1975 cells and the lowest in A549 cells (Fig. 2G–I and Supplementary Fig. S1B, C), and there was a clear positive correlation between protein and mRNA expression in different cell lines (Supplementary Fig. S1D, E).
Furthermore, the protein and mRNA levels of LPCAT3 and ACSL4 in different cell lines significantly positively correlated with the cell death, the rise of lipid ROS, MDA, and labile iron, and the decline of GSH caused by erastin (Fig. 2J–M and Supplementary Fig. S1F–M). These results suggested that similar to ACSL4, LPCAT3 is likely to be an indicator for ferroptosis sensitivity.
Overexpression of LPCAT3 and ACSL4 promotes ferroptosis in LUAD
We next evaluated the effects of overexpression of LPCAT3 and ACSL4 on LUAD cell lines. In A549 and H1975 cells, overexpression of LPCAT3 and ACSL4 (Fig. 3A, B and Supplementary Fig. S2A) was positively correlated with erasin-induced cell death (Fig. 3C–F), lipid ROS (Fig. 3G–J), MDA (Supplementary Fig. S2B, C), and labile iron elevation (Supplementary Fig. S2D, E). YAP increases ferroptosis sensitivity by stimulating the transcription of genes such as ALOXE3, ACSL4, and TFRC (Qin et al., 2021; Wu et al., 2019). We also found that overexpression of YAP was positively correlated with erasin-induced cell death and lipid ROS (Supplementary Fig. S2F–I).
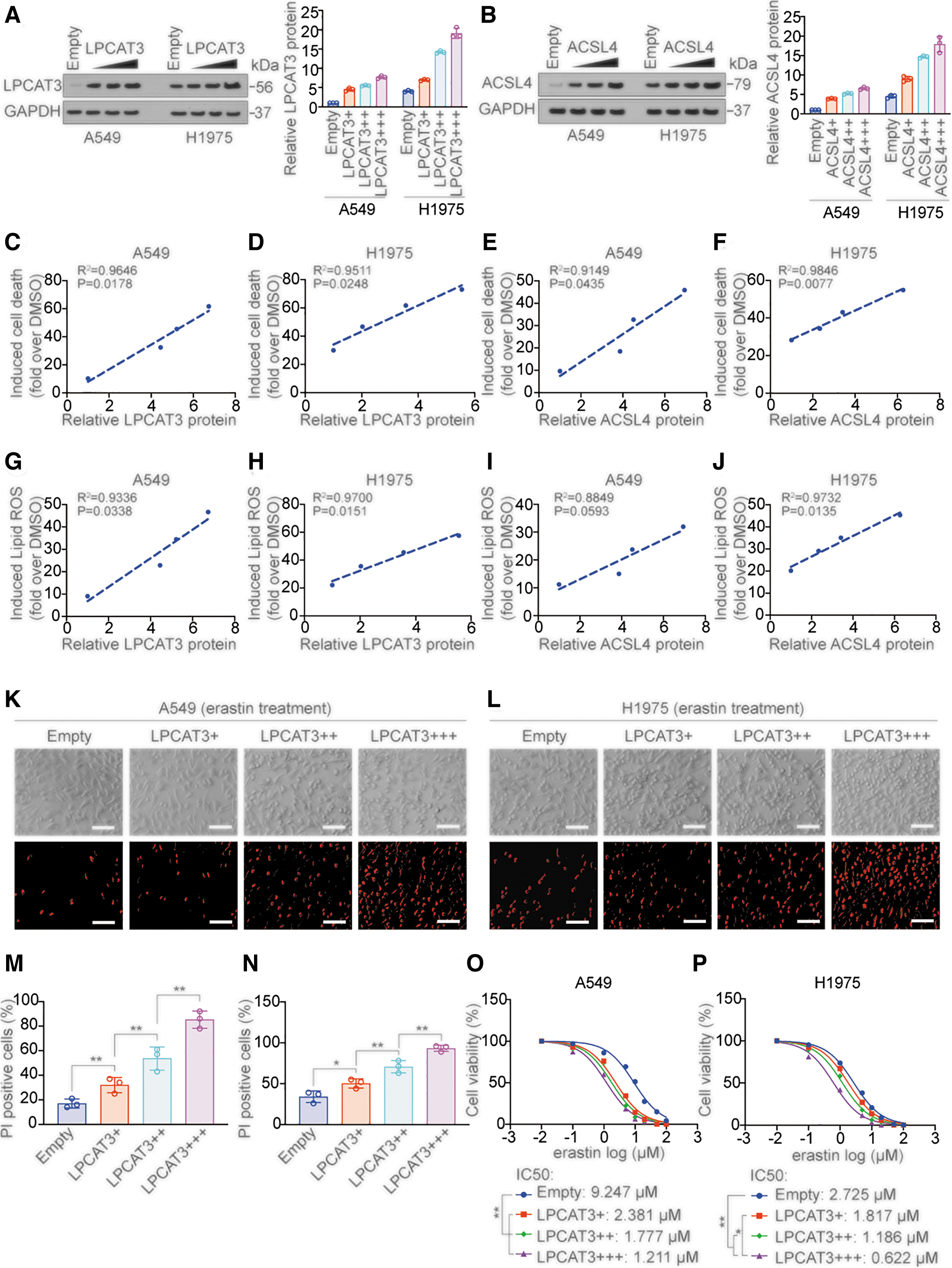
Furthermore, overexpression of LPCAT3 promoted erastin-induced cell death in a dose-dependent manner in A549 and H1975 cells by propidium iodide staining (which stains dead cells red) (Fig. 3K–N). We also found that overexpression of LPCAT3 or ACSL4 significantly decreased the IC50 of erastin in a dose-dependent manner in A549 and H1975 cells (Fig. 3O, P and Supplementary Fig. S2J, K). These results indicated that overexpression of LPCAT3, ACSL4, and YAP promoted ferroptosis sensitivity.
We then knocked out LPCAT3 and ACSL4 in various LUAD cell lines (Supplementary Fig. S3A, B), and found that after knocking out LPCAT3 and ACSL4, the rise of cell death (Supplementary Fig. S3C, D), lipid ROS (Supplementary Fig. S3E, F), MDA (Supplementary Fig. S3G, H), and labile iron (Supplementary Fig. S3I, J) caused by erastin decreased significantly, and the cell–cell difference was no longer obvious. These data suggested that knockout of ACSL4 and LPCAT3 inhibited erastin-induced ferroptosis in LUAD cells.
ZEB coodinates with YAP to stimulate the transcription of LPCAT3
A previous study has reported that ACSL4 is transcriptionally regulated by YAP (Wu et al., 2019). We further investigated the detailed mechanism how ACSL4 and LPCAT3 transcription was regulated. ACSL4 protein and mRNA expression were positively regulated by YAP, and similar results were observed with LPCAT3 protein and mRNA (Fig. 4A–D and Supplementary Fig. S4A). YAP is a cotranscription factor and does not function as a direct transcription factor (Zhu et al., 2019); we therefore used the LASAGNA database (
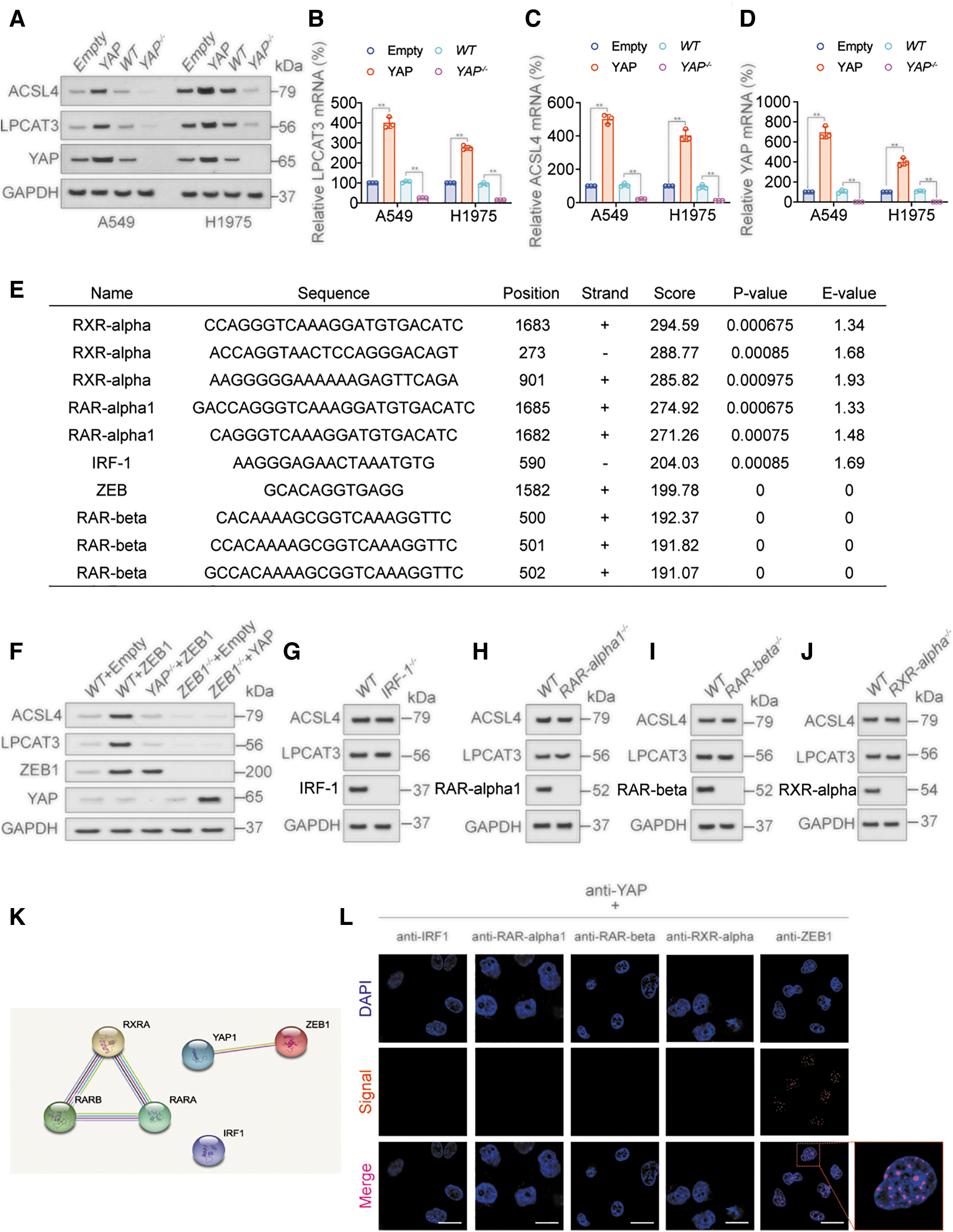
The results predicted retinoid X receptor (RXR)-α, retinoic acid receptor (RAR)-α1, interferon regulatory factor 1 (IRF-1), ZEB, and RAR-β as the most likely transcription factors functioning with YAP (Fig. 4E). Immunoblotting (IB) and quantitative real-time polymerase chain reaction (RT-qPCR) showed that ZEB1 positively regulated the expression of LPCAT3 and ACSL4 (Fig. 4F and Supplementary Fig. S4B–D), while IRF-1 (Fig. 4G), RAR-α1 (Fig. 4H), RAR-β (Fig. 4I), and RXR-α (Fig. 4J) had no similar effect.
The expression of LPCAT3 and ACSL4 mRNA and protein in each treatment group was close to the trend of ZEB activity, but had no correlation with YAP activity, and YAP and ZEB mRNA and protein expression (Fig. 4F and Supplementary Fig. S4B–D). We further identified a direct mutual binding between YAP and ZEB1 using the STRING database (
ZEB1 protein expression in LUAD cell lines was measured by IB, and confirmed to be positively correlated with LPCAT3 and ACSL4 (Supplementary Fig. S4F–H). These results suggested that YAP might promote the transcription of LPCAT3 and ACSL4 by cooperating with ZEB.
ZEB binds to LPCAT3 promoter to stimulate LPCAT3 transcription with YAP
We constructed luciferase reporters driven by the wild-type (WT) LPCAT3 promoter or ZEB-binding motif-mutated (Mut) promoter. ZEB1 expression significantly induced WT-LPCAT3 Luc reporter activity, and this effect was blocked by YAP knockout. Conversely, while ZEB1 knockout resulted in inhibited WT-LPCAT3 Luc reporter activity, this effect could not be reversed by YAP overexpression. These data suggested that YAP and ZEB are both indispensable for LPCAT3 transcription.
Furthermore, the effects of ZEB1 and YAP on LPCAT3 transcription were abrogated when the ZEB-binding site in the LPCAT3 promoter was mutated (Fig. 5A and Supplementary Fig. S5A). The reporter activity of WT-LPCAT3 Luc was unaffected by knockout of IRF-1, RAR-α1, RAR-β, and RXR-α (Fig. 5B and Supplementary Fig. S5B).
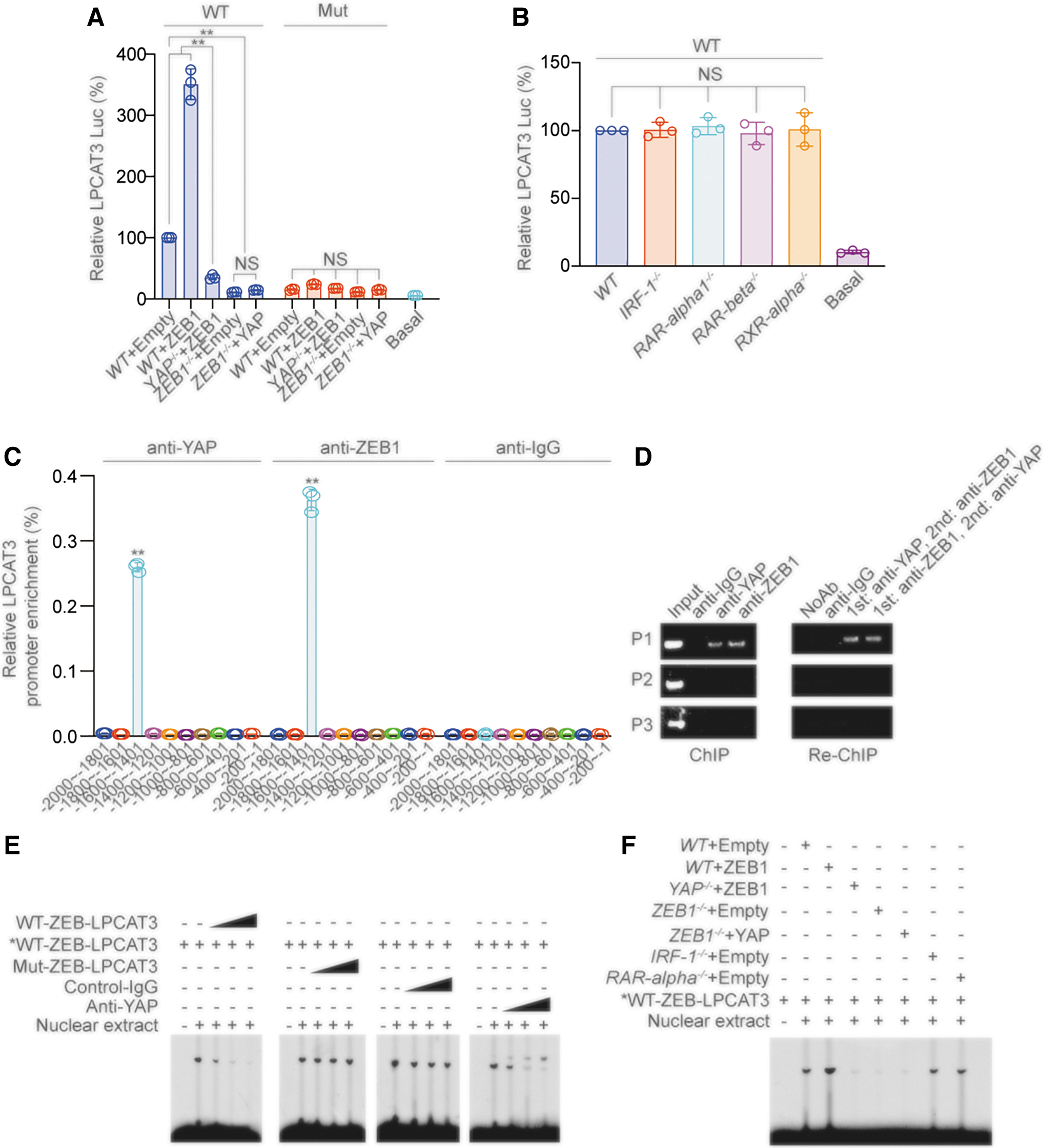
Chromatin immunoprecipitation (ChIP)-qPCR experiments confirmed that both YAP and ZEB1 bound to the −1600 to −1401 nt region of the LPCAT3 promoter (Fig. 5C and Supplementary Fig. S5C). In addition, combined ChIP and Re-ChIP experiments suggested that the LPCAT3 promoter was simultaneously bound by YAP and ZEB1 (Fig. 5D). We performed electrophoresis mobility shift assay (EMSA) using the LPCAT3 sequence containing the ZEB-binding motif.
We observed that excess unlabeled WT-LPCAT3 probe as competitor disrupted the DNA–protein complexes in a dose-dependent manner. In contrast, an excess of unlabeled Mut probe bearing the disrupted ZEB motif was unable to compete for the complex (Fig. 5E, left two images). We further examined whether YAP was involved in the interaction of ZEB with the LPCAT3 promoter.
Nonspecific control-IgG had no effects, whereas the addition of anti-YAP antibody abrogated the DNA–protein complexes in a dose-dependent manner (Fig. 5E, right two images). ZEB1 overexpression increased the intensity of the DNA–protein complexes, while YAP knockout reduced the intensity of the DNA–protein complexes.
Furthermore, ZEB1 knockout inhibited the formation of DNA–protein complexes, while YAP overexpression did not reverse this effect. The formation of DNA–protein complexes was not affected by IRF-1 or RAR-α knockout compared with control treatment (Fig. 5F). Collectively, these data suggested that the ZEB transcription factor directly bound to the LPCAT3 promoter, with YAP as the cotranscription factor.
The ZF domain of ZEB and WW domain of YAP mediate the ZEB/YAP interaction
We further explored the molecular mechanism of the binding between YAP and ZEB. First, we verified the binding between YAP and ZEB1 by coimmunoprecipitation (co-IP); in contrast, RXR-α, RAR-α1, IRF-1, and RAR-β did not bind to YAP and ZEB1 (Fig. 6A). ZEB1 contains three key domains: an N-terminal zinc-finger cluster, a central part (CP), and a C-terminal zinc-finger cluster. YAP contains two WW domains (WW1 and WW2) that often function in protein binding (Fig. 6B) (Lehmann et al., 2016; Zhang et al., 2017b).
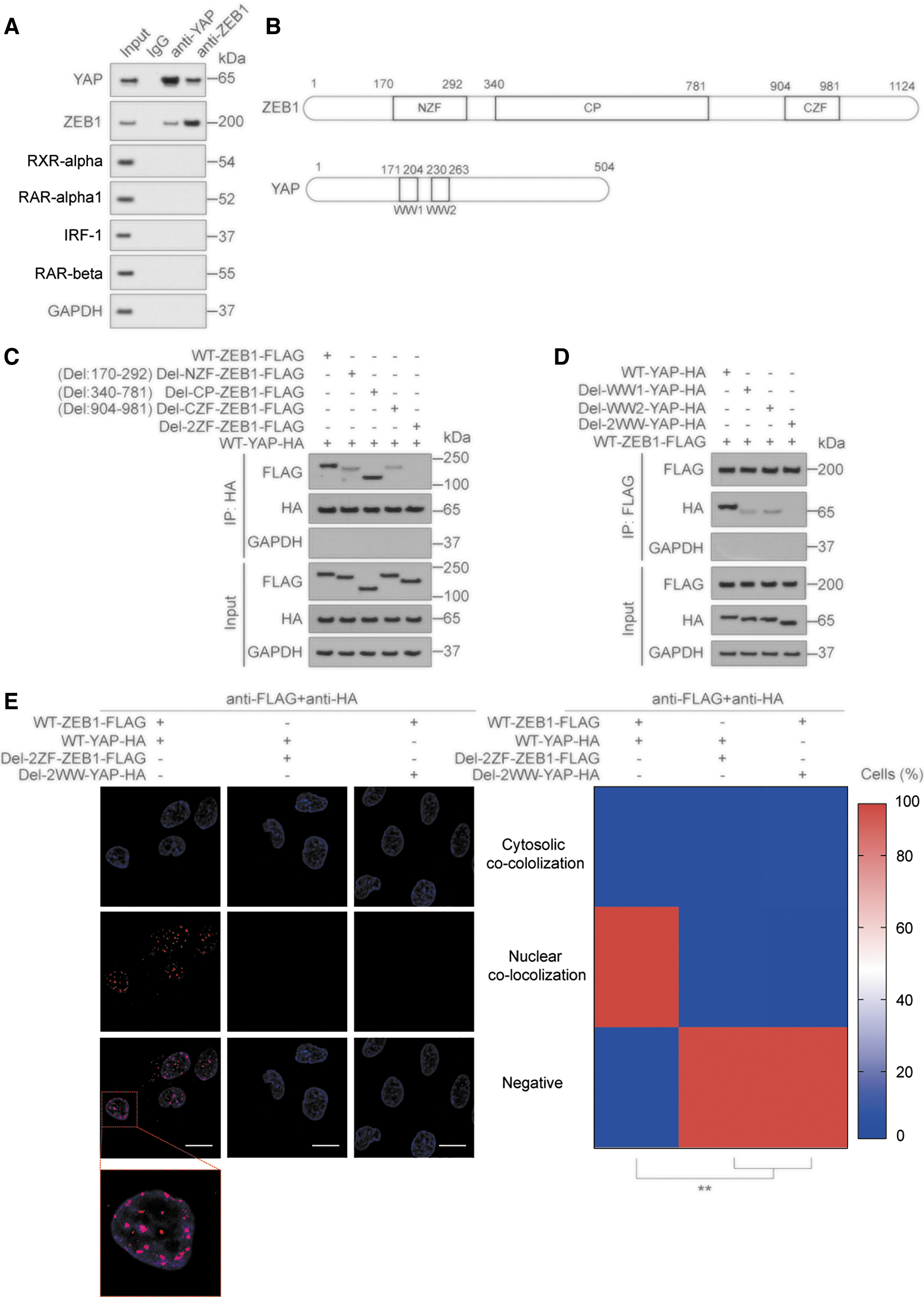
We found that when one of the ZF or WW domains was deleted, the interaction between YAP and ZEB1 was weakened; when both ZF and WW domains were deleted, the interaction between YAP and ZEB1 was eliminated (Fig. 6C, D). The results of PLA and immunofluorescence (IF) experiments further demonstrated the requirement of the ZF and WW domains for the binding between YAP and ZEB1 (Fig. 6E and Supplementary Fig. S6A, B). Deletion of the WW domains in YAP or ZF domains in ZEB1 significantly weakened their activity (Supplementary Fig. S6C, D). The above results suggested that YAP and ZEB interacted via their ZF and WW domains.
Removal of YAP phosphorylation promotes its interaction with ZEB
YAP is regulated by phosphorylation at two main sites, S127 and S397, and these phosphorylations are mediated by large tumor suppressor kinase. YAP S127 phosphorylation results in YAP binding to 14-3-3 and cytoplasmic retention, and YAP S397 phosphorylation leads to binding to CK1, which causes further phosphorylation on its phosphodegron, followed by βTrCP-mediated ubiquitination and degradation (Hu et al., 2017; Zhao et al., 2010). We constructed plasmids expressing YAP mutations at each or both of these sites: YAPS127A, YAPS397A, and YAPS127A+S397A.
We found that YAPS127A and YAPS397A could promote the binding between YAP and WT-ZEB1, and the promoting effect of YAPS127A+S397A is more obvious. When the two ZF domains of ZEB1 were mutated, the binding between YAP and ZEB1 disappeared (Supplementary Fig. S6E–G). Furthermore, we confirmed that YAPS127A, YAPS397A, and YAPS127A+S397A had higher activity compared with WT-YAP (Supplementary Fig. S6H). These results demonstrated that removing the phosphorylation site in YAP promoted the binding between YAP and ZEB.
EP300-dependent H3K27Ac stimulates YAP- and ZEB-induced LPCAT3 transcription
We further explored the mechanisms by which YAP and ZEB regulate LPCAT3 transcription. Transcriptional activation is regulated by histone modifications, such as methylation and acetylation (Hogg et al., 2021; Sankar et al., 2022). We used the STRING database (
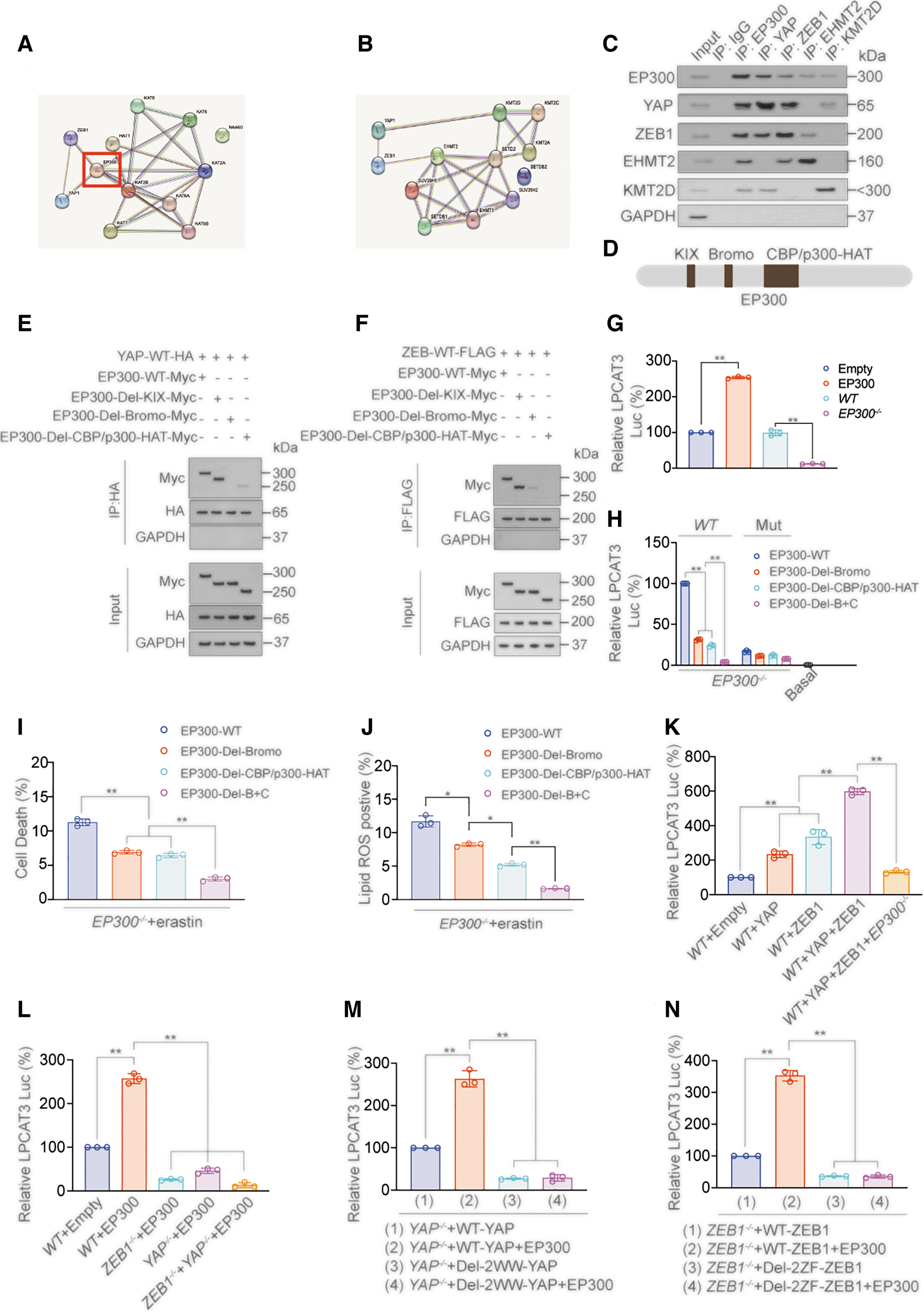
The results showed that the histone acetyltransferase EP300 might bind to YAP and ZEB1 simultaneously. Co-IP experiments confirmed that EP300 simultaneously interacted with YAP and ZEB1, whereas euchromatic histone lysine methyltransferase 2 (EHMT2) only bound with ZEB1, and lysine methyltransferase 2D (KMT2D) only bound with YAP (Fig. 7C). EP300 contains three domains: KIX, Bromo, and CBP/p300-HAT (Wang et al., 2021c) (Fig. 7D).
We constructed EP300 plasmids deleted for these domains and performed co-IP experiments to evaluate which domain was responsible for the interaction with YAP and ZEB1. We found that the deletion of the Bromo domain completely abolished the interaction between YAP and EP300, and the deletion of CBP/p300-HAT-Myc domain completely abolished the interaction between ZEB1 and EP300 (Fig. 7E, F). These results suggested that the YAP WW domain interacted with the ZEB ZF domain and YAP also bound to the EP300 Bromo domain, while ZEB bound to CBP/p300-HAT domain. However, we cannot rule out that these domains only play a role in promoting binding, but no direct binding occurs between the domains.
We next explored the promotion effect of EP300 on LPCAT3 transcription. We found that EP300 overexpression promoted the promoter activity of LPCAT3, while EP300 knockout inhibited promoter activity (Fig. 7G). In EP300-knockout cells transfected with EP300-WT or different EP300-domain-mutant plasmids, luciferase assays showed that knockout of the Bromo and CBP/p300-HAT domains alone inhibited the promoter activity of LPCAT3, while upon combined knockout of both Bromo CBP/p300-HAT domains, LPCAT3 promoter activity inhibition was more significant (Fig. 7H), and the change of cell death and lipid ROS induced by erastin in these cells also had a similar trend (Fig. 7I, J).
H3K27Ac is an EP300-induced histone acetylation event that can promote transcription (Ebrahimi et al., 2019); ChIP experiments found that H3K27Ac levels around the ZEB-binding motif in the LPCAT3 promoter were enriched in the presence of EP300-Myc, YAP, and ZEB1. However, when the Bromo and CBP/p300-HAT domains were deleted, H3K27Ac enrichment was significantly reduced (Supplementary Fig. S7A–E). We also found that YAP and ZEB1 promoted the transcription of LPCAT3, but this effect was dependent on EP300 (Fig. 7K).
Similarly, the effect of EP300 on promoting LPCAT3 transcription was dependent on YAP and ZEB1 (Fig. 7L). Moreover, the effect of EP300 on promoting LPCAT3 transcription was abolished when the binding between YAP and ZEB1 was abolished (Fig. 7M, N). These results showed that EP300 was crucial for the transcription of LPCAT3 promoted by YAP and ZEB, which was mediated by protein interactions via its Bromo and CBP/p300-HAT domains. YAP may interact with the Bromo domain of EP300, and ZEB may bind the CBP/p300-HAT domain of EP300, leading to the formation of a transcription-promoting complex.
Validation of ZEB and YAP-induced LPCAT3 transcription in primary LUAD cell and LUAD xenograft models
We next validated the ZEB and YAP-induced LPCAT3 transcription mechanism in primary cell and xenograft models. We selected five LUAD primary cell lines with low LPCAT3, ACSL4, and YAP expression (LPCAT3/ACSL4/YAP low expression lines) and five LUAD primary cell lines with high LPCAT3, ACSL4, and YAP expression (LPCAT3/ACSL4/YAP high expression lines). ZEB1 expression was significantly higher in the LPCAT3/ACSL4/YAP high expression lines compared with the low expression lines (Fig. 8A).
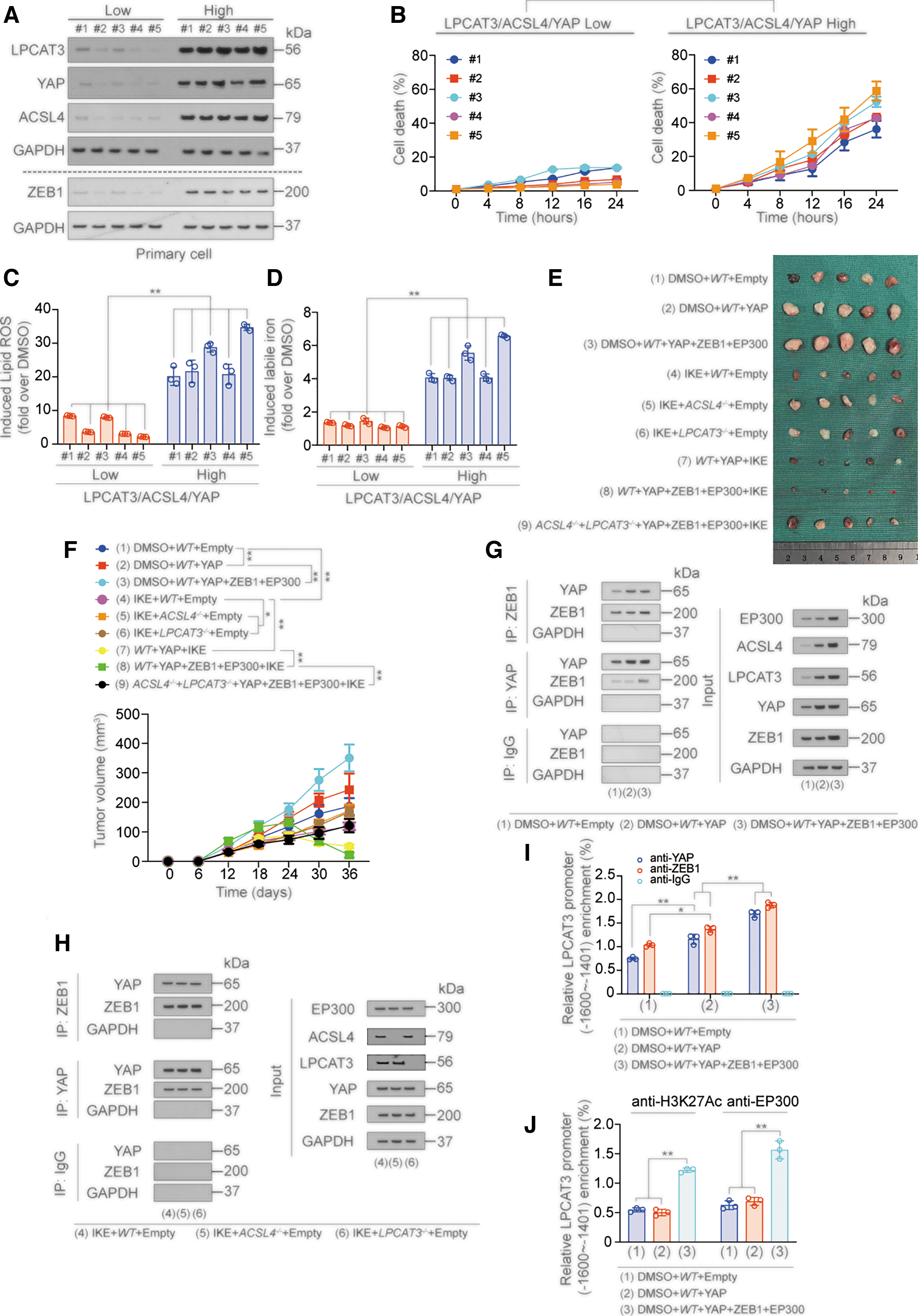
Compared with the LPCAT3/ACSL4/YAP low expression primary cell lines, the LPCAT3/ACSL4/YAP high expression primary cell lines were more sensitive to erastin, which was reflected in the rise in cell death rate, lipid ROS and labile iron, and the decline in GSH (Fig. 8B–D and Supplementary Fig. S8A, B). ChIP experiments showed that ZEB1, YAP, H3K27Ac, and EP300 were more enriched at the ZEB-binding motif in the LPCAT3 promoter in LPCAT3/ACSL4/YAP high expression lines, and the possibility that YAP and ZEB bound to −200 to −1 region was excluded (Supplementary Fig. S8C–J). These data indicated that LPCAT3/ACSL4/YAP high expression promoted ferroptosis sensitivity of primary cells.
We then constructed A549-based nude mouse xenograft models with the following treatments: Group 1: control group; group 2: YAP overexpression group; group 3: YAP/ZEB1/EP300 overexpression group; group 4: IKE treatment group; group 5: ACSL4 knockout with IKE treatment group; group 6: LPCAT3 knockout with IKE treatment group; group 7: YAP overexpression with IKE treatment group; group 8: YAP/ZEB1/EP300 overexpression with IKE treatment group; and group 9: ACSL4/LPCAT3 knockout combined with YAP/ZEB1/EP300 overexpression with IKE treatment group. IKE (dose: 50 mg/kg) was administered on day 18 once a day for 2 weeks.
We found that compared with the control group (group 1), the YAP overexpression group (group 2) exhibited increased tumor growth, and the overexpression of ZEB1 and EP300 (group 3) further promoted tumor growth compared with group 1. While IKE treatment (group 4) inhibited tumor growth, knockout of ACSL4 or LPCAT3 (group 5 and group 6) led to loss of this effect. However, YAP overexpression enhanced the tumor inhibition role of IKE (group 7), and this effect was further promoted by ZEB1 and EP300 (group 8), but reversed by knockout of ACSL4 and LPCAT3 (group 9). These data indicated that YAP, ZEB1, and EP300 promoted the effect of IKE in tumor shrinkage (Fig. 8E, F).
We also verified the mechanism by which YAP, ZEB1, and EP300 promoted LPCAT3 transcription in the xenograft model. First, the expressions of both ACSL4 and LPCAT3 were promoted by YAP, ZEB1, and EP300 overexpression (Fig. 8G, H and Supplementary Fig. S8K), and the interaction between YAP and ZEB1 was detected in all groups (Fig. 8G, H and Supplementary Fig. S8L).
Knockout of ACSL4 and LPCAT3 did not affect the expression of YAP and ZEB1 and their binding (Fig. 8H). Moreover, both YAP and ZEB1 bound to the −1600 to −1401 nt region of the LPCAT3 promoter, but could not bind to the −1400 to −1201nt and −800 to −601 nt regions of the LPCAT3 promoter (Fig. 8I and Supplementary Fig. S8M–O). We also found that H3K27Ac and EP300 were enriched in the −1600 to −1401 nt region of the LPCAT3 promoter (Fig. 8J and Supplementary Fig. S8P). These results suggested that YAP, ZEB, and EP300 also promoted LPCAT3 transcription in the xenograft model.
YAP, ACSL4, and LPCAT3 expression in LUAD samples
We next explored the relationship between YAP, ACSL4, LPCAT3, and clinicopathological indicators in 60 LUAD samples, respectively. Specimens were divided into low expression and high expression groups by immunohistochemistry (IHC) results (n = 30/group) (Fig. 9A). We found that YAP, ACSL4, and LPCAT3 were highly expressed in high-stage LUAD specimens, and their expression was positively correlated with each other (Fig. 9B and Supplementary Fig. S9A–C).
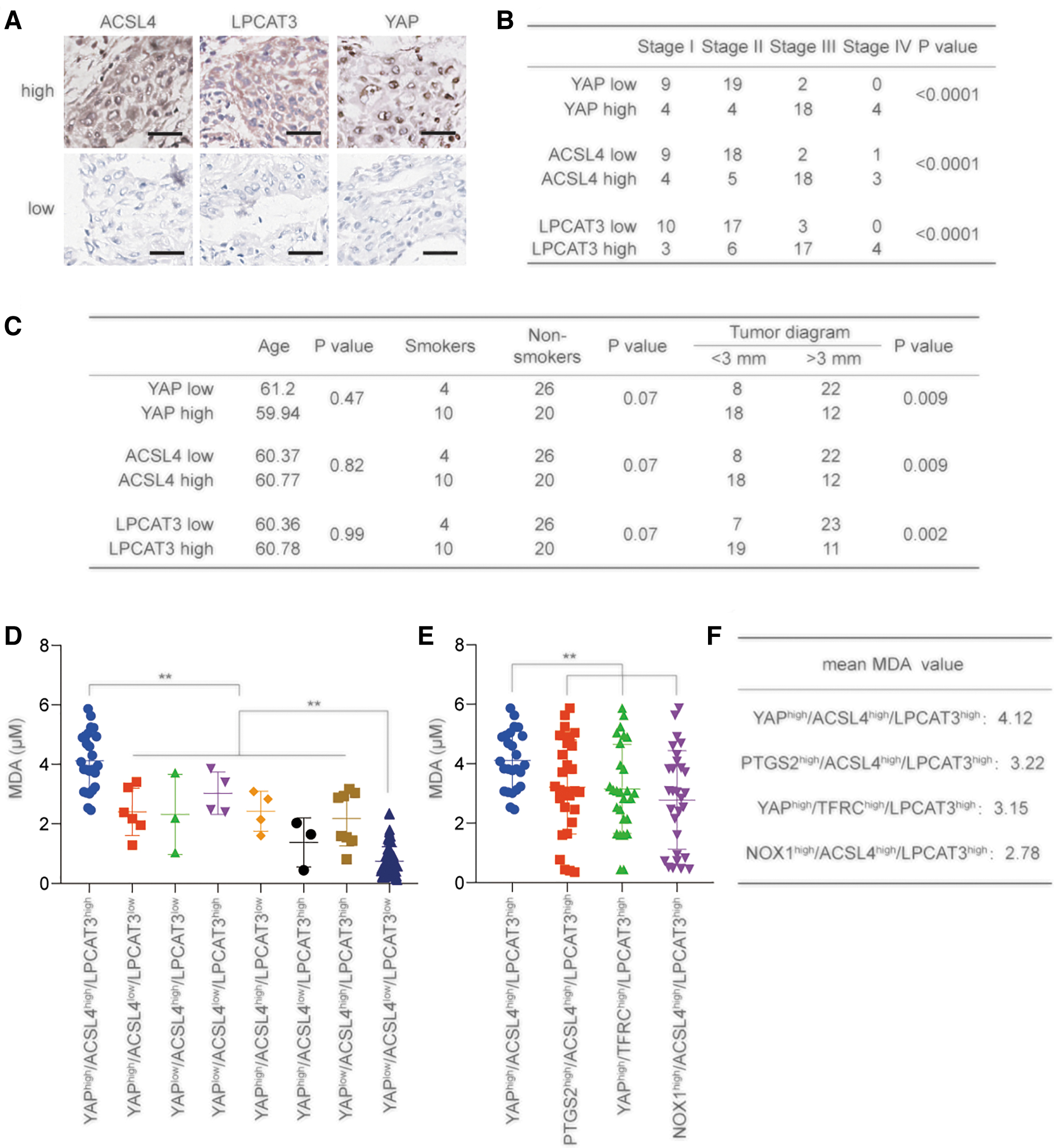
We also found that ZEB1 level and EP300 level were both positively associated with LPCAT3 and ACSL4 levels, respectively (Supplementary Fig. S9D–G). Their expression had no relationship with patient age and showed a trend of increase in smoking patients, but without significance; their levels were significantly increased in patients with larger tumor diameters (Fig. 9C). These results suggested that higher expression of YAP, ACSL4, and LPCAT3 corresponded to increased malignancy in LUAD.
ACSL4, LPCAT3, and YAP can jointly determine LUAD ferroptosis sensitivity
Finally, we cultured 120 cases of LUAD primary cells, divided them into 8 groups on the basis of the expression of YAP, ACSL4, and LPCAT3, and then treated them with erastin. We found that in the YAPhigh/ACSL4high/LPCAT3high group, MDA content was significantly higher than that in other groups, while MDA content was significantly lower in YAPlow/ACSL4low/LPCAT3low group than in other groups (Fig. 9D).
The MDA content in the YAPhigh/ACSL4high/LPCAT3high group was significantly higher than that in the PTGS2high/ACSL4high/LPCAT3high group, YAPhigh/TFRChigh/LPCAT3high group, NOX1high/ACSL4high/LPCAT3high group, YAPhigh group, LPCAT3high group, and ACSL4high group (Fig. 9E, F and Supplementary Fig. S9H). These findings indicated that the combination of YAP, ACSL4, and LPCAT3 might better determine the ferroptosis sensitivity compared with other indicators to a certain extent.
Discussion
The identification of ferroptosis biomarkers may aid in the future application of ferroptosis-related cancer treatments. PUFAs-containing phospholipid is the main objective of lipid peroxidation. Thus, lipid metabolism occupies an important position in the course of ferroptosis. In this study, we defined LPCAT3 as a new biomarker that indicated ferroptosis sensitivity of LUAD. We also found that the combination of three markers (LPCAT3, ACSL4, and YAP) indicated ferroptosis sensitivity of LUAD cells.
Several lipid metabolism-related enzymes have also been identified as ferroptosis sensitivity markers, such as arachidonic acid lipoxygenases (ALOXs), ADCY10, NADPH oxidase 1 (NOX1), TFRC, prostaglandin-endoperoxide synthase 2 (PTGS2), calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2), and glycogen synthase kinase-3beta (GSK3β) (Shintoku et al., 2017; Wang et al., 2022b; Wang et al., 2021a; Wu et al., 2021; Wu et al., 2019; Zhang et al., 2021; Zhu et al., 2021).
In this study, we compared the effectiveness of the LPCAT3, ACSL4, and YAP combination with NOX1, TFRC, and PTGS2 in evaluating ferroptosis sensitivity, and found that the combination of LPCAT3, ACSL4, and YAP was more effective to predict ferroptosis sensitivity of LUAD. However, because of the ferroptosis sensitivity of the patient LUAD cells need to be judged by adding ferroptosis inducers after the primary cells were successfully cultured. In this study, we performed extensive experiments in primary cells from 120 LUAD cases. Thus, the comparison with other indicators needs to be further completed by continuing to complete the culture of primary cells in subsequent studies.
Previous studies showed that inducing ferroptosis has a good effect on eliminating tumor cells with high metastasis tendency, immunotherapy resistance, chemotherapy resistance, and targeted therapy resistance (Wu et al., 2019; Zhang et al., 2022a; Zhang et al., 2021; Zhang et al., 2020). The content of ferroptosis-inducing material including labile iron, AA, and AdA is higher in tumor cells compared with normal cells, and ferroptosis is more likely to occur in tumor cells (Zhang et al., 2021). When certain cancer-promoting signaling pathways are activated, ferroptosis sensitivities are subsequently activated.
In this way, the treatment of cancer cells with ferroptosis inducers leads to high levels of ferroptosis (Grube et al., 2022; Wu et al., 2019; Zeng et al., 2023). Taking YAP signaling pathway as an example, activation of YAP promotes the proliferation, clonogenesis, and tumor angiogenesis; inhibits apoptosis, but also stimulates the transcription of ACSL4, ALOXE3, and TFRC to increase the ferroptosis sensitivity (Qin et al., 2021; Wu et al., 2019). We also found that YAP stimulated the transcription of LPCAT3 in LUAD to elevate the ferroptosis sensitivity, which is consistent with the results of previous studies.
YAP also decreases during ferroptosis. After system XC − inhibition, the accumulated glutamate suppresses YAP O-GlcNAcylation and expression via the ADCY10/PKA/GFPT1 axis, subsequently inhibits ferritin transcription, and leads to the release of labile iron to enhance the occurrence of ferroptosis. The above findings elucidated that YAP is a potential target for ferroptotic cancer treatment (Zhang et al., 2021).
Our results showed that highly malignant LUAD was prone to the high expression of YAP, ACSL4, and LPCAT3, which also suggested a higher ferroptosis sensitivity of these tumor cells. These findings provide further evidence for the positive correlation between tumor malignancy and ferroptosis sensitivity.
The YAP transcriptional regulator requires other transcription factors to exert its biological function because of the absence of a DNA-binding domain. YAP contains two WW domains (WW1 domain and WW2 domain) in its central region. WW domains typically recognize transcription factors containing a PPxY motif (P is proline, x is any amino, and Y is tyrosine). YAP can also bind to transcription factors in non-WW-PPxY ways. For example, we previously found that the WW domain of YAP can recognize and bind PSY motif on TFCP2, thereby recruiting a series of transcription factors (including FOXA1, FOXC1, POU3F2, NR1D1, MSC) that interact with the CC domain of YAP (Zhang et al., 2017b).
ZEB was initially identified as an epithelial–mesenchymal transition (EMT)–inducing transcriptional factor that activates the embryonic EMT program in cancer stem cells leading to early dissemination, metastasis, therapy resistance, and poor prognosis in different tumor types, including pancreatic, breast, and lung cancers (Choi et al., 2015). Recent studies showed that ZEB stimulates YAP-related target genes, such as connective tissue growth factor and the CYR61 genes (Feldker et al., 2020; Lobe et al., 2021). Here, we proved that upregulation of LPCAT3 transcription in LUAD cells was driven by ZEB in a complex with YAP, which bound to the LPCAT3 promoter region (−1600 to −1401 nt), and the formation of this transcriptional complex was via their zinc-finger and WW domains, respectively.
EP300 is a histone acetyltransferase that activates the transcription of downstream genes by increasing H3K27Ac levels (Durbin et al., 2022; Ebrahimi et al., 2019). EP300 can directly bind to transcription-related factors. For example, EP300 binds to CREB via its CBP/p300-HAT to form a transcription-promoting complex (Cui et al., 2018; Wang et al., 2021c). However, the interactions of EP300 with YAP and ZEB have not been previously reported.
We first predicted the interaction of EP300, YAP, and ZEB through STRING database analysis, and then confirmed the interactions between EP300, YAP, and ZEB by constructing a series of mutant plasmids and performing co-IP experiments. The EP300, YAP, and ZEB transcription complex plays an important role in stimulating the transcription of LPCAT3. The three proteins bound to each other through binding sites in each protein, so the stability is higher than that of a complex of two transcription factors. Further exploration of the role of the EP300-YAP-ZEB transcription complex in regulating tumors may lead to the discovery of new tumor therapeutic targets.
Previous studies showed that lipid metabolism-related enzymes, including ACSL4 and LPCAT3, were involved in the formation of phospholipids with PUFAs, which was associated with lipid peroxidation and ferroptosis. Here, we found that LPCAT3 and ACSL4 transcription was mediated by the Hippo/YAP pathway through the participation of ZEB1 and EP300 (Fig. 1). Using nude mouse xenograft and primary cell model, we found that tumor cells with high levels of YAP, ACSL4, and LPCAT3 not only corresponded to more malignant LUAD, but also these cells were sensitive to ferroptosis.
We also evaluated MDA levels in erastin-treated primary LUAD cells, and found that high expression of YAP, ACSL4, and LPCAT3 was accompanied with high concentrations of MDA (Fig. 9). These results suggested that LPCAT3 was closely associated with the sensitivity of ferroptosis, and the combination of related indicators could more accurately judge the sensitivity of ferroptosis. Inducing ferroptosis may be an effective measure to treat advanced cancer patients such as those with LUAD.
Materials and Methods
Cell culture
A549, H358, H1299, H1650, PC9, and H1975 cell lines were purchased from Fuheng Biotechnology (Shanghai, China). All the cell lines were validated by short tandem repeat analysis. Primary LUAD cells were established from LUAD tissues as previously described (Zhang et al., 2022b; Zhang et al., 2021).
In brief, tissues without necrosis cut in ∼1.0 cm3 pieces were washed with ice-cold Dulbecco's phosphate-buffered saline (DPBS) three times before resuspending in Dulbecco's modified Eagle's medium (DMEM) containing collagenase I (2 mg/mL; Solarbio, Shanghai, China) at 37°C for 4 h. After three washes with DMEM, cells were cultured in routine conditions (DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin).
Mouse experiments and tissue samples
We obtained 6-week-old athymic nude mice from Jiesijie (Shanghai, China).
For the generation of nude mouse xenograft models, A549 cells (5 × 106) with the indicated treatments were subcutaneously injected into mice. IKE was subcutaneously injected once a day from day 18 for 2 weeks. The tumor volume was calculated as 0.5 × length × width2. Animals were monitored and sacrificed on day 36. Tissue specimens were obtained from patients in Shanghai Chest Hospital (Shanghai, China) (mean age ± standard deviation, 60.57 ± 8.35 years; male:female ratio, 1.14:1) from March 2015 to November 2022. Informed written consent was obtained from all patients.
Reagents and plasmids
Erastin was obtained from Sigma (St Louis, MO).
LPCAT3, ACSL4, and ZEB1 expressing plasmids were purchased from Biovision (Shanghai, China). YAP expressing and knockout plasmids were described in previous studies (Wang et al., 2021b; Zhang et al., 2021). LPCAT3, ACSL4, ZEB1, EP300, IRF-1, RAR-α1, RAR-β, and RXR-α knockout plasmids were constructed using lentiCRISPR v2 vector. The YAP WW domain mutation and EP300 mutation plasmids were described in previous studies (Wang et al., 2021b, Wang et al., 2021c, Zhang et al., 2017b). The ZEB1 domain mutation and EP300-Del-Bromo+CBP/p300-HAT (B+C) plasmids were constructed using overlapping PCR and cloned into pcDNA3.1(+) vector. The primers are listed in Supplementary Table S1.
IHC, IF, and IB
IHC, IF, and IB were performed using conventional protocols. The primary antibodies used for IHC were anti-LPCAT3 (#ab232958; Abcam, Cambridge, MA), anti-ACSL4 (#ab155282; Abcam), anti-YAP (#ab205270; Abcam), anti-EP300 (#ab275378; Abcam), and anti-ZEB1 (#ab203829; Abcam).
The primary antibodies used for IF were anti-FLAG (#2368; CST, Boston, MA) and anti-HA (#ab18181; Abcam). The primary antibodies used for IB were anti-4-HNE (#ab243070; Abcam), anti-GAPDH (#5174 and #51332; CST), anti-LPCAT3 (#ab232958; Abcam), anti-ACSL4 (#ab155282; Abcam), anti-YAP (#ab52771; Abcam or#sc-101199; Santa Cruz Biotechnology, Santa Cruz, CA), anti-ZEB1 (#ab203829 and #ab181451; Abcam), anti-IRF-1 (#ab243895; Abcam), anti-RAR-α1 (#275745; Abcam), anti-RAR-β (#ab124701; Abcam), anti-RXR-α (#ab125001; Abcam), anti-FLAG (#8146 and #2368; CST), anti-HA (#ab9110 and #ab18181; Abcam), anti-Myc (#2040 and #3946; CST), anti-EP300 (#ab275378 and #ab14984; Abcam), anti-EHMT2 (#ab185050; Abcam), and anti-KMT2D (#ab213721 and #ab243044; Abcam).
Coimmunoprecipitation
Co-IP was performed as described previously (Yu et al., 2023; Zhang et al., 2017b). In brief, cell lysates were mixed with protein A/G-magnetic beads (Novex, Oslo, Norway) and incubated at 4°C overnight with the indicated antibodies. The next day, the beads were washed three times with Western/IP lysis buffer (Beyotime, Haimen, China), suspended in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer and separated by SDS-PAGE.
The antibodies used for co-IP were anti-ZEB1 (#ab181451 and #ab203829; Abcam), anti-IgG (#3900; CST), anti-FLAG (#2368; CST), anti-HA (#ab18181; Abcam), anti-YAP (#ab52771; Abcam), anti-Myc (#2040; CST), anti-EP300 (#ab275378; Abcam), anti-EHMT2 (#ab185050; Abcam), and anti-KMT2D (#ab213721; Abcam).
Proximity ligation assay
PLA was measured using the Duolink In Situ Red Starter Kit (Sigma) as previously described (Zhang et al., 2017a; Zhang et al., 2017b). Cells were seeded on glass cover slips in 24-well plates for 1 day. Cells were then fixed with 4% paraformaldehyde, blocked with blocking buffer, and incubated with primary antibodies overnight at 4°C. The primary antibodies used were anti-YAP (#ab52771; Abcam or #sc-101199; Santa Cruz), anti-ZEB1 (#ab203829 and #ab181451; Abcam), anti-IRF-1 (#ab243895; Abcam), anti-RAR-α1 (#275745; Abcam), anti-RAR-β (#ab124701; Abcam), anti-RXR-α (#ab125001; Abcam), anti-FLAG (#2368; CST), and anti-HA (#ab18181; Abcam).
On the third day, the PLA probe solution was added into each well, and the cells were incubated for 1 h at 37°C. The Ligase–Ligase solution was added into each well, and samples were incubated for 30 min at 37°C. After ligation, the amplification-polymerase solution was added into each well and incubated for 100 min at 37°C before the cells were subjected to confocal microscopic analysis.
Luciferase assays and YAP and ZEB activity assays
Luciferase vectors were cotransfected with a Renilla reporter plasmid into LUAD cells, and the reporter activities were measured using dual luciferase reagent (Promega, Madison, WI). YAP activity was measured using the pUAS-Luc/TEAD-Gal4 system (Zhang et al., 2017a). ZEB activity was measured using the ataxia telangiectasia mutated serine/threonine kinase and phosphofructokinase muscle isoform reporter (Zhang et al., 2018; Zhou et al., 2021).
Cell death and lipid ROS measurements
Cell death was analyzed by staining with SYTOX Green (Invitrogen, Carbsland, CA) followed by flow cytometry analysis. Lipid ROS was measured by staining C11-BODIPY (Invitrogen) followed by flow cytometry analysis.
Metabolite assay
MDA and labile iron were measured using the kits from Abcam. GSH was measured using a kit from Sigma.
Quantitative real-time polymerase chain reaction
Total RNA was extracted using Trizol (Ambion, Carlsbad, CA) and reverse-transcribed into complementary DNA using the PrimeScript™ RT reagent kit (Takara, Dalian, China). The SYBR premix Ex Taq kit (Takara) was used for RT-qPCR. The primers are listed in Supplementary Table S1.
ChIP and Re-ChIP
ChIP and Re-ChIP experiments were performed using kits from Active Motif (Carlsbad, CA) as previously described (Wang et al., 2021b; Zhang et al., 2017b). For ChIP experiments, cells (2 × 107) were fixed using 1% formaldehyde, washed using PBS, and lysed with lysis buffer. After sonication, protein–DNA complexes were incubated overnight with antibody-coupled protein G beads at 4°C. DNA was then eluted in 1% SDS/0.1 M NaHCO3, reversed crosslinked at 65°C, and purified via phenol/chloroform extraction and ethanol precipitation before analysis.
For Re-ChIP experiments, complexes were eluted by incubation for 30 min at 37°C in 10 mM DTT. After centrifugation, the supernatant was diluted 20 times with Re-ChIP buffer (1% Triton X-100, 2 mM ethylenediaminetetraacetic acid, 150 mM NaCl, 20 mM Tris–Hcl, pH 8.1) for further analysis. The primary antibodies used in ChIP and Re-ChIP experiments were anti-YAP (#ab52771; Abcam), anti-ZEB1 (#ab203829; Abcam), anti-Myc (#2040; CST), anti-H3K27Ac (#ab4729; Abcam), anti-EP300 (#ab275378; Abcam), and anti-IgG (#3900; CST).
Electrophoresis mobility shift assay
EMSA was performed as previously described (Chen et al., 2018; Wang et al., 2021b). Nuclear extracted proteins were prepared using the kit from Active Motif, and incubated in reaction buffer with or without DNA competitors (either WT or mutant) on ice before adding biotin-labeled probes (synthesized and 5′ labeled by Sangon, Inc., Shanghai, China).
Antibodies against YAP (#14074; CST) or IgG (#3900; CST) were also added to the mix. DNA–protein complexes were resolved by electrophoresis on 5% native polyacrylamide gels and blotted to Immobilon-My+transfer membranes (Millipore, Billerica, MA). The probes used are listed in Supplementary Table S1.
Statistical analysis
Differences between groups were analyzed by Student's t-test, one-way and two-way ANOVA, and the χ2 test. The correlation between two groups was evaluated by Spearman's rank-correlation analysis. p < 0.05 was considered statistically significant.
Footnotes
Authors' Contributions
J.C. contributed to experimental operation (lead); data analysis (lead); and writing—original draft (supporting). Y.W. performed experimental operation (supporting); data analysis (supporting); and writing—original draft (equal). X.T. assisted with experimental operation (supporting); writing—review and editing (lead). Y.M. conceptualized experimental operation (supporting); writing—review and editing (supporting). L.M., C.Z., and X.X. contributed to writing—review and editing (equal). J.W. and W.F. provided experimental design (supporting); writing—original draft (supporting). X.Z. assisted with experimental design (lead); writing—original draft (lead).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Natural Science Foundation of China (82273099), The Innovative Research Team of High-level Local Universities in Shanghai (SHSMU-ZLCX20212302), Shanghai Rising Star Program (22QA1408400), Project of Clinical Research Supporting System, Clinical Medicine First-class Discipline, Talent training plan of Shanghai Chest Hospital (to Xiao Zhang), and Excellent Talents Nurture Project of Shanghai Chest Hospital (2021YNZYY01). We asked Elsevier English Language Editing Services to proofread and carefully check the English grammar and usage in our article.
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.