
Abstract
Significance:
Nicotinamide adenine dinucleotide (NADH) represents the reduced form of NAD+, and together they constitute the two forms of the nicotinamide adenine dinucleotide whose balance is named as the NAD+/NADH ratio. NAD+/NADH ratio is mainly involved in redox reactions since both the molecules are responsible forcarrying electrons to maintain redox homeostasis.
Recent Advances:
NADH acts as a reducing agent, and one of the most known processes exploiting NADH function is energy metabolism. The two main pathways generating energy and involving NADH are glycolysis and oxidative phosphorylation, occurring in cell cytosol and in the mitochondrial matrix, respectively.
Critical Issues:
Although NADH is primarily produced through the reduction of NAD+ and consumed by its own oxidation, several are the biosynthetic and consumption pathways, reflecting the NADH role in multiple cellular processes. This review gathers all the main current data referring to NADH incorrelation with metabolic and cellular pathways, such as its coenzyme activity, effect in cell death, and on modulating redox and calcium homeostasis.
Future Directions:
Gene expression control, as well as the potential impact on neurodegenerative, cardiac disorders and infections, suggest NADH application in clinical
Introduction
Human cells contain organelles called mitochondria, which are responsible for the production of energy through adenosine triphosphate (ATP) generation. Therefore, mitochondrial health and function are key elements in the maintenance of cellular homeostasis, and these organelles are involved in multiple essential processes. As a result, it is not surprising that mitochondrial dysfunction is linked to disruption of cellular activities that can lead to cellular necrosis and apoptosis (Edeas and Weissig, 2013; Lopez-Otin et al., 2013), as well as organs' failure (Monsalve et al., 2007).
The Krebs cycle, also known as the citric acid cycle or the tricarboxylic acid (TCA) cycle, is a fundamental process in cellular respiration occurring within the mitochondria of eukaryotic cells (Fig. 1). This cycle serves as a central hub in the breakdown of carbohydrates, fats, and proteins, generating energy in the form of ATP and producing crucial electron carriers nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2).
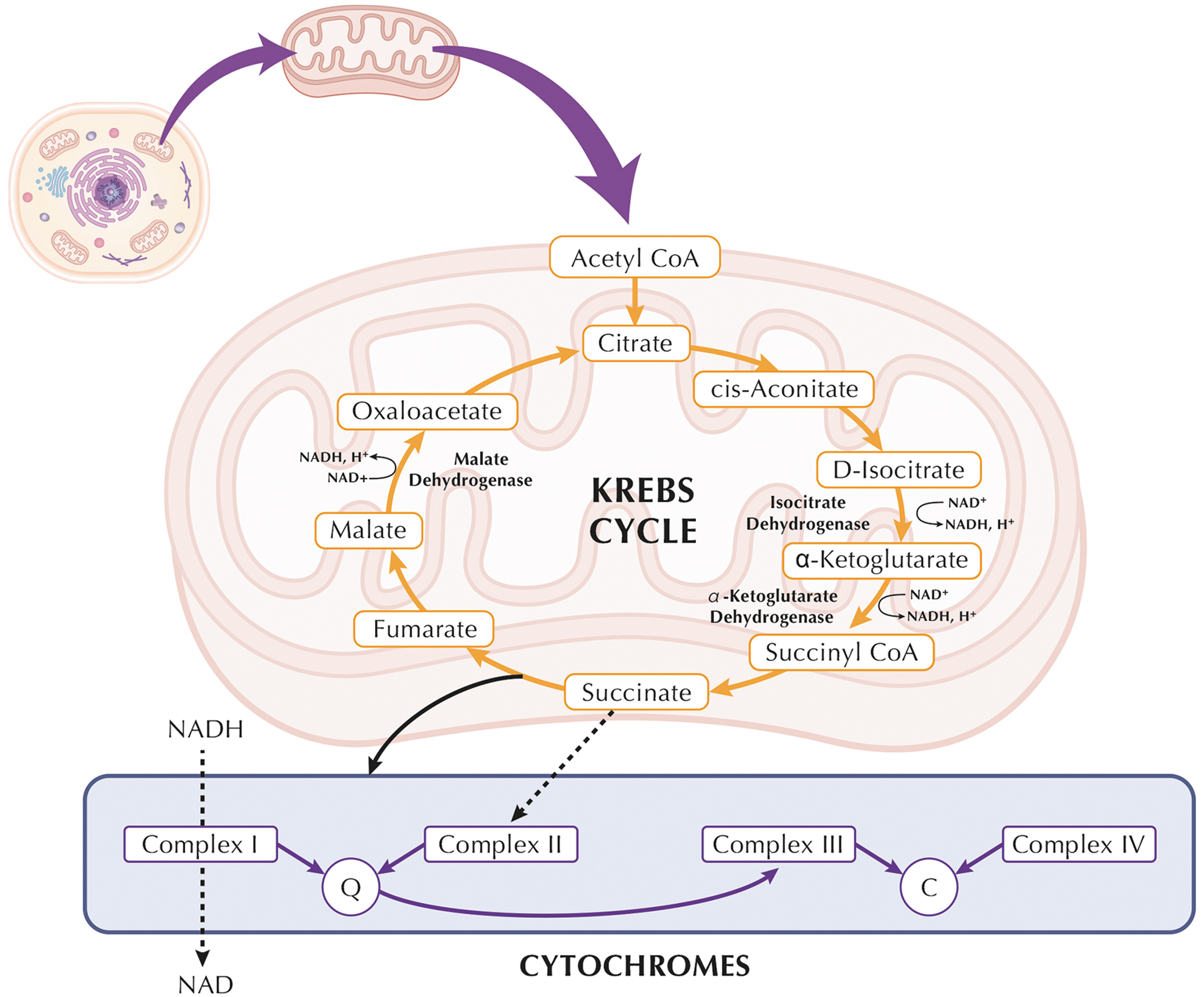
The cycle begins with the acetyl-CoA molecule entering the cycle and combining with oxaloacetate to form citrate, initiating a series of enzymatic reactions that result in the regeneration of oxaloacetate. Throughout these reactions, NADH and FADH2 are produced as electron carriers that will later fuel the electron transport chain (ETC), enabling the synthesis of ATP through oxidative phosphorylation. These electron carriers hold high-energy electrons that are utilized in the final steps of cellular respiration, playing a pivotal role in the production of ATP.
The maintenance of the mitochondrial redox balance is controlled by nicotinamide nucleotide translocase (NNT), a mitochondrial protein involved in transferring reducing equivalents across the inner mitochondrial membrane. NNT couples the transfer of reducing equivalents with the transport of ADP and ATP, impacting the NADH/NAD+ ratio and influencing cellular redox homeostasis.
In parallel, the 2-ketoglutarate dehydrogenase complex (KGDHC) catalyzes the decarboxylation of 2-ketoglutarate, an intermediate in the TCA cycle, to succinyl-CoA, a substrate for the subsequent enzymatic steps, leading to the regeneration of oxaloacetate, which can then re-enter the cycle, produce and channel NADH (Wagner et al., 2020). Specifically, the KGDHC has been suggested to channel NADH directly toward NNT. Isocitrate dehydrogenase (IDH) is an enzyme involved in the TCA cycle, which takes place within the mitochondrial matrix. While IDH primarily functions to catalyze the conversion of isocitrate to alpha-ketoglutarate, it also generates NADH during this process.
The close spatial organization of IDH with other enzymes in the TCA cycle, such as alpha-ketoglutarate dehydrogenase (KGDC) and malate dehydrogenase (MDH), allows for efficient channeling of NADH and intermediates between these enzymes (Qi et al., 2008). The mechanism of NADH channeling from D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to L-lactate dehydrogenase (LDH) is a crucial aspect of glycolysis. In glycolysis, GAPDH catalyzes the conversion of glyceraldehyde-3-phosphate (GAP) to 1,3-bisphosphoglycerate (1,3-BPG) while generating NADH from NAD+. This NADH, a potent reducing agent, serves as a carrier for high-energy electrons.
The subsequent step in glycolysis involves the conversion of 1,3-BPG to pyruvate, a reaction catalyzed by phosphoglycerate kinase and pyruvate kinase (PK), yielding ATP. However, under certain conditions such as hypoxia or during intense exercise, when the demand for ATP surpasses oxygen availability, pyruvate is converted into lactate by LDH, regenerating NAD+ in the process.
The channeling mechanism facilitates the direct transfer of NADH generated by GAPDH to LDH, ensuring the efficient regeneration of NAD+ required for the continuation of glycolytic flux. This coordination between GAPDH and LDH exemplifies the concept of metabolic channeling (Svedruzic et al., 2020), where intermediates are efficiently shuttled between enzymes within a pathway, optimizing energy production under varying physiological conditions. This spatial proximity enhances the efficiency of substrate conversion and electron transfer within the cycle, optimizing energy production through the generation of NADH. This orchestrated channeling of NADH ensures its prompt utilization in subsequent oxidative phosphorylation reactions, contributing to the overall efficiency of cellular energy production.
The concept of channeling underscores the intricacies of metabolic pathways and the finely tuned mechanisms that cells employ to optimize their energy metabolism. The notion of physical enzyme–enzyme complexes, named metabolon, was introduced in 1985 (Srere, 1985), a transient structural–functional complex formed by consecutive enzymes of a metabolic pathway, a concept known as metabolic channeling, metabolite channeling, or substrate channeling. This process involves the retention of intermediates within the metabolon, with their conversions catalyzed by sequential enzymes.
Intracellular ATP, and indirectly NADH, that acts as an indirect regulator by influencing the ATP/ADP ratio, accounts for cellular energy status, which is sensed by ATP-sensitive potassium (KATP) channels. KATP are specialized ion channels found in the cell membranes of various tissues, including pancreatic beta cells, neurons, and cardiac muscle cells. Under conditions of cellular energy depletion, a decrease in ATP concentration leads to the opening of KATP channels, controlled by PEP (phosphoenolpyruvate), allowing the efflux of potassium ions and leading to cell hyperpolarization (Foster et al., 2022). This modulation of KATP channels plays a crucial role in regulating cellular excitability and functions, such as insulin secretion in pancreatic beta cells and protection against ischemic stress in the heart.
TCA cycle is characterized by peculiar processes that account for the removal and addition of intermediates to the cycle. The reactions, cataplerotic and anaplerotic pathways, are crucial to maintaining the cycle's activity, especially when intermediates are withdrawn for biosynthetic purposes or other metabolic pathways. Cataplerotic pathways involve the removal of TCA cycle intermediates for biosynthetic reactions (e.g., the conversion of oxaloacetate to phosphoenolpyruvate) or export from the mitochondria. Anaplerotic pathways, on the other hand, replenish TCA cycle intermediates (e.g., the conversion of pyruvate to oxaloacetate; the carboxylation of certain compounds such as pyruvate or phosphoenolpyruvate) that are diverted for biosynthesis or lost.
The alteration of specific reactions within the TCA cycle, such as truncating the cycle at citrate synthase or substituting the isocitrate dehydrogenase 3 reaction with the nicotinamide adenine dinucleotide phosphate (NADPH)-dependent isocitrate dehydrogenase 2, coupled with the export of isocitrate or citrate, decreases the yield of NADH produced by the TCA cycle. Various transport and cooperation mechanisms with cytosolic enzymes are involved in these alterations, leading to modified metabolic fluxes. For instance, the substitution of isocitrate dehydrogenase 3 with isocitrate dehydrogenase 2, favoring NADPH generation, affects the redox balance and alters the NADH/NADPH ratio within the mitochondria.
In addition, the export of isocitrate or citrate from the mitochondria affects the availability of TCA cycle intermediates for downstream reactions. Some of these transport shuttles might be disrupted, as the pyruvate–citrate pathway serves as a crucial channel for the movement of metabolites between cellular compartments, particularly involving the transport of pyruvate and citrate. Any disturbance in this pathway can disrupt the finely tuned balance of metabolic intermediates within the cell.
NAD/NADH molecules
NAD is an essential molecule involved in regulating numerous biochemical processes. If “NAD” refers to the chemical backbone, its oxidized and reduced forms are constituted by “NAD+” and “NADH,” respectively. The NAD+/NADH ratio is crucial for maintaining cellular reduction–oxidation (redox) homeostasis and also for modulating energy metabolism (Yang and Sauve, 2016). In fact, the pyridine nucleotides are well known to play pivotal role in ATP production and oxidation–reduction reactions of the cell. The ratio of NAD+/NADH is therefore an indicator of cell metabolism.
In the glycolytic pathway, there are different key enzymes responsible for generating NADH from NAD+; one of these is GAPDH, followed by pyruvate dehydrogenase complex, whereby the actual enzyme catalyzing NADH formation is dihydrolipoamide dehydrogenase (Yan et al., 2007). Specifically, the NADH produced in glycolysis can be utilized in reactions catalyzed by LDH or transported to the mitochondria for oxidation in the ETC (Gladden, 2004), to maintain the redox homeostasis (NAD+/NADH) in cytosol and mitochondria (Fig. 2).
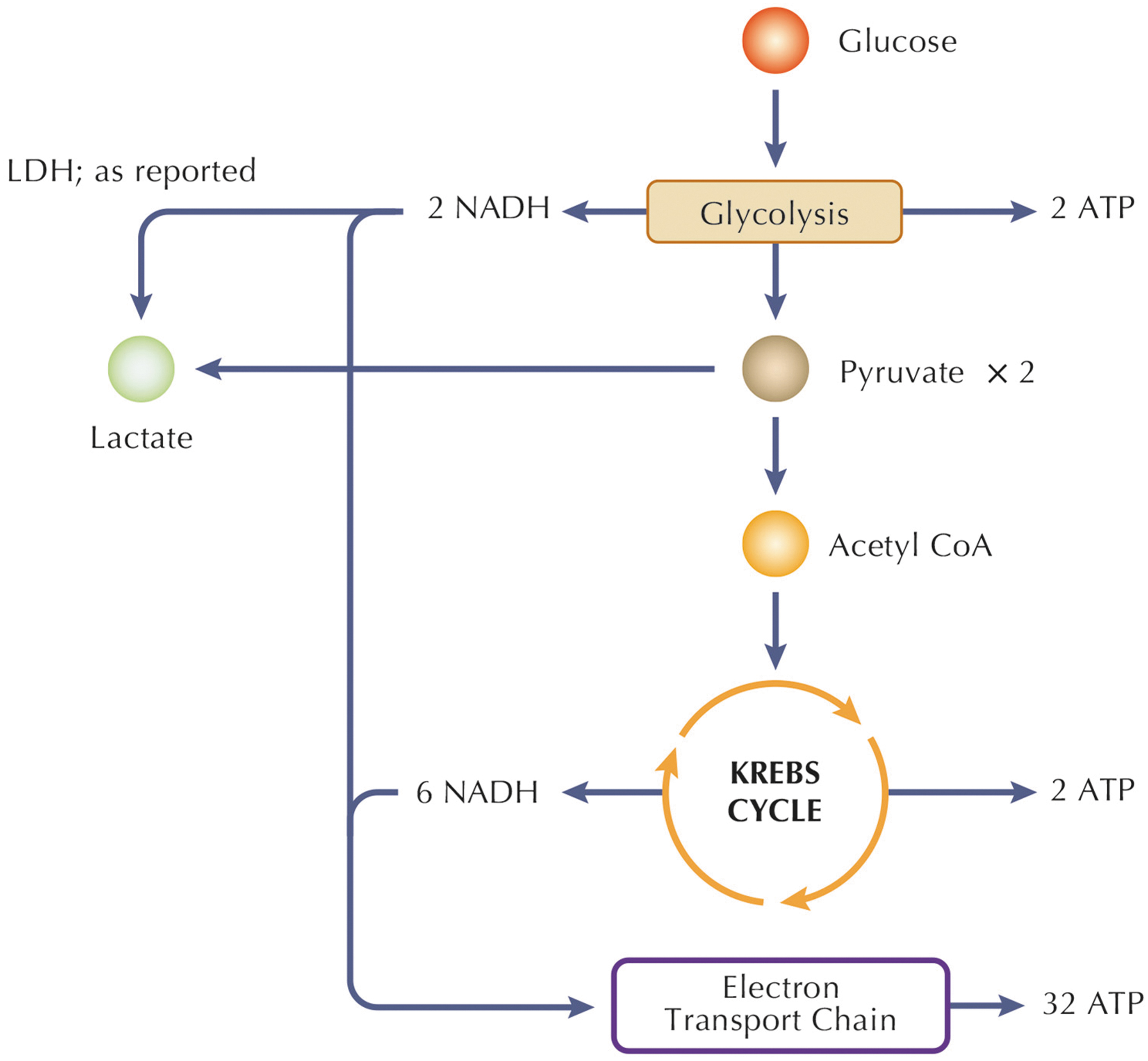
Redox homeostasis, particularly in the context of NAD+/NADH, plays a pivotal role in cellular function, energy production, and overall metabolic balance within both the cytosol and mitochondria. NAD+ and its reduced form, NADH, serve as key redox pairs in cellular respiration. In the cytosol, NAD+ is a crucial coenzyme involved in glycolysis, where it facilitates the conversion of glucose into pyruvate, generating NADH in the process. The produced NADH is shuttled to the mitochondria, where it contributes to oxidative phosphorylation. Within the mitochondria, NADH donates electrons to the ETC, driving the production of ATP.
The dynamic balance between NAD+ and NADH in both cellular compartments is essential for maintaining cellular redox equilibrium, efficient energy production, and preventing oxidative stress. Disruptions in this redox homeostasis are implicated in various diseases, highlighting the critical role of NAD+/NADH in cellular health and function. NADH effect on the mitochondria is carried out by specialized shuttle systems (e.g., malate-aspartate or glycerol-3-phosphate), classified as indirect systems since they transfer metabolites reduced in the cytosol and oxidized inside the mitochondria.
The malate–aspartate shuttle (Mal-Asp shuttle) requires the presence of MDH and aspartate amino transferase in both cytosolic and mitochondrial compartments (Abbrescia et al., 2012). The combined activity of these enzymes with two carrier systems catalyzes, respectively, the malate/α-ketoglutarate and the glutamate/aspartate exchange, promoting two cycling fluxes. One flux moves aspartate and malate into and out of the mitochondria, and the other, shown in Figure 3, moves glutamate in and α-ketoglutarate out in the opposite manner.
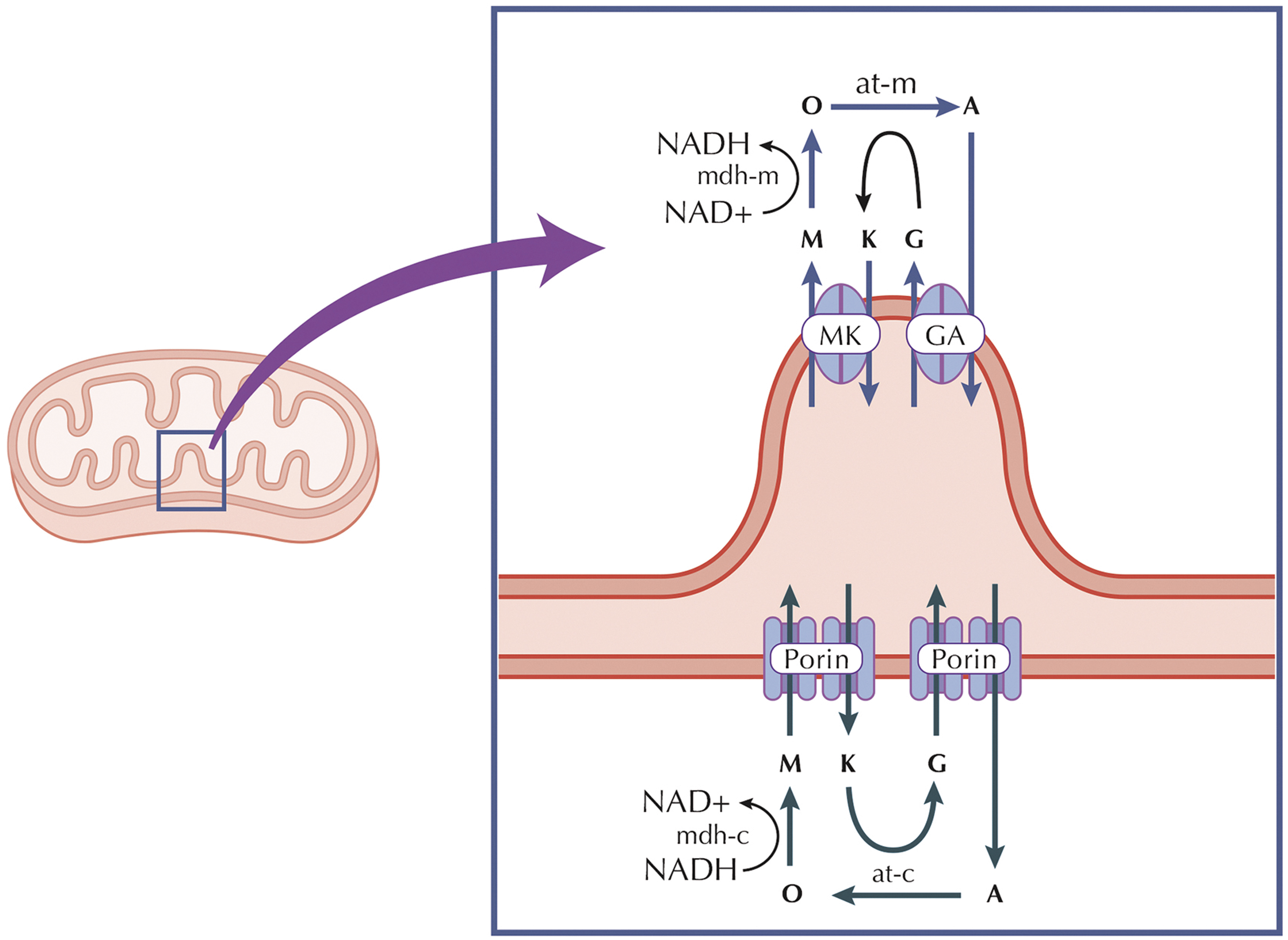
The out/in electrochemical proton gradient caused by the respiratory chain's activity controls the direction of the Mal-Asp shuttle (Abbrescia et al., 2012). The shuttle systems contribute to nonequilibrium distribution of NADH and NAD+ between the two different compartments of cytosol and mitochondria (Mason et al., 2020; Ren et al., 1988). The compartmentalized distribution of the NAD+/NADH and NADP+/NADPH redox couples is ensured by the specific localization of NAD+ and NADP+ biosynthetic enzymes and the bioavailability of NAD+ precursors (Xiao et al., 2018).
Nevertheless, these largely distinct redox states in cytosol and mitochondria play different roles in metabolic regulation (Powers and Schrager, 2022; Zhou et al., 2005). Recently, mammalian mitochondria were demonstrated to absorb complete NAD+ molecules by SLC25A51, a transporter of metabolites across the mitochondrial inner membrane (Girardi et al., 2020; Kory et al., 2020; Luongo et al., 2020; Merrins et al., 2022; Ouyang et al., 2021; Ziegler et al., 2021). When SLC25A51 is lost, there is a reduction in mitochondrial NAD+ levels, while the overall cellular NAD+ content remains unchanged. This loss negatively impacts mitochondrial respiration and hinders the intake of NAD+ by isolated mitochondria.
Besides ATP synthesis, the glycolytic and oxidative phosphorylation pathways can be implied in glucose metabolization (Bouche et al., 2004), and can stimulate insulin secretion by β cells (Prentki et al., 2013). As pancreatic beta cells respond to elevated blood glucose levels, insulin is released to facilitate glucose uptake by cells. This process is intricately linked to cellular respiration and, consequently, the generation of NADH. NADH synthesis resulting from this insulin secretion contributes to the cellular redox balance and the facilitation of various metabolic reactions, further underscoring the intricate connections between energy metabolism, hormonal regulation, and cellular function.
In fact, once inside the cells, glucose is phosphorylated by glucokinase to yield glucose-6-phosphate (G-6-P) (Matschinsky, 2002), which is then converted to two molecules of pyruvate by the glycolytic pathway. In the mitochondria, pyruvate carboxylase serves as a key enzyme in beta cells, converting pyruvate, derived from glucose metabolism through glycolysis, into oxaloacetate. This conversion is essential as it replenishes TCA cycle intermediates, sustaining the flux of metabolites in the cycle.
By channeling pyruvate to oxaloacetate, pyruvate carboxylase ensures a continuous supply of intermediates in the TCA cycle, supporting the generation of reducing equivalents like NADH (Merrins et al., 2022; Sugden and Holness, 2011). Moreover, the activity of pyruvate carboxylase in beta cells allows for an important anaplerotic pathway. This pathway facilitates the reverse MDH reaction, enabling the conversion of malate back into pyruvate within the mitochondria. This reversal helps recycle TCA cycle intermediates and recover pyruvate, mitigating the loss of one NADH molecule per turn of the TCA cycle (Fu et al., 2021).
Therefore, since both Krebs cycle and glycolytic pathways can be heavily fluxed by glucose under hyperglycemic conditions (Jankovic et al., 2015), NADH can be unbalanced in pathological conditions characterized by high-glucose levels such as diabetes (Luo et al., 2015), causing the alteration of redox homeostasis (Kowaltowski et al., 2009; Yan, 2014; Yarian et al., 2006).
Another critical element of energy metabolism involves ensuring ATP stability postenergy expenditure. The pathways responsible for ATP synthesis quickly engage to match the rate at which ATP is used (Li et al., 2020). These pathways encompass anaerobic glycolysis in the cytosol and oxidative phosphorylation within the mitochondria, implicated in interconverting NADH and NAD+ (Huttemann et al., 2007). Thus, due to the considerable surge in metabolic activity and subsequent NADH production during exercise, preserving the equilibrium of the NAD+/NADH ratio between the cytosol and mitochondria becomes exceedingly crucial to prevent oxidative stress (Lasorsa et al., 2003).
NAD+ is also converted into NADH during both fatty acid and amino acid oxidation, highlighting how the redox cycling of NAD between its oxidized (NAD+) and its reduced form (NADH) takes place in different metabolic pathways (Lin et al., 2021). A brain study conducted on aging and Alzheimer's disease (AD) mouse models revealed that redox-dependent bioenergetic enzymes possess the capability to produce NADH, instigating a metabolic shift.
An elevated proportion of NAD+/NADH redox states can serve as a signal prompting this metabolic shift aimed at generating additional NADH. This shift occurs through mechanisms involving epigenetics, redox-sensitive transcription factors, redox-sensing cysteines, nitration, and acylation of the KGDC, or by constraining the availability of the NAD+/NADH substrate. Consequently, this phenomenon leads to the generation of NADH, fulfilling the energy deficit and preserving the equilibrium of redox levels (Dong and Brewer, 2019).
Methods
This perspective reviews the major current data concerning NADH involvement in many biological processes and functions, as well as its correlation with aging and aging-related disorders with the purpose to evaluate a possible beneficial impact of the molecule on multiple clinical conditions. The selection of data and the sources of information have followed eligibility criteria according to the reviewed topic.
We have employed a set of electronic databases (Medline/PubMed, Scopus, Web of Sciences (WOS), Cochrane Library) for a systematic investigation of literature until May 2023. Combined text words and Medical Subject Headings (MeSH) terminology have been used, searching for MeSH keywords/terms, such as “NADH,” “NAD+/NADH ratio,” “NADH/NAD+ ratio,” “energy metabolism,” “aging,” “aging-related disorders,” and “therapies,” without applying date or language restriction criteria to include as much data as possible.
This study is in compliance with the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) statements (Moher et al., 2015). The assessment of title, abstract, and full texts of all the reported studies has been performed by two independent reviewers. In cases of duplicate information, the data were checked and combined to avoid overlapping. Studies reporting NAD+/NADH ratio as well as NADH have been evaluated, hence the selection of the publications has been conducted by typing specific keywords (i.e., NAD+/NADH ratio, NADH, energy metabolism, aging, aging-related disorders, etc.) also according to the date of publication (not older than 1969) and for the fulfillment of the topic of this review.
The reported studies exclude case reports and commentaries. The retrieval of the data from the selected studies has been performed by two reviewers separately, which have considered key characteristics of the studies such as publication year, author, type of study, country, sample size, and laboratory findings. A quantitative synthesis of the included studies has been performed. The funnel plot and Egger's regression test were used to assess publication bias (van Enst et al., 2014).
NADH Biosynthetic Pathways and Physiological Levels
Since NADH is the reduced form of NAD+, its production is strictly related to NAD biosynthesis as well (Fig. 4). In mammals, NAD/NADH biosynthesis can proceed via four main pathways: (i) de novo synthesis from tryptophan (TRP), synthesis from either form of (ii) vitamin B3, (iii) nicotinamide (NAM), or nicotinic acid (NA); or (iv) conversion of nicotinamide riboside (NR) (Fig. 5) (Imai, 2009). In mammals, De novo NAD biosynthesis from the amino acid TRP proceeds via the kynurenine pathway.
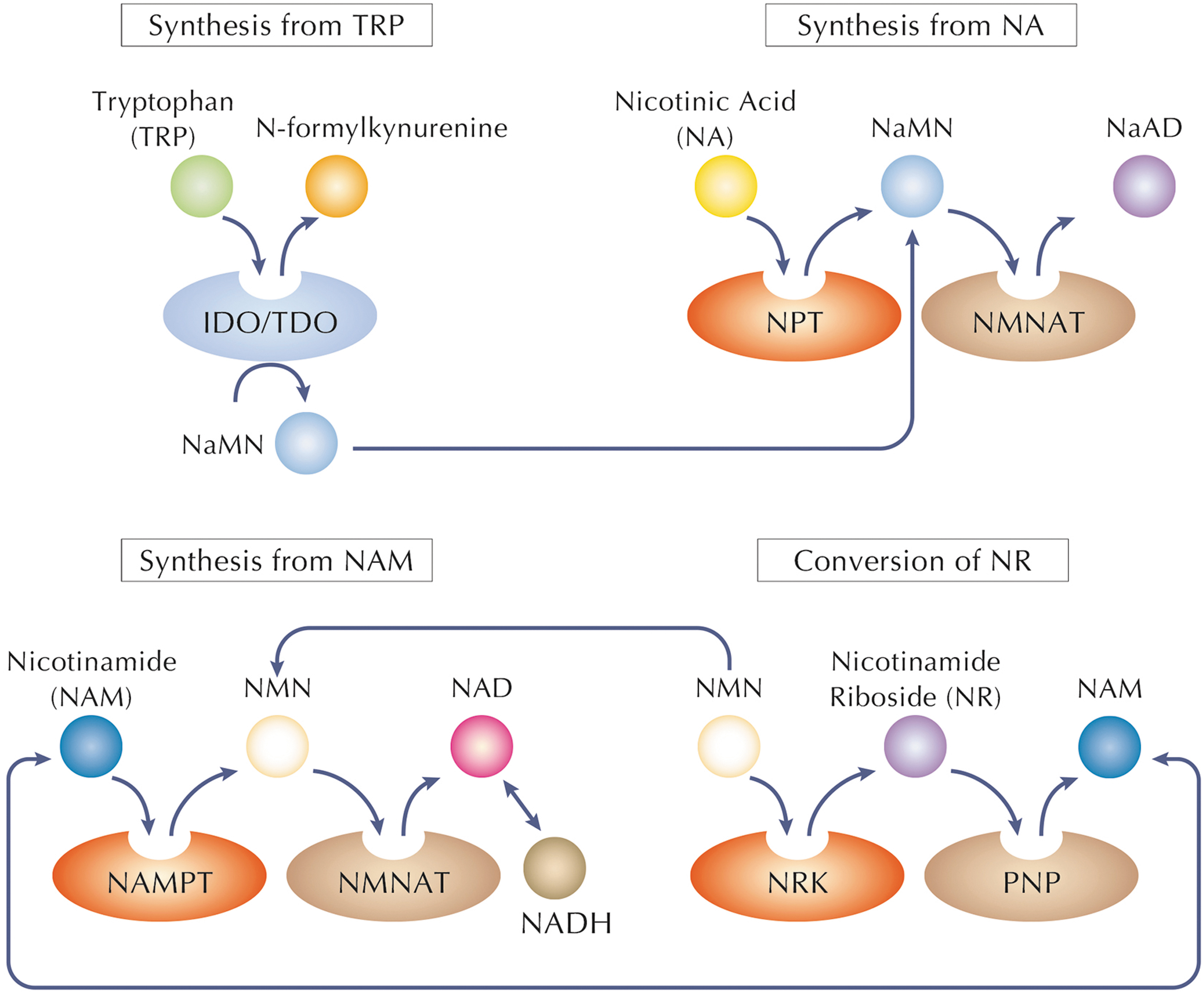
In particular, tryptophan dioxygenase and indoleamine-2,3-dioxygenase constitute the first and rate-limiting enzymes of this pathway to produce N-formylkynurenine (Penberthy and Tsunoda, 2009). The latter, through a series of enzymatic steps, is converted to nicotinic acid mononucleotide (NaMN), which is subsequently transformed into NAD by the sequential reactions of NMNAT and NADS (Fig. 5) (Penberthy and Tsunoda, 2009).
In cells, vitamin B3 undergoes enzymatic conversion to nicotinamide mononucleotide (NMN) through various intermediates (Makarov et al., 2019). NMN is further transformed into NAD through the action of specific enzymes, such as nicotinamide mononucleotide adenylyl transferases (NMNATs). This conversion is crucial for maintaining adequate cellular NAD levels, critical for processes such as glycolysis, the citric acid cycle, and oxidative phosphorylation.
NAM has a great ability to stimulate NAD biosynthesis (Trammell et al., 2016). Nicotinamide phosphoribosyltransferase (NAMPT), which is the rate-limiting enzyme in the NAD biosynthetic pathway from NAM, converts NAM and 5-phosphoribosyl-pyrophosphate to NMN. NMN is then adenylated to form NAD by NMNAT (Fig. 5) (Houtkooper et al., 2010; Revollo et al., 2007; Sauve, 2008). Accordingly, elevated gene expression of NAMPT but not NMNAT raises NAD levels (Nikiforov et al., 2011; Yang et al., 2007).
Conversion of NA to NAD takes place via the Preiss–Handler pathway (Bogan and Brenner, 2008). Nicotinic acid phosphoribosyltransferase converts NA to NaMN, which is then adenylated to nicotinic acid adenine dinucleotide (NaAD) by NMNAT (Bogan and Brenner, 2008; Hara et al., 2007). NaAD is amidated to NAD by NAD synthetase (NADSYN1 or NADS) (Fig. 5) (Bogan and Brenner, 2008).
Two pathways use NR to synthesize NAD (Bogan and Brenner, 2008). In the first one, ATP can be used to phosphorylate NR, generating NMN, in a reaction catalyzed by two isoforms of nicotinamide riboside kinase (NRK1, 2) (Bieganowski and Brenner, 2004). In the second pathway, purine nucleoside phosphorylase can break the glycosidic linkage of NR, so the NAM molecules liberated from this reaction are reused as starting point of the other biosynthetic pathway (Belenky et al., 2007; Belenky et al., 2009). All of these NAD biosynthetic pathways merge at the last step of the dinucleotide formation, catalyzed by NMNATs (Fig. 5).
In mammals, there are three NMNAT isoforms (NMNAT1–3), located in the nucleus, the golgi complex, and the mitochondria, respectively (Berger et al., 2005; Emanuelli et al., 2001; Schweiger et al., 2001). All the three NMNAT isoforms can use both the oxidized and reduced (NMNH) forms of NMN for NAD or NADH synthesis (Berger et al., 2005). The catalytic efficiency of NMNAT1 [Km = 20.1 mM (Revollo et al., 2004)] is much higher than the other isoforms, and mitochondrial NMNAT3 is the least efficient (Sorci et al., 2007).
Thus, the final production of NAD through these multiple routes constitutes the fundamental requirement to guarantee the following synthesis of NADH molecules via the different pathways previously explained.
In the cytoplasm of mammalian cells, the estimated concentration of NAD+ has been reported to range from ∼0.2 to 0.5 mM, while NADH concentrations are significantly lower, usually in the submicromolar range. The NAD+/NADH ratio in the cytoplasm typically ranges from 10:1 to 700:1, highlighting the predominance of the oxidized form, NAD+, in maintaining redox balance and supporting cellular processes. In mitochondria, where a significant portion of cellular energy production occurs, the NAD+/NADH ratio varies across compartments.
The mitochondrial matrix exhibits a higher NAD+/NADH ratio compared with the intermembrane space. Estimates suggest a matrix NAD+/NADH ratio ranging from 7:1 to 15:1, emphasizing the oxidized state's prevalence to facilitate oxidative phosphorylation and ATP synthesis. The levels of the free NADH fraction correspond to the overall NADH pool size, encompassing both free and bound NADH. Consequently, the absolute concentrations of free NADH may surpass alterations in the free NADH fraction of the total pool (Dong et al., 2019).
Generally, the levels of NADH are minimally impacted by the physiological concentrations of free NAD+ (Zhao and Yang, 2016), while they directly correlate with a cell's energy demands. As a result, organs requiring substantial energy, such as the heart, muscles, brain, and liver tissue, show elevated NADH levels, containing ∼90, 50, 40, and 11 mg/kg tissue, respectively (Birkmayer, 2009). Specifically, in hepatocytes, the estimated levels of mitochondrial and cytoplasmic NADH are 0.64 and 0.27 mM, respectively (Olek et al., 2004).
During specific cellular processes or in specialized cell types, alterations in NAD+ and NADH concentrations can occur. For instance, in situations of heightened energy demand, such as in exercising muscle cells or during periods of metabolic stress, the NAD+/NADH ratio may fluctuate, favoring NADH production to meet increased energy needs. In certain disease states, such as cancer or neurodegenerative disorders, disruptions in NAD+ metabolism have been observed. Lowered NAD+ levels or alterations in the NAD+/NADH ratio can impact cellular functions and contribute to disease progression.
Changes in the cytosolic and mitochondrial NADH levels are affected by several factors, such as NADH transport, ETC function, malate-aspartate shuttle activity, glucose metabolism, hypoxia, and redox environment (Zhao and Yang, 2016). For instance, blocking the malate-aspartate shuttle or Complex II will result in decreased mitochondrial NADL level, while it increases with the Complexes I, II, and IV inhibitors (Zhao et al., 2011). Metabolic rates can greatly affect NADH levels as well. For example, intensive physical exercise may lead to excessively elevated cytosolic NADH, resulting in cytotoxicity (Ying, 2006).
An extremely high intracellular NADH level may also lead to reductive stress because of its activity in inducing release of ferrous iron from ferritin leading to great oxidative damage, or due to the reactive oxygen species (ROS) generation, which is produced by the xanthine oxidase/xanthine dehydrogenase (Xiao et al., 2018; Ying, 2006). NADH can also induce cellular ROS production acting as a substrate for the NADPH oxidases (NOX) family proteins (NOX1-7) that produce H2O2 and O2 •− (Bedard and Krause, 2007). Therefore, a higher activity of G-6-P dehydrogenase leads to the increase of cellular NADH levels and also to the upregulation of NOX gp91phox and p22phox subunit mRNA expression, resulting in ROS production and oxidative damage in mouse pancreatic β cells and thymic lymphoma cells (Lee et al., 2011).
Understanding the concentrations and ratios of NAD+ and NADH is crucial in delineating cellular redox state, energy metabolism, and various metabolic pathways. Recent advancements in metabolomics, exemplified by previous studies (Chen et al., 2016), have provided valuable insights into estimating these concentrations across different cellular contexts.
The researchers outlined a technique to swiftly and precisely separate mitochondria, which they employed alongside a database called “MITObolome,” predicting mitochondrial metabolites. This combined approach enabled the measurement of >100 metabolites within the mitochondrial matrix across different respiratory chain (RC) functional states. These estimations provide critical insights into cellular redox balance, energy metabolism, and the regulatory role of NAD+ in physiological and pathological conditions.
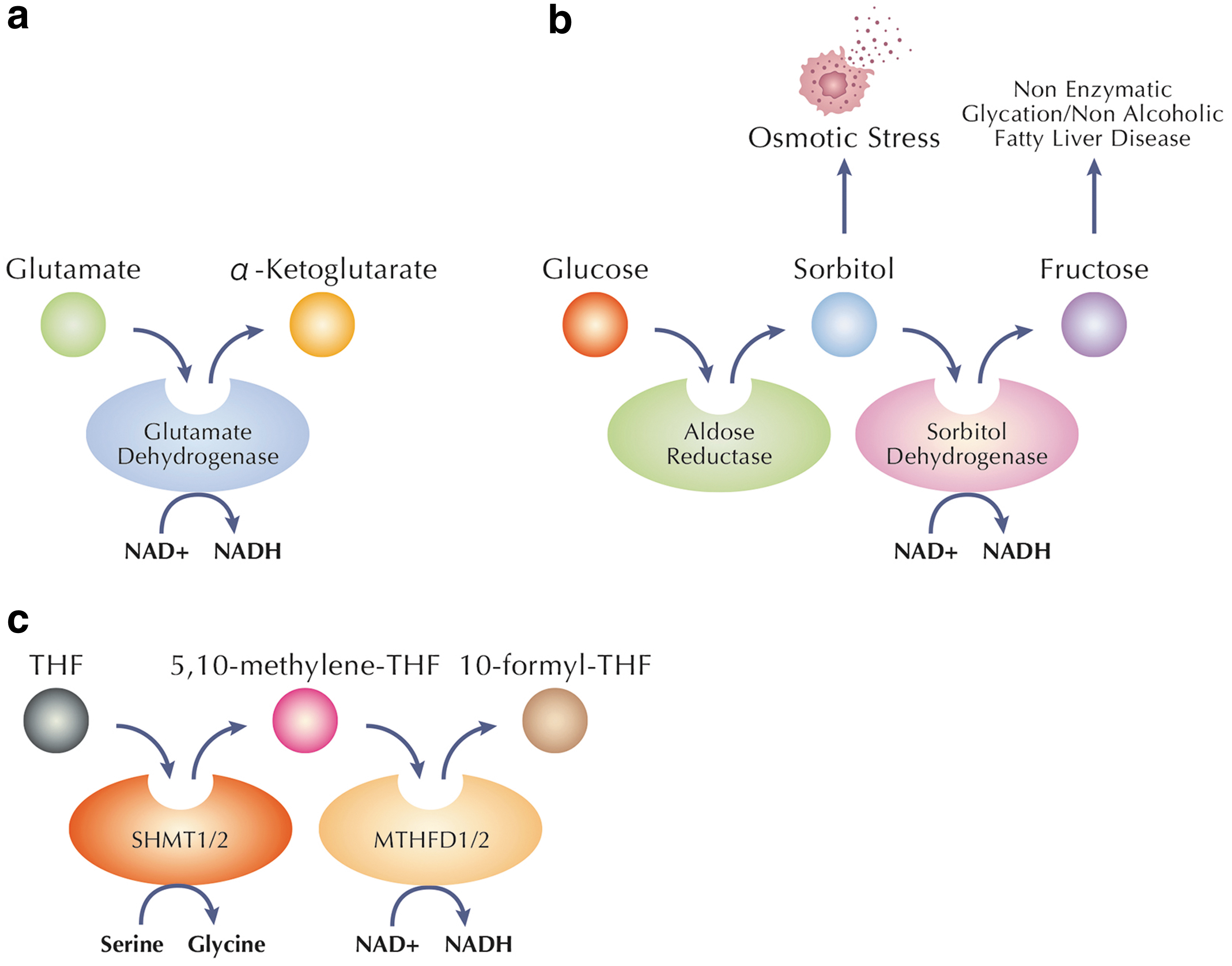
To maintain NADH physiological level, the central pathways involved in complete glucose breakdown and electron storage in NADH are the glycolytic pathway and the Krebs cycle. An alternative pathway generating NADH from NAD+ involves glutamate dehydrogenase (GDH), a central enzyme participating in α-ketoglutarate formation from glutamate (Gohring and Mulder, 2012; Li et al., 2014; Otter and Lammert, 2016) (Fig. 4a).
α-Ketoglutarate (AKG), also known as 2-oxoglutarate, is an essential metabolite because it participates in a variety of biological processes, including antioxidative defense, energy production, signaling mechanisms, and genetic modification. AKG can be transformed into glutamate by the enzyme GDH, and the resulting glutamate can be further metabolized into glutamine by glutamine synthase (Legendre et al., 2020; Radzki et al., 2009; Wagner et al., 2020). Since glutamate is an essential neurotransmitter, the GDH interaction is crucial for the physiological neuronal function, resulting in NADH production as a direct consequence. On the contrary, when glutamate is produced from glutamine, ammonia, which is a toxic compound, is also released leading to an increased risk of neurological damages (Bosoi and Rose, 2009).
An additional NADH biosynthetic route is the polyol pathway, including two consecutive reactions that are catalyzed by aldose reductase and sorbitol dehydrogenase, respectively (Fig. 4b). Under euglycemic conditions, this pathway is generally quite inactive (Dunlop, 2000), but under diabetic hyperglycemia it can become a highly active pathway for the disposal of excess glucose (Boesten et al., 2015; Yan, 2014). The creation of NADH, sorbitol, and fructose is the primary characteristic of this process (Chung and Chung, 2005; Dunlop, 2000; Iwata et al., 2007).
Yang et al. (2020) provided evidence that mitochondrial one-carbon metabolism can also produce NADH (Fig. 4c). One-carbon metabolism is an essential cellular process directly involved in nucleotide synthesis, cellular methylation, and redox balance (Ducker and Rabinowitz, 2017). Serine is the primary substrate of the one-carbon metabolism, and it transfers a carbon moiety to tetrahydrofolate in a reaction that occurs either in the cytoplasm or in the mitochondria through the enzymes SHMT1 or SHMT2, respectively.
In mitochondrial one-carbon metabolism, the two enzymes, named MTHFD2 and MTHFD2L, can catalyze the production of either NADPH or NADH (Shin et al., 2017). The study published here by Yang et al. (2020) reveals MTHFD2(L)'s ability to produce NADH in both physiological and pathological conditions, suggesting a new role for mitochondrial one-carbon metabolism in the synthesis of NADH.
NADH Consumption Routes
To maintain the physiological NAD+/NADH ratio, besides the various biosynthetic pathways, the consumption routes play a significant role, showing a great impact on both forms.
In particular, pyruvate is involved in redox cycling between NADH and NADPH (Cline, 2011; Heart et al., 2009; Jitrapakdee et al., 2006), due to the three pyruvate cycling pathways across the mitochondrial membranes. The first is pyruvate-malate pathway, in which pyruvate carboxylase converts pyruvate into oxaloacetate. The latter is then converted to malate by the enzyme mitochondrial MDH. Malate is shuttled out of mitochondria to the cytosol, to be converted back to pyruvate.
The whole process results in the consumption of NADH to form NADPH. The second consumption pathway is the pyruvate
Cytosolic NADH consumption is represented by its oxidation to NAD+ through the NADH shuttles, the LDH-catalyzed pyruvate-lactate conversion, and other dehydrogenase-catalyzed reactions. The modulation of the NADH shuttles may dramatically affect cellular functioning due to its impact on the fate of cytosolic NADH, since these routes may have significant implications on cellular energy metabolism and other cellular functions. In addition, alterations of the NADH shuttles could have significant pathological consequences (Wang et al., 2016).
Since NAD+ and NADH exert the role of coenzymes in numerous dehydrogenase-mediated reactions, multiple families of enzymes catalyze various reactions by consuming NAD+, consequently reducing the probability of NADH production as well. These reactions can significantly affect multiple biological functions, and result in degradation of NAD+ into nicotinamide and other products containing ADP ribose.
The major NAD+-consuming enzymes include the following: (i) poly(ADP-ribose) polymerases (PARPs) that consume NAD+ to produce nicotinamide and poly (ADP-ribose) on target proteins (d'Amours et al., 1999; Virag and Szabo, 2002; Ying et al., 2005); (ii) mono (ADPribosyl) transferases, a family of enzymes that uses NAD+ as a substrate to produce nicotinamide and mono-ADP-ribosylation of proteins (Corda and Di Girolamo, 2003; Di Girolamo et al., 2005); (iii) NAD+-dependent histone deacetylases; that is, the Sir2 family proteins that deacetylated histones by consuming NAD+ (Blander and Guarente, 2004); (iv) bifunctional ADPribosyl cyclases/cyclic ADP-ribose hydrolases, which can consume NAD+ to both generate cyclic ADPribose (cADPR) and hydrolyze cADPR into free ADPribose (Ziegler, 2000). The most powerful NAD+-consuming enzyme known to date is PARP-1 (d'Amours et al., 1999), which can lead to the decrease of intracellular NAD+ by ∼70% (Alano et al., 2004; d'Amours et al., 1999; Ying et al., 2005).
A different route of NADH consumption can involve NADH kinase that phosphorylates NADH to form NADPH by using ATP as a phosphoryl donor, representing the only pathway for the de novo NADPH biosynthesis and therefore resulting in the “consumption” of NADH molecules (Shi et al., 2009).
From the study of Pseudomonas fluorescens survival strategies in oxidative environment, it has emerged that NAD(H) kinase plays a key role in controlling NAD(H)/NADP(H) balance, especially NADPH from NADH, to be able to adapt to environmental variance (Lemire et al., 2008; Singh et al., 2007; Singh et al., 2008). This evidence suggests the critical role of NAD(H) kinase in adjusting NAD(H)/NADP(H) coenzyme in the metabolic networks of different species affecting NADH level. In particular, the P. fluorescens exposure to oxidative stress induces the activation of other enzymes involved in disparate metabolic modules, whose activities converge in converting NADH into NADPH.
At the same time, an upregulation of NAD kinase, malic enzyme, together with pyruvate carboxylase (PC) of gluconeogenesis and MDH of TCA cycle, has been observed, ensuring the cyclic supplying of NADPH from NADH. Conversely, the phosphoenolpyruvate carboxykinase (PEPCK) is downregulated, while the PK is upregulated. PEPCK and PK regulation is responsible for enhancing the acceleration of oxaloacetate, which contributes to NADH oxidation and, together with all the previously mentioned regulations, can result in effective synthesis of NADPH (Singh et al., 2008).
Several are the routes of NADH consumption due to the multiple effects exerted by this molecule.
Biological Functions of NADH
NADH exerts several biological functions and takes part in multiple processes such as in antioxidant defenses, by supporting the regeneration of reduced glutathione (GSH), protecting cells from oxidative stress. The multifaceted functions of NADH underscore its significance in cellular energy production, redox balance, and the orchestration of diverse metabolic processes essential for cellular function and survival.
Metabolism
Foremost, NADH is a cofactor for various enzyme reactions, further emphasizing its crucial role in numerous cell functions (Pelzmann et al., 2003). It produces biological effects by regulating numerous NAD+/NADH-dependent enzymes (Walker and Tian, 2018), as shown in Table 1. As a cofactor, NADH serves as an electron carrier, shuttling high-energy electrons derived from metabolic reactions to the ETC in the mitochondria. This involvement is particularly prominent in processes such as glycolysis and the TCA cycle, where NADH is generated during the oxidation of glucose-derived substrates.
Major Enzymatic Reactions in Substrate Metabolism That Are Dependent Upon NAD+/NADH Ratio
↓, decreased ratio; ↑, increased ratio.
Moreover, NADH acts as a cofactor for dehydrogenase enzymes in various biosynthetic pathways, including fatty acid synthesis. Its role extends to supporting antioxidant defenses by participating in the reduction of GSH, contributing to cellular protection against oxidative stress. The versatility of NADH as a cofactor highlights its fundamental importance in facilitating diverse enzymatic reactions critical for energy production, metabolic regulation, and the maintenance of cellular redox balance.
Mitochondrial functions
NADH is primarily found in mitochondria and, together with FADH2, is one of the essential coenzymes that benefit the mitochondrial state as it contributes to generating ATP (Lee et al., 2022). In addition, NADH improves the mitochondrial membrane potential due to its property of inhibiting voltage-dependent anion channels (VDACs) (Fig. 6) in the mitochondria's outer membrane. NADH interacts with VDAC, influencing the channel's permeability and contributing to the regulation of mitochondrial function, influencing the flux of metabolites across the mitochondrial outer membrane, affecting processes such as oxidative phosphorylation and apoptosis.
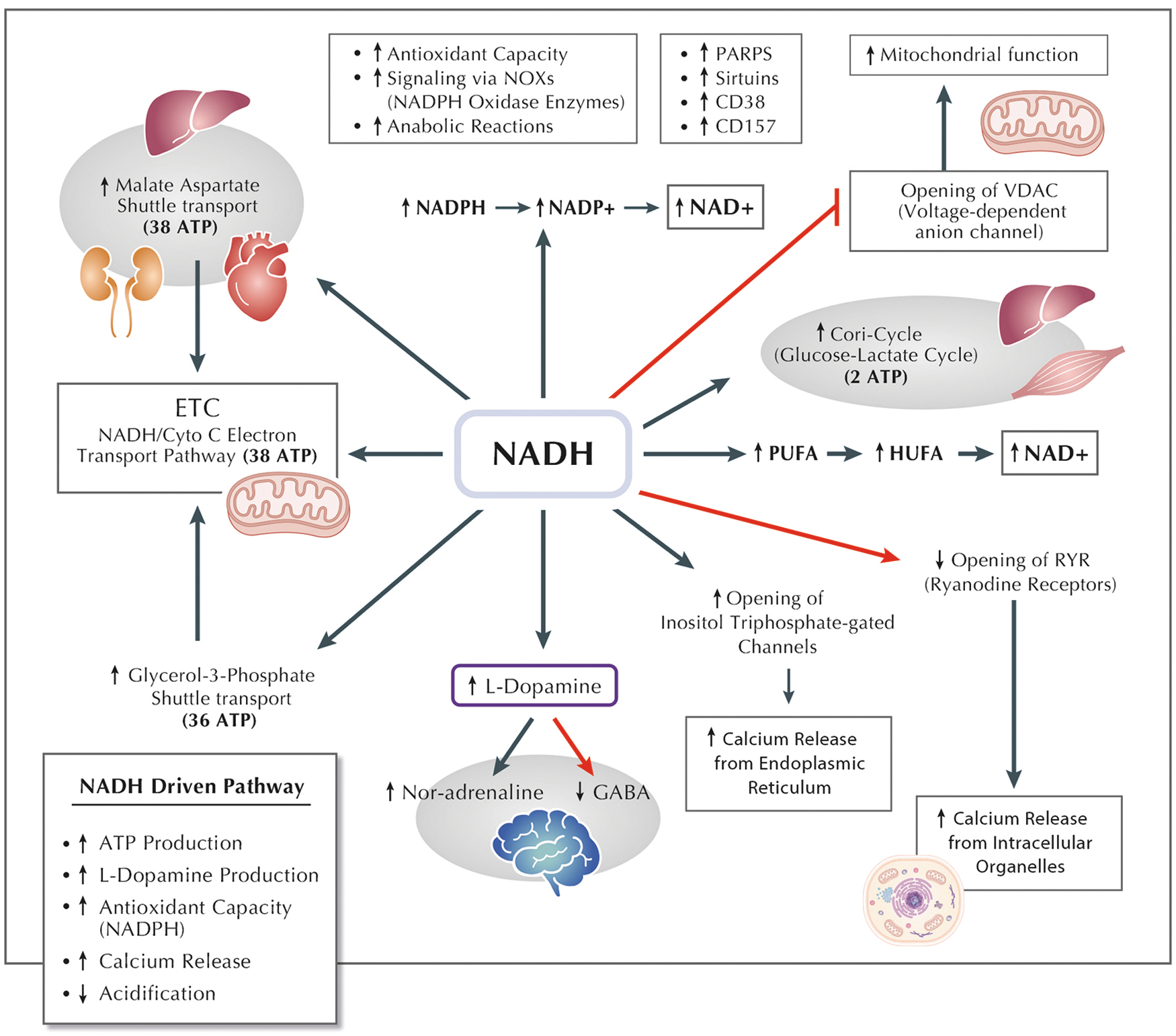
The improvement of the mitochondrial membrane potential is also controlled through cytochrome oxidase, which can directly utilize the extramitochondrial NADH. Cytochrome oxidase is a key enzyme in the ETC located in the inner mitochondrial membrane. Extramitochondrial NADH, generated outside the mitochondria, can serve as a substrate for cytochrome oxidase. This utilization of NADH by cytochrome oxidase not only contributes to the maintenance or improvement of the mitochondrial membrane potential but also represents a dynamic connection between cellular compartments.
The delicate balance of the NAD+/NADH ratio emerges as a crucial factor in orchestrating mitochondrial functions, particularly in the context of the mitochondrial permeability transition (MPT) and calcium homeostasis. The NAD+/NADH ratio serves as a metabolic indicator, reflecting the cellular redox state and energy status. Perturbations in this ratio have been associated with the modulation of MPT, a phenomenon where the mitochondrial inner membrane becomes permeable, influencing the release of various molecules. A balanced NAD+/NADH ratio helps regulate MPT, preventing the undesired opening of MPT pores. Furthermore, this ratio indirectly influences calcium homeostasis within the mitochondria, as the activity of enzymes involved in calcium regulation often depends on NADH availability (Ying, 2006).
Calcium homeostasis
NADH plays a crucial role in regulating calcium homeostasis, primarily by influencing intracellular Ca2+ channels (Fig. 6), particularly the inositol 1,4,5-triphosphate (IP3)-gated Ca2+ channels, and by inhibiting ryanodine receptors (Patterson et al., 2005; Ying, 2006). The activity of GAPDH can impact local NADH levels, thereby potentially regulating IP3R activity in response to signals during respiratory metabolism. This modulation can lead to the release of Ca2+, entering mitochondria to enhance oxidative respiration or release cytochrome c (Patterson et al., 2005).
NADH potency in modulating IP3-gated Ca2+ channels is four times that of NAD+, displaying a fivefold increase in its maximal effect (Lahiri et al., 2003), and the half-maximal augmentation of IP3R calcium release occurs at 50 μM NADH (Patterson et al., 2005). In addition, hypoxia-induced increases in NADH directly contribute to Ca2+ release from IP3-gated Ca2+ channels in specific cellular contexts (Fig. 6) (Lahiri et al., 2003). Furthermore, NADH inhibits ryanodine receptors in cardiac muscle (Fig. 6), possibly due to its NADH oxidase activity in the cardiac sarcoplasmic reticulum (Ying, 2006).
Gene expression
When NADH is not used as a substrate for ATP production, it is transported to the cytosol to modulate protein acetylation and gene expression (Marcu et al., 2014; Ying, 2006).
The mechanism through which NADH is responsible for regulating gene expression can involve other molecules implicated in the transcriptional pathway. For instance, both NAD+ and NADH have a direct effect on the function of the corepressor carboxyl-terminal binding protein (CtBP), which binds cellular and viral transcriptional repressors, with the overall effect of silencing the expression of specific genes implicated in development, cell cycle regulation and transformation. It has been demonstrated that increasing levels of NADH can stimulate CtBP binding to its targets, leading to the improvement of CtBP-mediated transcriptional repression (Zhang et al., 2002).
The regulation of gene expression can also occur at an epigenetic level, thanks to the role of molecules such as Sirtuins in regulating chromatin condensation and transcription. Sirtuins, a class of highly conserved NAD+-dependent protein deacetylases, play a central role in regulating cellular processes associated with aging (Covarrubias et al., 2021), metabolism, and stress response. Named after the silent information regulator 2 (Sir2) protein in yeast, sirtuins have been implicated in diverse biological functions, including DNA repair, apoptosis, and energy metabolism.
There are seven sirtuin isoforms (SIRT1-7) in mammals, each with distinct cellular localization and substrates (Tang, 2016). SIRT1, the most extensively studied isoform, is involved in various pathways such as the deacetylation of transcription factors such as p53 and forkhead box O (FOXO), contributing to enhanced DNA repair and increased stress resistance. Sirtuins influence metabolic homeostasis by regulating key metabolic pathways, including gluconeogenesis, fatty acid oxidation, and mitochondrial function. Furthermore, their dependence on NAD+ for activity links sirtuins to cellular energy status, making them crucial sensors of metabolic health (Xie et al., 2020).
The modulation of sirtuin activity has garnered significant interest in the field of longevity research, and various compounds that boost NAD+ levels, such as resveratrol, have been explored for their potential sirtuin-activating effects. An increased NAD+/NADH ratio, indicative of higher cellular energy levels, positively influences sirtuin activity. NADH, being the reduced form of NAD+, competes with NAD+ for binding to sirtuins, potentially inhibiting their deacetylase activity (Anderson et al., 2017). Therefore, the balance between NAD+ and NADH emerges as a crucial determinant of sirtuin function, impacting cellular responses to stress, DNA repair, and metabolic regulation.
Cell death
NAD+ and NADH forms have a role in cell death, such as necrosis, apoptosis, and ferroptosis.
Necrotic cell death is the typical consequence of severe acute cellular injury (Trump and Berezesky, 1995), characterized by the formation of protrusions of the plasma membrane called blebs (Lemasters et al., 1983). When these plasma membrane blebs break, irreversible injury occurs (Lemasters et al., 1987), resulting in inflammation. Different findings reported that necrosis can be induced by SIRT2 silencing, a deacetylase (Milne and Denu, 2008) member of the sirtuin family proteins (Dali-Youcef et al., 2007), already involved in various biological functions such as gene regulation (Ying and Xiong, 2010).
Since NAD+ and NADH are regulators of sirtuins, both the molecules could participate in inducing necrosis. A recent study has highlighted the main histological changes observed in pancreatic islets pretreated with a unique NADH intraperitoneal injection in a streptozotocin-induced diabetes model, to evaluate the function of NADH toward necrosis in pancreas. At a histological level, the NADH pretreatment revealed a decrease in beta cell death reducing necrosis in favor of apoptosis, with the aim to prevent inflammation with further beta cell destruction (Abdellatif et al., 2023).
Apoptosis is a form of programmed cell death that occurs without causing inflammation. At the early stages of apoptosis, there is a depletion of NADH/NADPH, and the concentration of NAD(P)H is reduced (Petit et al., 2001). This reduction in NAD(P)H can be detected before several other characteristic changes associated with late apoptosis. These changes include increased superoxide production, exposure of phosphatidylserine on the cell surface, loss of cytosolic potassium (K+), decreased cytoplasmic pH, nuclear DNA fragmentation, cell shrinkage, loss of viability, and the appearance of the mitochondrial antigen APO2.7.
Elevated NADH concentration has been observed to inhibit the opening of the permeability transition (PT) pore (Batandier et al., 2004), and in specific circumstances it may prevent apoptosis. Conversely, NAD(P)H depletion leads to PT pore opening, resulting in uncoupling of the RC, interruption of electron transfer, and the release of cytochrome c with the overproduction of superoxide anions. The NAD+/NADH ratio is considered a significant index of cellular reducing power, influencing MPT and mediating apoptosis under various conditions (Kharechkina et al., 2021). The relationship between NADH depletion, loss of the inner mitochondrial transmembrane potential, caspase 3 activation, and changes in cellular redox processes, including GSH depletion, underscores the critical role of NADH in apoptotic pathways (Trachootham et al., 2008).
Ferroptosis, a form of regulated cell death characterized by lipid peroxidation, has the NAD+/NADH ratio as a critical determinant. Perturbations in this ratio can influence ferroptotic cell death through several mechanisms. NAD+ is essential for the activity of certain enzymes involved in antioxidant defenses, such as sirtuins and PARPs, which are implicated in modulating ferroptotic pathways (Tang et al., 2021). In addition, NADPH, generated through NADH oxidation, is crucial for the function of the GSH and thioredoxin systems (Zheng and Conrad, 2020), which are central to cellular antioxidant defense mechanisms against lipid peroxidation and ferroptosis. Alterations in the NAD+/NADH ratio can disrupt the redox balance, diminish the antioxidant capacity, and potentially promote susceptibility to ferroptosis.
Due to the critical roles of oxidative stress in cell death, NADH may modulate cell life span by influencing oxidative stress and causing death. Since the redox couple plays key roles in numerous redox reactions, it is already well known that NAD+/NADH ratio is an index of cellular reducing potential. In particular, some studies have suggested that NADH can produce direct antioxidation effects (Kirsch and De Groot, 2001; Mazzio and Soliman, 2003; Olek et al., 2004), even if it is noteworthy that excessive intracellular NADH can produce “reductive stress,” which may result from the capacity of xanthine oxidase/xanthine dehydrogenase to generate ROS by oxidizing NADH (Sohn et al., 2003), or from the capacity of NADH to induce release of ferrous iron from ferritin (Wu et al., 2016).
Redox homeostasis
NADH acts as a potent antioxidant directly in both enzymatic and nonenzymatic reactions, as illustrated in Figure 6. Unlike other forms of nicotinamide nucleotides, NADH, being in its reduced form and possessing hydrophilic properties, exhibits the ability to inhibit lipid peroxidation (Castro-Marrero et al., 2021; Olek et al., 2004). Mitochondrial NNT under normal physiological conditions regenerates NADPH and utilizes NADH as a substrate to fuel NADPH-dependent antioxidative processes.
Interestingly, activating or overexpressing NNT has been found to reduce the NAD+/NADH ratio, converting high levels of NADH to NAD+. This shift contributes to a decrease in ROS levels, demonstrating the potential role of NNT in mitigating oxidative stress (Olgun, 2009). These findings highlight the dual role of NADH as an antioxidant and as a substrate in maintaining cellular redox balance through NNT activity (Bertero and Maack, 2018).
The antioxidant properties of NADH are observed through enhanced levels of lymphocyte GSH and catalase activity, coupled with reduced levels of malondialdehyde and carbonyl proteins (Bouamama et al., 2017). In addition, in vitro treatment of PC12 cells with 1 mM of NADH has shown improvements in the levels of nuclear factor erythroid 2-related factor 2, catalase activity, and total GSH. This improvement is attributed to NADH's influence on sirtuins, particularly SIRT2, as depicted in Figure 6 (Bouamama et al., 2017).
In an in vitro study, NADH demonstrated its capacity to reduce oxidative stress by rescuing the human liver cell line LO2 from damage caused by X-ray irradiation. NADH exerted cryoprotection effects by preventing the induction of apoptosis, thereby maintaining cell viability through the activation of autocrine IL (interleukin)-4 production (Bouamama et al., 2017; Liu and Zhang, 2003). Furthermore, NADH exhibited protective effects by upregulating the expression of the antiapoptotic Bcl-2 protein and downregulating the expression of proapoptotic proteins, including p53, bax, Fas, and Fasl.
Another study observed similar protective effects of NADH on DNA damaged by doxorubicin, attributing the repair mechanism to the modulation of crucial cell division-related proteins such as cyclin A, cyclin B1, p53, and Bcl-2 (Birkmayer, 2009). NADH also restored the antioxidant properties of coenzyme Q10, an essential component of the ETC crucial for mitochondrial ATP production (Birkmayer, 2009; Castro-Marrero et al., 2021). Notably, a human study administering 20 mg of NADH daily in combination with CoQ10 for 12 weeks resulted in reduced fatigue in individuals with myalgic encephalomyelitis, commonly known as chronic fatigue syndrome (Castro-Marrero et al., 2021).
Interestingly, there is a direct relationship between NADH and the formation of superoxide at Complex I of the ETC (Kussmaul and Hirst, 2006). When Complex I is impaired or overloaded, an increased level of reduced NADH contributes to electron leakage, leading to the production of superoxide radicals. This phenomenon underscores the intricate relationship between cellular redox balance, NADH as an antioxidant, and the delicate equilibrium between ROS production and scavenging systems, shedding light on the interconnectedness of these processes in maintaining cellular homeostasis.
Complex I is a major source of ROS production within mitochondria, primarily generating superoxide radicals as a by-product of electron transfer reactions. Over time, the oxidative damage inflicted by ROS can impair mitochondrial function, leading to a decline in cellular energy production and increased cellular dysfunction. Dysfunction of Complex I itself can further exacerbate ROS generation, creating a vicious cycle of oxidative stress and cellular damage. Thus, Complex I occupies a pivotal position in the mitochondrial free radical-dependent aging (Barja, 2013), serving as both a contributor to ROS production and a target for oxidative damage, highlighting its significance in the aging process and age-related diseases.
Similarly, NADPH serves as a critical player in cellular antioxidant defenses, primarily through its involvement in maintaining the reducing environment necessary for the function of various antioxidant systems. It powers the regeneration of antioxidants such as GSH and thioredoxin, essential for scavenging ROS and countering oxidative stress. Conversely, NOX constitute a family of enzymes responsible for ROS generation by transferring electrons from NADPH to molecular oxygen. This dual role of NADPH and NADH highlights their significance in redox balance regulation.
Consequently, the interplay between NAD+/NADH and NADH/NAD+ ratios is pivotal in maintaining cellular homeostasis. Disruptions in this equilibrium can impact redox-sensitive signaling pathways, leading to oxidative stress and cellular damage. Oxidative stress, characterized by an imbalance in ROS production and antioxidant defenses, often arises from alterations in NAD+ metabolism. It can trigger detrimental effects on cell function, contributing to aging, neurodegenerative diseases, and metabolic disorders. Strategies aimed at restoring redox balance, such as enhancing NAD+ levels through supplementation or modulating NAD+/NADH ratios through metabolic interventions, hold promise in mitigating oxidative stress and preserving cellular health, highlighting the critical role of redox homeostasis in overall physiological well-being.
Nutrition and ketosis
Diet and food intake influence NADH levels acting as NADH regulators, by alternative increase or decrease in its amount, affecting NAD+/NADH ratios.
The ketogenic diet, a high-fat, low-carbohydrate diet created to mimic the effects of calorie restriction, is not only an extremely efficacious treatment for medically intractable epilepsy (Rubenstein and Vining, 2004; Ye et al., 2022), but it also induces several metabolic changes, notably an increase in serum concentrations of the ketones β-hydroxybutyrate (BHB) and acetoacetate (ACA) (Denny et al., 2006).
Ketones are by-products of fat metabolism, produced in hepatic mitochondria (Fukao et al., 2004) from the precursor acetyl coenzyme A (acetyl CoA), which is formed by β-oxidation of free fatty acids in the liver. Acetoacetate is reduced to BHB by β-hydroxybutyrate dehydrogenase using NADH as a reductant (Veech, 2004). Therefore, the presence and ratio of BHB to acetyl CoA reflect the redox state within the mitochondrial matrix (Veech, 2006) and the cellular pool of NAD+/NADH (Stern et al., 2023).
In particular, the conversion of acetoacetate to beta-hydroxybutyrate is coupled with the oxidation of NADH to NAD+, directly impacting the cellular NAD+/NADH ratio. This metabolic shift promotes a more reduced cellular environment, influencing various cellular processes. In addition, ketones have been associated with the activation of certain NAD+-dependent enzymes, such as sirtuins, which play roles in cellular metabolism, longevity, and stress response. Several studies have stated that ketones inhibit the increased mitochondrial production of ROS associated with excitotoxic injury by enhancing NADH oxidation (i.e., increased NAD+/NADH ratios) in the ETC, affecting directly the level of NADH (Maalouf et al., 2007).
NADH in Aging-Related Disorders
The dysregulation of oxidative stress and NADH levels is believed to significantly impact the aging process by contributing to its primary hallmarks and influencing pathological pathways associated with various age-related diseases (Luo et al., 2020). Notably, studies have shown that the plasma NAD+ metabolome undergoes dysregulation during “normal” aging (Clement et al., 2019). This observation implies that the balance of the NAD+/NADH pool plays a crucial role in a diverse array of cellular processes, including the aging process and, consequently, the development of aging-related disorders.
Schwarzmann et al. (2021) revealed that human plasma contains low micromolar concentrations of total NAD, with a higher prevalence of the oxidized form, NAD+, compared with the reduced form, NADH. However, an age-related decline in the difference of the plasma NAD+/NADH ratio between men and women has been noted, indicating a potential influence of age on the regulation of extracellular molecules. This is further supported by markers of biological age showing a sex-related difference in the plasma NAD+/NADH ratio and its decline with higher biological age compared with chronological age (Clement et al., 2019).
Investigations into the impact of NADH on aging brains demonstrated enhanced cognitive functions in old rats upon NADH administration (Rex et al., 2004). Other studies highlighted age-associated differences in NADH-dependent responses to anoxia, with adult rats showing a 36% increase and old rats a 10% increase in NADH levels, suggesting age-related variations in NADH-dependent processes (Zarchin et al., 2002).
In addition, observations in aged rats indicated lower NADH oxidase activity in nonsynaptic mitochondria (Genova et al., 1997). The decline in the energetic NADH pool with age is closely associated with the development of various age-related diseases (Ying, 2006). This decrease is attributed to a reduced capacity to regenerate free NADH necessary for fueling oxidative phosphorylation (Dong and Brewer, 2019). Restoring free NADH levels in aging neurons to those of young age is suggested as a potential strategy to counter age-associated diseases and increase life span.
The association between Sir2, the yeast homolog of SIRT1, and the regulation of life span has sparked interest in exploring the modulation of Sir2 activity through NADH as a potential strategy for extending life span, particularly influenced by caloric restriction, as proposed by Tissenbaum and Guarente in 2001. Caloric restriction has been linked to increased longevity, and Sir2 has been identified as a key player in this process.
Although in vitro studies have suggested that NADH might inhibit sirtuin activities, particularly at nonphysiological millimolar levels, as observed in research by Madsen et al. (2016) and Walker and Tian (2018), in vivo reports indicate a more nuanced relationship. Instead of directly sensing NADH, sirtuins are suggested to require NADH as a cosubstrate for their enzymatic activity, as highlighted in studies by Madsen et al. (2016).
Among aging-related disorders, cardiovascular diseases, infections, and cognitive impairments like Parkinson's disease (PD) and AD are particularly noteworthy. NADH administration has shown potential benefits in alleviating symptoms of cognitive impairment, possibly by increasing intracellular ATP levels and decreasing PARP-mediated cell death (Ma et al., 2015; Ying, 2007). In Parkinson's patients, NADH supplementation has been used to boost endogenous dopamine production, showing positive effects in clinical trials (Birkmayer et al., 1989; Kuhn et al., 1996).
Similarly, studies have reported improved cognitive functions in AD patients after NADH treatment (Demarin et al., 2004). An interesting in vivo study explored the reversibility of age-related oxidized free NADH redox states in AD neurons through external cysteine (Cys)/cystine (CySS) redox shifts. The results suggested the potential reversibility of free NADH levels in old age and AD with external oxidative or reductive stress, offering a perspective for innovative therapeutic approaches (Dong et al., 2019). The effect of NADH in age-related diseases may be also connected to the ability of NADH to enter astrocytes.
It has been observed that extracellular NADH, at a concentration of 10 μM, entered astrocytes and exhibited protective effects by mitigating poly(ADP-ribose) polymerase-1-induced astrocyte death (Lu et al., 2007; Zhu et al., 2005). This entry into astrocytes is facilitated through the P2X7 receptor (P2X7R), which also influences the entry of NAD+ into cortical neurons (Ying, 2006; Ying, 2013). Notably, even low concentrations of NADH treatment, as low as 1 μM, significantly reduced the number of surviving C6 glioma cells, leading to an increase in PARP activity (Fig. 6). Moreover, the effects of NADH on glioma cell survival were diminished when PARP inhibitors were introduced, emphasizing the significant roles played by oxidative stress mediation and PARP activation in these observed effects (Ma et al., 2011).
The increase in susceptibility to infections as an age-related disease is a well-documented phenomenon associated with the aging process. Aging is characterized by a gradual decline in the efficiency of the immune system, a phenomenon referred to as immunosenescence. This decline involves alterations in both the innate and adaptive immune responses, leading to a reduced ability to mount robust defenses against pathogens.
In the context of infectious diseases, the relationship between infections and NAD+/NADH is under exploration. The molecule PARP-1, involved in NAD+/NADH metabolism, plays a crucial antiviral role through ADP-ribosylation of the viral genome. However, certain viral families can inhibit PARP-1, leading to excessive NAD+ consumption (Andrabi et al., 2014; Zhu et al., 2016). Understanding this association could potentially aid in mitigating the severity of aging-related microbial infections.
Aging-related decline in SIRT activity has prompted investigations into NAD+ and NADH biosynthetic precursors as potential interventions for cardiovascular disturbances (Fang et al., 2017), with compounds such as NR and NMN currently undergoing clinical trials.
NAD+ and NADH precursors are theorized to confer cardioprotective effects through the activation of SIRT1 in the context of heart failure, as suggested by studies such as those conducted by Hsu et al. (2010) and Nadtochiy et al. (2011). SIRT1, a member of the sirtuin family of proteins, is known for its role in regulating cellular processes associated with longevity, metabolism, and stress response. In the setting of heart failure, where the heart's ability to pump blood efficiently is compromised, activating SIRT1 through NAD+ and NADH precursors is thought to have beneficial effects. These precursors, which may include compounds like NR and NMN, are believed to enhance cellular NAD+ levels and, in turn, stimulate SIRT1 activity.
Therapeutic Effects of NADH
NADH has garnered attention for its potential therapeutic applications due to its crucial role in cellular energy production and various cellular processes. As a coenzyme involved in redox reactions, NADH participates in the transfer of electrons during metabolic pathways, contributing to the synthesis of ATP—the primary energy currency of cells. Given its role in supporting mitochondrial function, NADH has been explored as a potential treatment for conditions characterized by mitochondrial dysfunction, such as certain neurodegenerative disorders. Studies have investigated the use of NADH supplementation in diseases such as Parkinson's and Alzheimer's, where disruptions in cellular energy metabolism are implicated (Amjad et al., 2021).
Furthermore, NADH's antioxidant properties make it a candidate for combating oxidative stress, a factor linked to aging and various pathological conditions. Emerging research also suggests that NADH may play a role in modulating sirtuin activity, influencing cellular processes associated with longevity (Mahajan et al., 2011). While further clinical studies are needed to establish the efficacy and safety of NADH as a therapeutic molecule, these initial findings highlight its potential in addressing conditions related to energy metabolism and cellular stress.
In addition to its potential role in mitochondrial disorders, NADH has shown promise in supporting cognitive function and alleviating fatigue. Some studies have explored NADH supplementation as a means to enhance mental alertness and alleviate symptoms associated with conditions like chronic fatigue syndrome (Castro-Marrero et al., 2015). NADH's involvement in neurotransmitter synthesis and its ability to influence cellular energy levels suggest potential benefits for mental and cognitive well-being.
Moreover, as an antioxidant, NADH may contribute to reducing oxidative damage in neural tissues, a factor implicated in age-related cognitive decline (Tan et al., 2018). The therapeutic use of NADH is a multifaceted area of research, encompassing its impact on cellular energy dynamics, antioxidant properties, and potential influence on key regulatory pathways. While cautious optimism surrounds the therapeutic potential of NADH, ongoing research will provide a more comprehensive understanding of its applications, mechanisms of action, and any potential limitations or side effects associated with its use in various clinical contexts.
Numerous studies suggest that NADH may have beneficial effects on cholesterol levels and blood pressure. In two separate in vivo studies, rats were given a daily supplementation of 5 mg NADH for 8 weeks, resulting in a significant decrease in total cholesterol by ∼30% and 10% in the respective studies. In addition, these studies revealed an increase in aortic ring contraction, indicating a potential influence on vascular function (Birkmayer, 2009).
Another study focused on hypertensive rats, where a 5 mg NADH supplementation led to a notable reduction in systolic blood pressure after 11 weeks. The treatment group exhibited an average systolic blood pressure of 184 mm Hg ±2.8 (standard error mean), compared with the placebo group with 201 mm Hg ±2.8 (SEM) (Birkmayer, 2010). These findings suggest a potential role for NADH in modulating cardiovascular parameters (Nadtochiy et al., 2018), including cholesterol levels and blood pressure, warranting further exploration in clinical contexts.
Various in vitro studies conducted on cancerous cells have revealed significant growth inhibition upon the addition of NADH. These studies (Bagchi and Preuss, 2004; Kelley and HD, 2006; Moore et al., 2021) demonstrated the inhibitory effects of NADH on the growth of diverse cancer cell types. Human breast, colon, larynx, cervix, and skin cancer cells exhibited suppressed proliferation in response to NADH supplementation. The findings suggest that NADH may influence cellular mechanisms involved in cancer cell division and growth regulation. While the precise molecular pathways underlying this inhibition warrant further investigation, these in vitro results highlight the potential of NADH as a candidate for further exploration in cancer research and therapeutic development.
Conclusion
This review provides a comprehensive overview of the multifaceted roles of NADH in various biological processes. Primarily, NADH, along with its oxidized form NAD+, plays a crucial role in ATP production and cellular oxidation–reduction reactions, essential for energy metabolism. Processes such as the ETC, oxidative phosphorylation, and glycolysis contribute to ATP production and NADH synthesis. The Krebs cycle supplies NADH as an electron carrier to the ETC, while glycolysis-produced NADH can be used by LDH or transported to the mitochondria for redox homeostasis.
Beyond ATP and NADH generation, these metabolic pathways are integral to glucose metabolization for biomolecule formation. In pathological conditions like diabetes, hyperglycemia can lead to NADH overproduction, disturbing redox homeostasis. In addition, NADH serves as a cofactor for various enzyme reactions and coenzymes crucial for mitochondrial functions, linking it to aging and diseases such as Alzheimer's, Parkinson's, heart disease, and infections.
NADH supplementation has demonstrated diverse effects, lowering cholesterol and blood pressure, inhibiting cancer cell growth, and improving cognitive functions. Restoring free NADH levels through extracellular reductive conditions shows promise in counteracting age-related diseases. Notably, NADH treatment in Parkinson's and Alzheimer's patients has shown positive clinical outcomes, potentially linked to its impact on dopamine synthesis and ATP levels.
Future Perspectives
The future perspective of utilizing NADH as a therapeutic tool holds immense promise across various medical domains. As our understanding of NADH intricate roles in cellular processes deepens, there is growing interest in harnessing its potential for targeted interventions (Rajman et al., 2018). NADH supplementation has exhibited positive effects on energy metabolism, cognitive functions, and overall health in preclinical and clinical studies. The ability of NADH to influence redox homeostasis, mitochondrial functions, and enzymatic reactions positions it as a versatile therapeutic candidate.
Emerging research suggests that manipulating the NAD+/NADH ratio through NADH supplementation could offer novel strategies for addressing age-related diseases, neurodegenerative disorders, cardiovascular conditions, and metabolic dysregulations. Ongoing investigations aim to elucidate the molecular mechanisms underlying NADH's therapeutic effects, refine dosages, and explore its applicability in personalized medicine.
The possible strategies to modulate NADH levels can include the following: (i) NADH precursors: providing the cell with compounds that can be readily converted into NADH is one approach. For instance, precursors such as NR and NMN can be metabolized to generate NADH; (ii) NADH dehydrogenase inhibitors: compounds that inhibit NADH dehydrogenase, an enzyme involved in the ETC, can affect NADH levels. However, the use of these inhibitors needs to be carefully regulated as they can impact overall mitochondrial function and cellular respiration; (iii) diets rich in certain nutrients, such as niacin (vitamin B3), can impact NADH metabolism; (iv) mitochondrial targeting agents: compounds designed to specifically target mitochondria and modulate NADH levels are also under investigation. These may include antioxidants and agents that affect mitochondrial biogenesis; (v) supplementation with exogenous NADH.
The potential of NADH to enhance cellular resilience, counteract oxidative stress, and modulate key metabolic pathways hints at a future where NADH-based therapeutics play a pivotal role in promoting health span and mitigating various disease states. However, comprehensive clinical trials and further exploration of NADH's long-term effects will be essential to establish its efficacy and safety, paving the way for its broader adoption as a therapeutic tool in the medical arsenal.
Authors' Contribution
R.R. and M.N. contributed to conceptualization; G.S. and D.L. performed data curation; R.R., M.N., G.S., D.L., and J.C. assisted with writing, original draft preparation. All authors have read and agreed to the published version of the article.
Footnotes
Acknowledgment
We thank Yumiko Kajiwara for figure preparation.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.