
Abstract
Significance:
This review investigates how radiation therapy (RT) increases the risk of delayed cardiovascular disease (CVD) in cancer survivors. Understanding the mechanisms underlying radiation-induced CVD is essential for developing targeted therapies to mitigate these effects and improve long-term outcomes for patients with cancer.
Recent Advances:
Recent studies have primarily focused on metabolic alterations induced by irradiation in various cancer cell types. However, there remains a significant knowledge gap regarding the role of chronic metabolic alterations in normal cells, particularly vascular cells, in the progression of CVD after RT.
Critical Issues:
This review centers on RT-induced metabolic alterations in vascular cells and their contribution to senescence accumulation and chronic inflammation across the vasculature post-RT. We discuss key metabolic pathways, including glycolysis, the tricarboxylic acid cycle, lipid metabolism, glutamine metabolism, and redox metabolism (nicotinamide adenine dinucleotide/Nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADP+)/NADPH). We further explore the roles of regulatory proteins such as p53, adenosine monophosphate-activated protein kinase, and mammalian target of rapamycin in driving these metabolic dysregulations. The review emphasizes the impact of immune-vascular crosstalk mediated by the senescence-associated secretory phenotype, which perpetuates metabolic dysfunction, enhances chronic inflammation, drives senescence accumulation, and causes vascular damage, ultimately contributing to cardiovascular pathogenesis.
Future Directions:
Future research should prioritize identifying therapeutic targets within these metabolic pathways or the immune-vascular interactions influenced by RT. Correcting metabolic dysfunction and reducing chronic inflammation through targeted therapies could significantly improve cardiovascular outcomes in cancer survivors. Antioxid. Redox Signal. 43, 92–114.
Introduction
Radiation is broadly categorized into Irradiation (IR) and non-IR, each with distinct energy levels and biological effects. IR originates from unstable atoms or human activities and produces ions by dislodging electrons from atoms. It includes neutrons, alpha (α), beta (β), and gamma (γ) particles, along with X-rays emitted by radioactive materials. Among these, α-particles have low penetration capacity, β-particles penetrate deeper, while γ-rays and X-rays exhibit the highest penetration ability. In contrast, non-IR, with lower energy levels, includes radiofrequency radiation, infrared radiation, visible light, ultraviolet (UV) radiation, and extremely low-frequency radiation, each with distinct applications and biological effects (Donya et al., 2014).
Radiation therapy (RT), which accounts for 98% of public radiation exposure, has revolutionized cancer treatment. Initially utilized for breast cancer and Hodgkin’s lymphoma, RT now benefits over half of all patients with cancer (Yang et al., 2021a). Despite its efficacy in improving cancer survival rates, RT poses dose-dependent risks to noncancerous tissues, particularly the heart, significantly increasing the risks of cardiovascular disease (CVD) in cancer survivors. For example, RT in breast cancer therapy often exposes the heart to radiation, leading to various conditions and heightened CVD risk. This risk is further amplified in patients treated for head and neck cancers (Lin et al., 2022), where RT induces microvascular dysfunction and impairs salivary gland function. These adverse effects are mediated by elevated levels of reactive oxygen species (ROS) and ceramides, which contribute to tissue damage and inflammation (Cherukuri et al., 2022; van Nimwegen et al., 2015; Wang et al., 2022; Weintraub et al., 2010) (Fig. 1).
Radiation-induced CVD (RICVD) was first identified in survivors of the atomic bomb (Cullings, 2014; Shimizu et al., 2010) and subsequently observed in individuals exposed to low-dose occupational radiation and patients undergoing medical treatments (Baskar et al., 2012; Emami et al., 1991). Over the decades, RICVD has emerged as a significant concern for cancer survivors treated with RT, particularly those with breast cancer, lymphoma, and lung cancer. These patients face increased morbidity and mortality risks due to RT exposure, especially to critical cardiac structures situated near the treatment field (Armenian et al., 2016; Hooning et al., 2007; Lally et al., 2007; Singal and Iliskovic, 1998; Swerdlow et al., 2007). Table 1 summarizes the effects of RT on CVD in cancer survivors, consolidating findings from human studies. The proximity of the heart to the radiation field during RT remains a major challenge, with cardiac exposure acting as a key risk factor for cardiovascular complications. To mitigate this risk, various techniques have been developed to minimize cardiac radiation exposure. Among these, breathing control methods, such as deep inspiration breath-hold (DIBH), have demonstrated efficacy in reducing the mean heart dose (MHD), thereby potentially lowering the likelihood of radiation-induced cardiac injury. Since 2014, advancements in RT techniques have significantly reduced MHD during whole breast irradiation, although substantial variability persists. Despite these improvements, the mean dose to the left anterior descending artery remains as high as at 12.4 Gy in some cases, highlighting the persistent challenge of cardiac exposure (Drost et al., 2018; Maraldo et al., 2015; Wihlm et al., 1997). While advancements in radiation oncology have reduced MHD and enhanced precision, clinical concerns over radiation-induced vascular injury-a phenomenon first described a century ago-remain. Many patients with cancer still face substantial cardiac radiation exposure, underscoring the need for further innovation in RT techniques and the development of protective strategies to mitigate RICVD risk.
Risk of Cardiovascular Complications in Cancer Survivors After Radiotherapy
CVD, cardiovascular disease; RT, radiation therapy.
The relationship between radiation exposure and CVD is influenced by factors such as dose, exposure duration, affected tissues, and individual susceptibility. Radiation dose is measured in Gy, which quantifies energy deposited in tissues, and Sieverts (Sv), which account for biological effects, accounting for energy absorption (Ikeda et al., 2021).
A meta-analysis revealed that CVD risk increases with radiation dose, particularly at moderate doses (<0.5 Gy) or low-dose rates (<5 mGy/h), suggesting heightened cardiovascular sensitivity to fractionated exposures. This inverse dose and fractionation effect was most notable for ischemic heart disease and overall CVD. Region-specific excess absolute risks per Gy, such as 2.33% in England and 3.66% in Germany, highlight differences in baseline CVD mortality rates (Little et al., 2023).
In RT, an increase of 1 Gy in MHD raises the likelihood of major coronary events by 7.4%, reaching 16.3% per Gy within 4 years posttreatment (Darby et al., 2013; Zhou et al., 2013). High doses (>30–40 Gy) are strongly correlated with acute and long-term cardiac complications, including pericarditis, cardiomyocyte toxicity, cardiomyopathy, valvular dysfunction, and heart failure (Adams et al., 2004; Menezes et al., 2018). Hodgkin lymphoma survivors exposed to cumulative doses above 20 Gy face two to three times the risk of secondary malignancies compared with those receiving lower doses (van Nimwegen et al., 2017). While prenatal IR exposure can disrupt fetal brain development (Jain, 2021), doses below 0.5 Gy may enhance certain cellular functions, demonstrating a dose-dependent duality in radiation effects (Lumniczky et al., 2021; Xu et al., 2022a).
Research findings on radiation and CVD cardiovascular risk vary significantly. Some studies suggest no definitive increased risk below 2.6 Gy, while others indicate an elevated risk of heart disease even above 1 Gy (Azimzadeh et al., 2022; Azizova et al., 2010; Baselet et al., 2016; Hoving et al., 2008; Kreuzer et al., 2015; Little, 2010; Little et al., 2012, 2021, 2023; Nagane et al., 2021; Schollnberger et al., 2018, 2019; Yuan et al., 2021). Radiation doses ranging from 0.5 to 10 Gy are associated with vascular aging and RICVD due to the accumulation of senescent vascular cells (Andreassi et al., 2015; Benedicto et al., 2021; Darby et al., 2013; Gorgoulis et al., 2019; Kim et al., 2023; Lin et al., 2012; McRobb et al., 2017; Rombouts et al., 2013; Tabasso et al., 2019). Doses exceeding 10 Gy cause extensive DNA damage and can trigger apoptosis in certain cell types, particularly endothelial cells (ECs) (Ballarini et al., 2011; Bonner, 2003; Elhamiasl and Nuyts, 2020; Eriksson and Stigbrand, 2010; Hietanen, 2006; Huang et al., 2003; Hussien and Rashed, 2023; Jiao et al., 2022; Khanson and Zhivotovskiĭ, 1990; Masuda et al., 2015; Ootsuyama, 2016; Rey et al., 2021; Sia et al., 2020; Suzuki and Yamashita, 2012; Tan et al., 2013; Tsai et al., 2022).
Table 2 summarizes the effects of radiation on CVD in mice models, focusing on key experimental findings and implications. In mice, radiation exposure has been linked to weight loss, hair loss, and reduced saliva production. Notably, treatment with a ROS scavenger partially alleviated these effects, though it did not affect lysozyme levels in saliva, indicating selective modulation of radiation-induced changes (Mizrachi et al., 2016). Studies on ApoE knocked out (KO) mice demonstrate that low-dose IR affects atherosclerosis, with dose rate being a critical factor (Joven et al., 2007). Chronic low-dose IR impacts monocyte levels and plaque stability (Ebrahimian et al., 2018). Interestingly, some studies suggest that low-dose IR at low-dose rates may slow plaque progression (Mitchel et al., 2011, 2013), whereas acute irradiation at 300 mGy exacerbates it (Mancuso et al., 2015).
Risk of Cardiovascular Abnormalities After Irradiation Exposure in Animal Study
BCA, brachiocephalic artery; SMC, smooth muscle cell.
A study investigating the immunomodulatory response in ApoE KO mice revealed that low-dose IR might reduce atherosclerotic plaque formation by selectively decreasing pro-inflammatory monocytes, impairing adhesion molecule expression, and reducing vessel wall inflammation without significantly affecting splenic T lymphocytes. Interestingly, radiation-induced responses often exhibit nonlinear behavior, with significant reductions in aortic gene expression observed at intermediate doses but not at extreme doses (Rey et al., 2021). Chronic low-dose IR enhances plaque stability by reducing CD68+ foam cell and promoting anti-inflammatory cytokine production (Ebrahimian et al., 2018; Gabriels et al., 2014; Le Gallic et al., 2015; Rodel et al., 2012; Williams et al., 2003). Total body γ-irradiation at 12 Gy, followed by bone marrow reconstitution in ApoE KO mice, reduced smooth muscle cell investment in specific arteries, excluding the aortic root and abdominal aorta (Newman et al., 2018). Additionally, IR suppresses low-density lipoprotein (LDL) accumulation in the intima of the aortic arch, delayed lipid and monocyte accumulation, and reduced foam cell coalescence. Single-cell transcriptomic analysis suggested that radiation redirected LDL uptake by ECs to lysosomal degradation and reverse cholesterol transport, reducing intimal lipid accumulation and affecting lesion growth without altering paracellular leakage (Ikeda et al., 2021).
Three major theories have been proposed to explain the effects of radiation on blood vessels: metabolic, neuromuscular, and cellular theories. The metabolic theory suggests that radiation indirectly impacts blood vessels by inducing metabolic changes in surrounding tissues. These effects were initially attributed to histamine-like substances released by irradiated tissues. However, this theory has been criticized because nonhistamine agents can elicit similar vascular responses and there are notable differences between radiation-induced erythema and histamine-mediated effects, attributing radiation effects to histamine-like mechanisms may be misleading. The neuromuscular theory proposes that radiation directly affects blood vessels, leading to either constriction or dilation through neuromuscular elements. Despite its plausibility, the precise primary target-whether nerves or muscle cells-remains contentious. Radiobiological and anatomical evidence indicates that the doses required to induce phenomena such as erythema do not significantly affect nerves or muscles, suggesting their low radiosensitivity. Additionally, erythema caused by visible and ultraviolet light likely involves photochemical mechanisms, while X-rays and radium may act through photoelectric effects, further complicating the understanding of these processes. The cellular theory, which is supported by substantial evidence, identifies ECs as the primary targets of radiation-induced vascular damage. ECs, which line the interior of blood vessels, are highly radiosensitive. Radiation-induced damage or dysfunction of ECs results in compromised vascular function, including increased vascular leakage due to disrupted integrity, impaired blood flow, reduced oxygen supply, and diminished angiogenesis (Venkatesulu et al., 2018).
This review aims to bridge the gap in understanding by elucidating the relationship between IR-mediated metabolic alterations and their implications for vasculopathy.
Cellular Metabolism and Homeostasis
Cellular metabolism is fundamental for energy production and homeostasis (Gomes and Blenis, 2015), supporting processes like DNA repair and mitochondrial function (Chandel, 2021; Judge and Dodd, 2020). Glucose undergoes glycolysis to produce pyruvate, which is converted to acetyl-CoA and enters the tricarboxylic acid (TCA) cycle for adenosine triphosphate (ATP), metabolite, and NADPH production (Chandel, 2021; Judge and Dodd, 2020). Under oxygen-limited conditions, glucose is redirected to anaerobic metabolism, leading to lactate production, which influences histone modifications and epigenetic regulation. Glycolytic intermediates, such as glucose 6-phosphate (G6P), can enter the pentose phosphate pathway (PPP) to generate NADPH for antioxidant defense and nucleotide biosynthesis (Judge and Dodd, 2020; TeSlaa et al., 2023). This flexibility allows cells to adapt to stress by scavenging ROS and synthesizing nucleotides for DNA repair and survival (Bailleul et al., 2023; De Saedeleer et al., 2012; Huang et al., 2023; Zhang et al., 2019; Zhao et al., 2017).
Fatty acid (FA) synthesis relies on glucose-derived acetyl-CoA, with key enzymes like ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and FA synthase (FASN) catalyzing the process (Batchuluun et al., 2022). Synthesized FAs and lipid droplets are utilized via β-oxidation in mitochondria to produce acetyl-CoA, Nicotinamide adenine dinucleotide (NADH), and FADH2, fueling the TCA cycle and electron transport chain (ETC) (Ma et al., 2018).
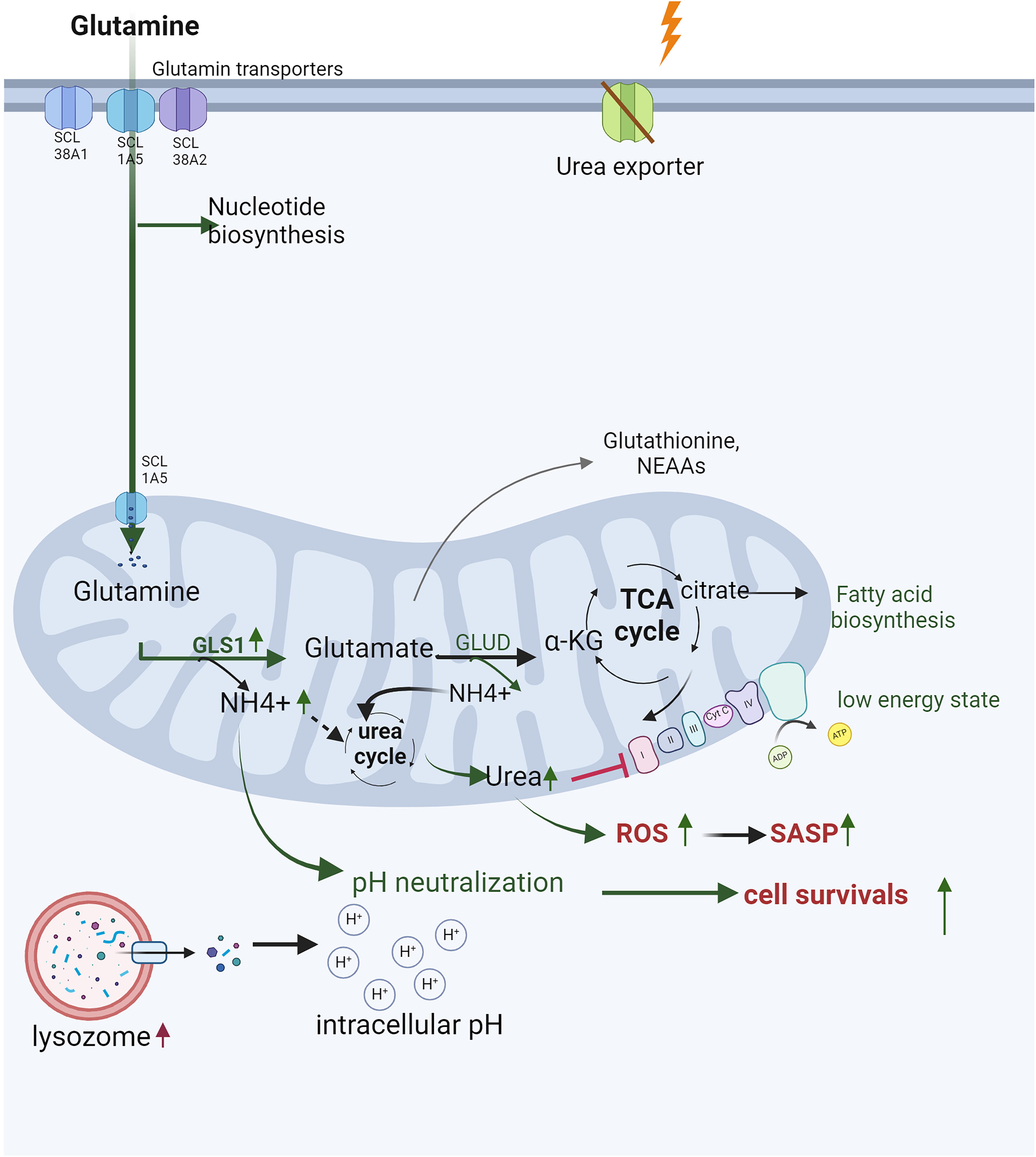
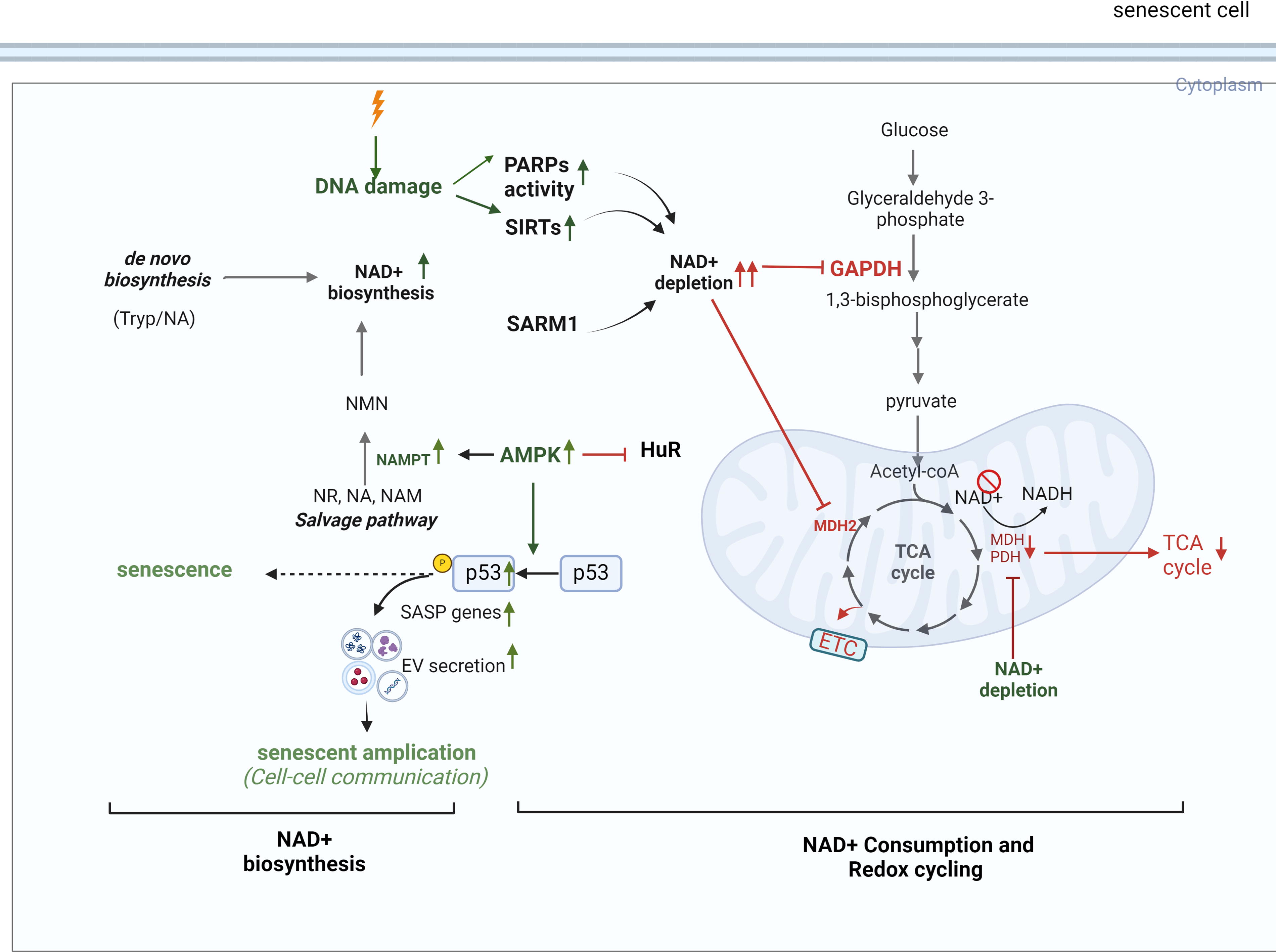
Nicotinamide adenine dinucleotide (NAD+) plays a crucial role in maintaining metabolic and redox balance, DNA repair, and epigenetic modifications (Amjad et al., 2021), which regulate genomic integrity and cellular processes (Kalous et al., 2016; Man et al., 2019; Rouleau-Turcotte and Pascal, 2023; Tseng et al., 2013). NAD+ regenerates salvage pathways, de novo biosynthesis, and the malate-aspartate shuttle (Katayoshi et al., 2021; Lee et al., 2012), but its depletion impairs the TCA cycle, reduces ATP production, and contributes to mitochondria dysfunction, DNA damage, and (Luna et al., 2013).
Metabolic Alterations and Cellular Senescence
Aging induces structural and functional changes in blood vessels, including the accumulation of senescent cells, altered vascular tone, and compromised DNA repair mechanisms (Ghebre et al., 2016; Saz-Lara et al., 2023; Su et al., 2021). These vascular changes significantly contribute to the development and progression of CVD (Dhingra and Vasan, 2012; Watanabe et al., 2023). Consequently, aging individuals, as well as those with premature aging syndromes such as Hutchinson–Gilford progeria syndrome (HGPS), face an elevated risk of CVD (Banerjee et al., 2023; Jain et al., 2023; Nabel, 2012). In HGPS, the accumulation of progerin disrupts nuclear morphology, impairs cellular functions, and induces cellular senescence (Olive et al., 2010; Xu et al., 2022b).
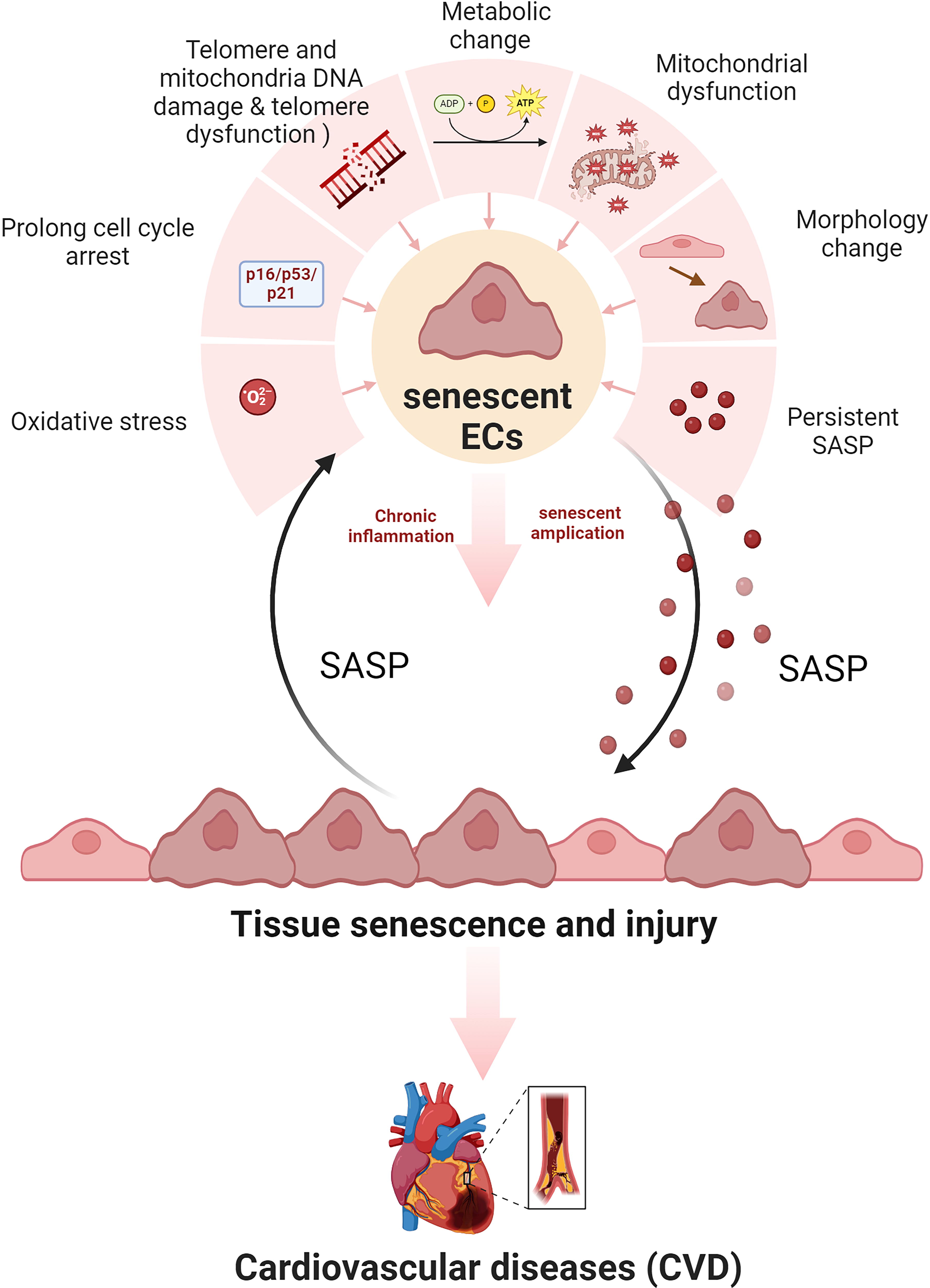
Before entering senescence, cells undergo profound metabolic alterations. These include increased glycolysis, reduced TCA cycle activity, reliance on glutaminolysis as the primary carbon source for the TCA cycle, and diminished PPP activity due to inhibition of 6-phosphogluconate dehydrogenase (Banerjee et al., 2023; Feng et al., 2020; Jain et al., 2023; Le, 2023a; Lin et al., 2015; Wiley and Campisi, 2021; Yang et al., 2021b; Zimmer, 1992) (Fig. 2). FA accumulation, depletion of NAD+, and dysfunction of the ETC exacerbate metabolic stress, resulting in endoplasmic reticulum (ER) stress, mitochondrial ROS production, and succinate accumulation. Remarkably, these metabolic disruptions persist even when oxidative phosphorylation (OXPHOS) and glycolysis are suppressed by low doses of IR (Kotla et al., 2021).
Upon entering senescence, metabolic enzyme dysfunction leads to increased expression of glycolytic enzymes in human diploid fibroblasts (Bittles and Harper, 1984; Sohn et al., 2022). Additionally, senescent cells secret senescence-associated secretory phenotype (SASP) factors (Coppé et al., 2010), including pro-inflammatory cytokines, growth factors, and matrix components. SASP factors impair tissue function, promote fibrosis, facilitate endothelial–mesenchymal transition (Banerjee et al., 2023; Ramadhiani et al., 2021; Su et al., 2021; Tominaga, 2015; Ungvari et al., 2020), and fuel inflammation. They also sustain the survival of senescent cells and create a positive feedback loop that perpetuates SASP activity (Childs et al., 2016; Lee et al., 2022; Li et al., 2020; López-Otín et al., 2013; Matejka and Reindl, 2019; Peng et al., 2020; Sadhu et al., 2021; Tang and Loganovsky, 2018; Tchkonia et al., 2013; Vaitiekus et al., 2020; Wang et al., 2021; Wang and Bennett, 2012; Xu et al., 2022a). This chronic SASP activity fosters the accumulation of senescent cells and drives persistent vascular inflammation, contributing to CVD progression and EC dysfunction (Darby et al., 2005; Li et al., 2023; Tchkonia et al., 2013) (Fig. 2).
Impacts of Radiation on Metabolic Alterations
Radiation induces the generation of free radicals that damage DNA and oxidize cellular molecules, leading to genotoxicity and oxidative stress. These effects initiate metabolic reprogramming to support DNA damage repair and oxidative stress defense mechanisms (Childs et al., 2015, 2016; González-Gualda et al., 2021; Huang et al., 2022; Khavinson et al., 2022; Little et al., 2023; López-Otín et al., 2013; Masuda et al., 2015; Matejka and Reindl, 2019; Owens et al., 2021; Peng et al., 2020; Philipp et al., 2017; Read et al., 2022; Sadhu et al., 2021; Stojanovic et al., 2020; Tabasso et al., 2019; Vaitiekus et al., 2020; Verdin, 2015; Wang and Bennett, 2012; Yang et al., 2021a). The metabolic shifts triggered by radiation differ from those directly activated by p53 and exhibit unique temporal dynamics (Bailleul et al., 2023; Mullarky and Cantley, 2015; Zhang et al., 2019; Zhao et al., 2017) (Figs. 2–8). In cancer cells, radiation enhances glycolysis while suppressing pyruvate dehydrogenase activity (Sattler et al., 2010; Shimura et al., 2014; Viale et al., 2014), promoting glucose diversion into alternative pathways, such as the PPP, serine synthesis pathway, and lactate metabolism via upregulated lactate dehydrogenase (Bailleul et al., 2023; Tang et al., 2018; Zhang et al., 2019; Zhao et al., 2017). This metabolic shift aligns with the Warburg effect, characterized by a preference for aerobic glycolysis over OXPHOS, which supports tumor growth, survival, and resistance to RT (Gatenby and Gillies, 2004; Khodarev et al., 2004; Kondoh et al., 2005; Lall et al., 2014; Liberti and Locasale, 2016; Moeller et al., 2004; Pitroda et al., 2009; Racker, 1972; Yogalingam et al., 2013; Zhao et al., 2017; Zhong et al., 2013). For example, glioblastoma cells redirect glucose into the PPP postradiation for antioxidant benefits, regulated by pyruvate kinase M2 (PKM2) (Bailleul et al., 2023) while triple-negative breast cancer cells increase glucose uptake and lactate production following radiation (Zhang et al., 2019).
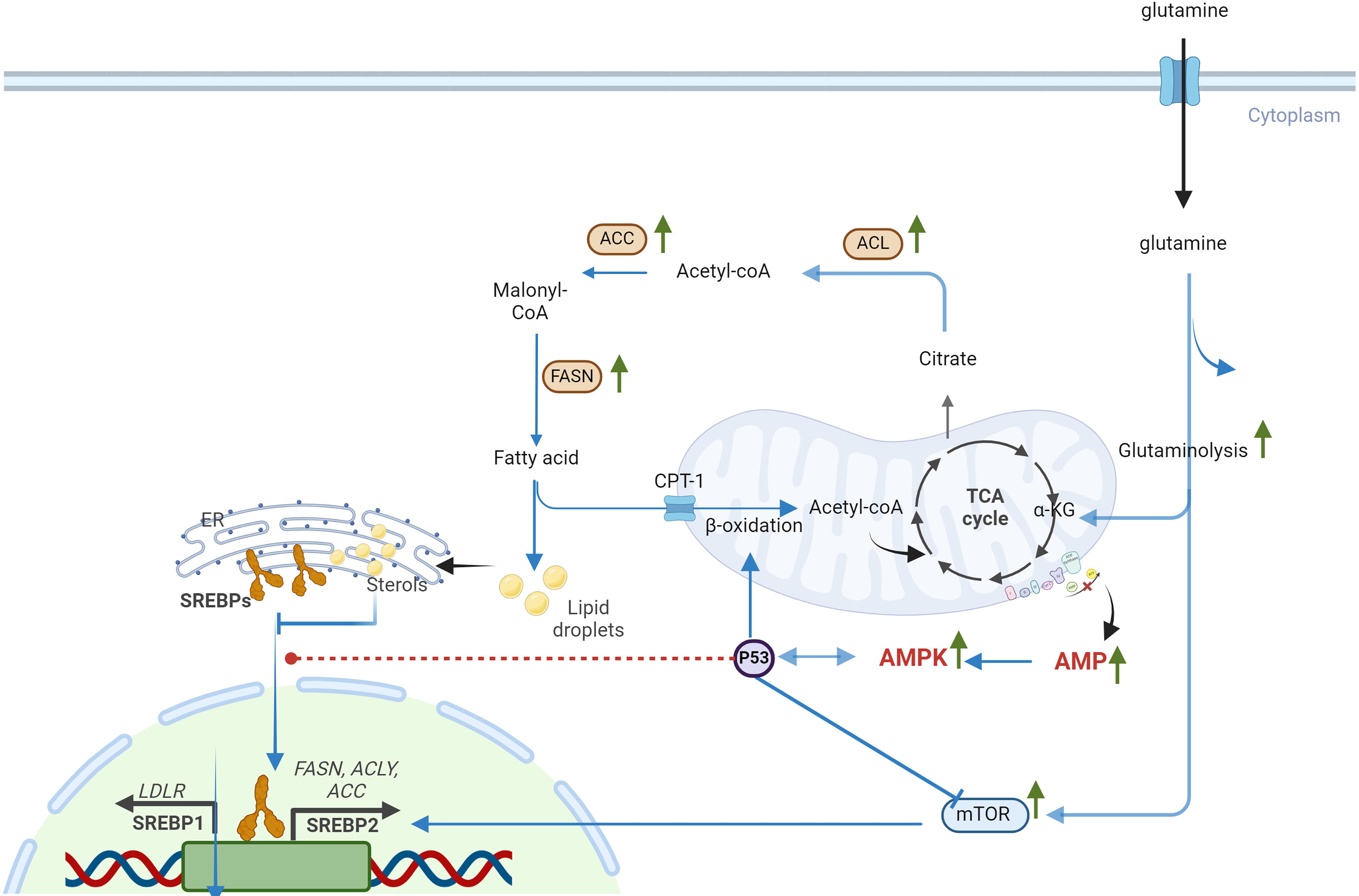
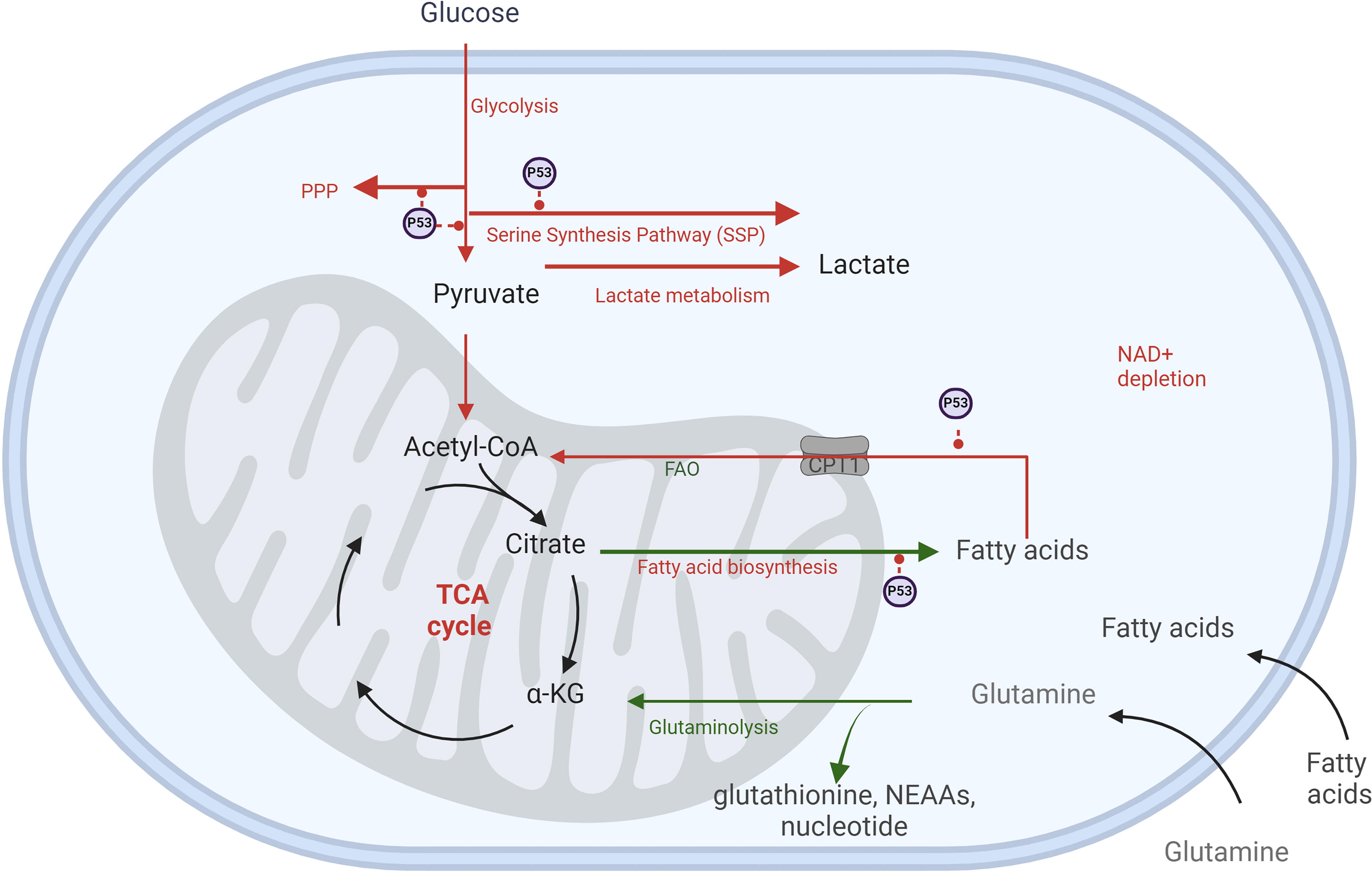
Radiation also influences lipid metabolism by enhancing lipid biosynthesis and promoting FA accumulation. These processes are linked to increased glucose breakdown and acetyl-CoA production, fueling lipogenesis via upregulated lipogenic enzymes, such as ACLY, ACC, and FASN (Horton et al., 2002; Jin et al., 2021; Levis et al., 1975; Zhao et al., 2022). Furthermore, mitochondrial β-oxidation is upregulated to meet the elevated energy demands required for DNA repair (De Martino et al., 2023; Gao et al., 2015; Ginini et al., 2022; Liu et al., 2022; Martinez-Outschoorn et al., 2017).
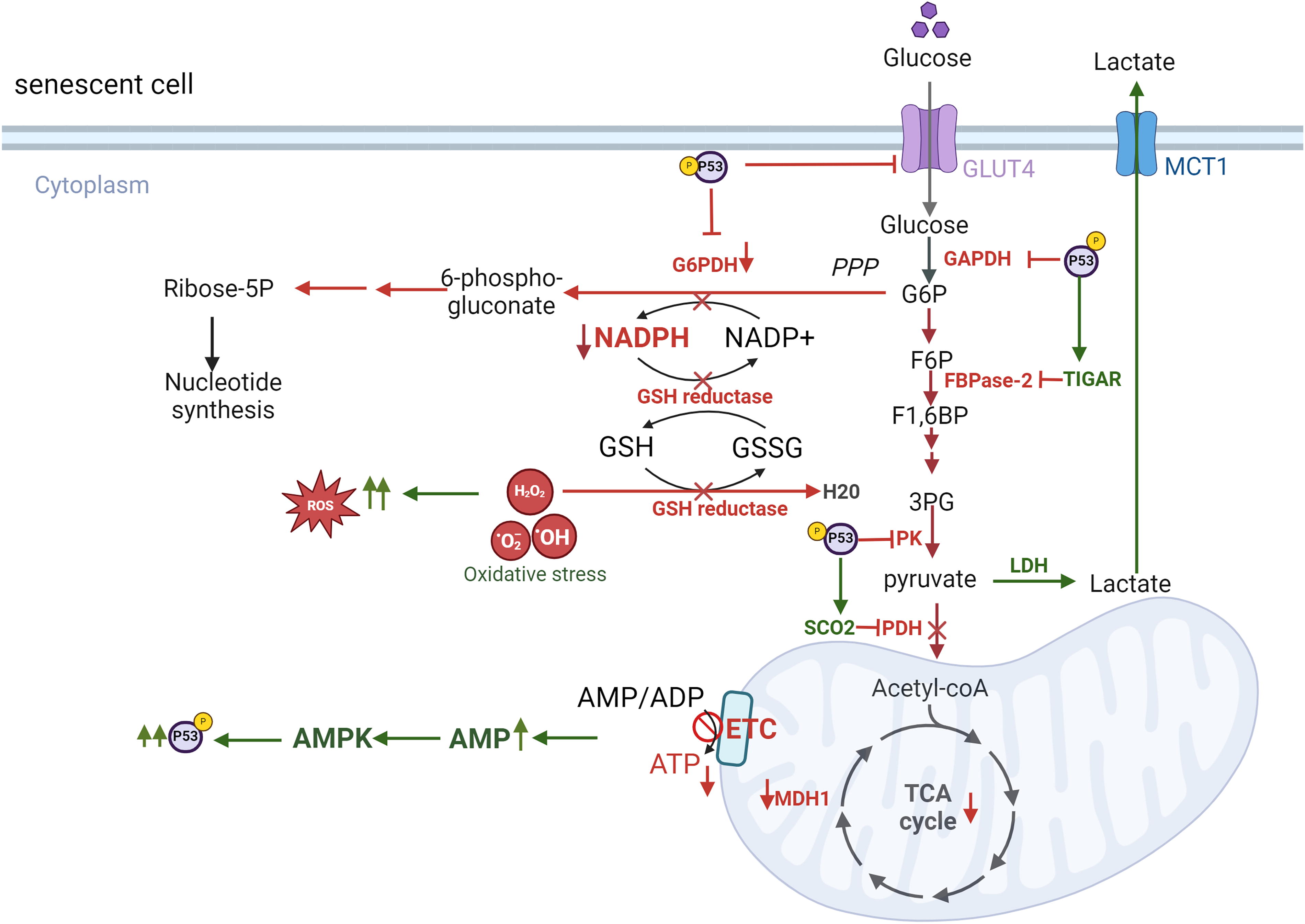
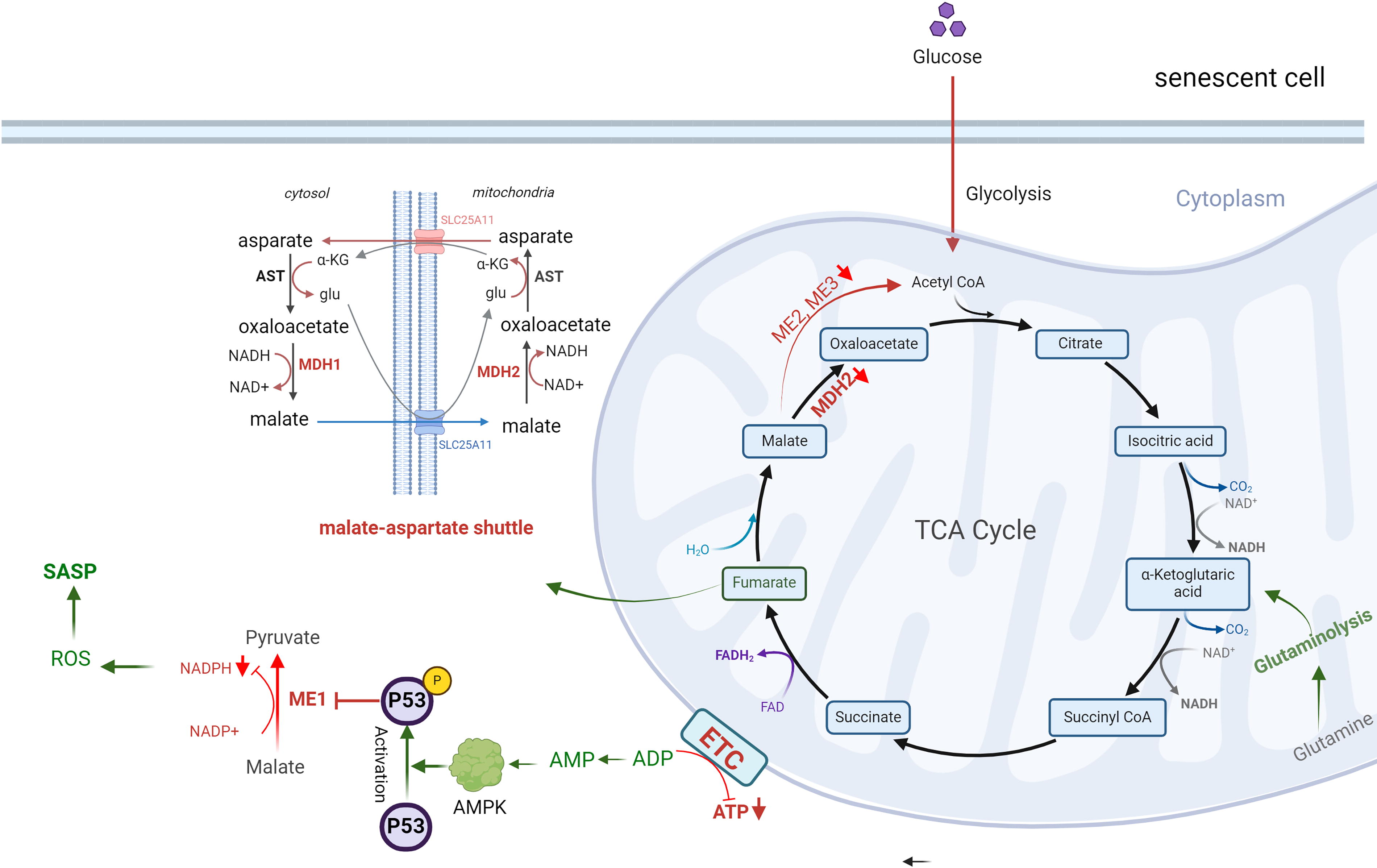
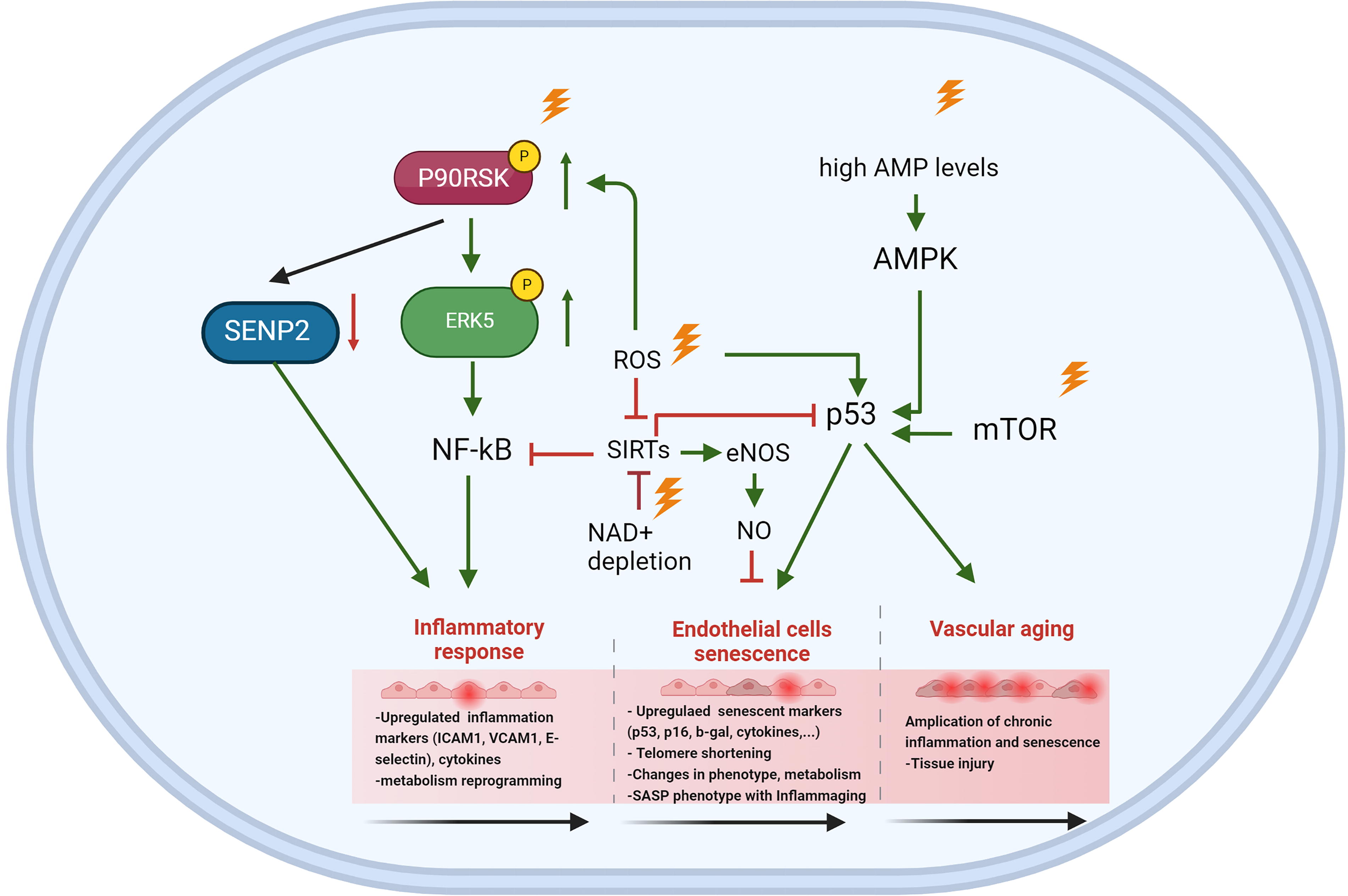
Radiation-induced metabolic changes are also mediated by p53 activation, which play a pivotal role in promoting cellular senescence (Bailleul et al., 2023; Jiang et al., 2011; Kuehne et al., 2015; Mullarky and Cantley, 2015; Zhang et al., 2019; Zhao et al., 2017) (Figs. 2 and 3). A key regulator in this process is adenosine monophosphate-activated protein kinase (AMPK), which is activated by elevated levels of adenosine monophosphate (AMP) and ROS. AMPK exhibits a dual role in cellular senescence, acting as both an inhibitor and an inducer depending on the context (He et al., 2014; Hinchy et al., 2018). In hydrogen peroxide-induced senescence of NIH3T3 fibroblasts, AMPK activation suppresses senescence by inhibiting mammalian target of rapamycin, preventing autophagy dysregulation, and restoring NAD+ levels to facilitate DNA repair (Han et al., 2016; Rabinovitch et al., 2017; Salminen and Kaarniranta, 2012). AMPK can also phosphorylate nicotinamide phosphoribosyl transferase (NAMPT), a critical enzyme in the NAD+ salvage pathway, to maintain NAD+ homeostasis under radiation-induced stress (Liao et al., 2022). Conversely, in other contexts, AMPK activation contributes to senescence by enhancing p53 activity. Activated p53 inhibits G6P dehydrogenase, a key enzyme in the PPP, suppressing NADPH production. Reduced NADPH impairs FA synthesis, glucose uptake, glutaminolysis, and antioxidant defenses, leading to elevated ROS levels and oxidative stress (Jiang et al., 2011; Stanton, 2012; Vermot et al., 2021). p53 also impacts glycolysis by downregulating glucose transporter GLUT1 and GLUT4 and inhibiting glycolytic enzymes. This occurs via the upregulation of p53-dependent regulatory proteins such as TP53-induced glycolysis and apoptosis regulator (TIGAR) and synthesis of cytochrome c oxidase 2 (SCO2). As glycolysis and PPP activity decline, cells increasingly rely on TCA cycle-associated malic enzymes for NADPH production (Jiang et al., 2011). However, p53 suppresses these malic enzymes (Jiang et al., 2011, 2013), further exacerbating ROS accumulation and cellular oxidative stress. The accumulation of ROS and metabolic dysregulation ultimately leads to inflammation, with NF-κB signaling playing a central role in this process (Lingappan, 2018) (Fig. 4).
In cells exposed to radiation, ROS-mediated activation of AMPK can have dual effects. On the one hand, AMPK activation can counteract the SASP by activating DNA damage response enzymes and promoting NAD+-dependent repair pathways. On the other hand, chronic AMPK activation driven by sustained AMP accumulation and ROS overload can induce senescence and exacerbate the SASP, thereby accelerating atherosclerosis and CVD. Furthermore, AMPK activation is linked to Mitochondrial Dysfunction Associated Senescence, highlighting its complex role in cellular responses to stress (Morsczeck et al., 2023). A critical function of AMPK is its ability to phosphorylate NAMPT, a key enzyme in the NAD+ salvage pathway, thereby regulating NAD+ homeostasis under stress conditions (Liao et al., 2022). However, radiation exposure disrupts this balance by depleting intracellular NAD+ levels, likely due to the increased consumption of NAD+ by DNA repair enzymes, sirtuins (SIRTs), and other metabolic pathways activated in response to oxidative damage (Childs et al., 2015, 2016; González-Gualda et al., 2021; Huang et al., 2022; Khavinson et al., 2022; Little et al., 2023; López-Otín et al., 2013; Masuda et al., 2015; Matejka and Reindl, 2019; Owens et al., 2021; Peng et al., 2020; Philipp et al., 2017; Read et al., 2022; Sadhu et al., 2021; Stojanovic et al., 2020; Tabasso et al., 2019; Vaitiekus et al., 2020; Verdin, 2015; Wang and Bennett, 2012; Yang et al., 2021a). For example, UVA/B irradiation has been shown to reduce NAD+ levels within one hour, severely impacting cell viability after 24 h (Katayoshi et al., 2021). Additionally, radiation-induced activation of p53 can exacerbate NAD+ depletion. p53 downregulate mean heart dose-1, a key enzyme in the malate-aspartate shuttle, impairing NAD+ regeneration from NADH (Lee et al., 2009). This disruption affects critical NAD+-dependent processes, including DNA repair, mitochondrial function, and ATP production, thereby contributing to cellular aging (Lee et al., 2012). Radiation also directly inhibits specific enzymes in glycolysis and the TCA cycle, further impairing NAD+ production. This imbalance between NAD+ biosynthesis and consumption exacerbate radiation-induced NAD+ depletion after radiation, emphasizing its critical role in the progression of senescence.
Impacts of Radiation on Inflammasome Activation
Delayed CVD following radiation exposure is partly mediated by the activation if the NLRP3 inflammasome. Accumulating evidence demonstrates that radiation induced the upregulation of inflammasome components, marked by increased levels of pro-inflammatory cytokines IL-1β and IL-18 in various cell types and tissues (Janko et al., 2012; Wijerathne et al., 2021). Once secreted, IL-1β drives inflammatory responses, leading to vascular injury and cardiac dysfunction by damaging microvascular ECs or inducing cellular senescence (Wijerathne et al., 2021). For example, Rao et al. (2023) showed robust activation of the NLRP3 inflammasome in lung epithelial cells following exposure to radiation at a dose rate of 4 Gy/min. This activation was accompanied by a significant increase in ROS levels, imbalances in ATP and the nicotinamide adenine dinucleotide phosphate (NADP+)/NADPH ratio, and fibroblast activation (Rao et al., 2023). Similarly, Smith et al. (2021) demonstrated that γ radiation exposure in microvascular ECs upregulated the NOD-like receptor family inflammasome, including NLRP1 and NLRP3, leading to the activation of caspase-1 and subsequent production of IL-1β and Il-18 (Smith et al., 2021). Mechanistically, radiation triggers the release of damage-associated molecular patterns (DAMPs) from dying or dead cells. These DAMPs are sensed by Toll-like receptors on neighboring healthy cells, which, in combination with radiation-induced intracellular ROS production, activate the NLRP3 inflammasome. Activation of the NLRP3 inflammasome results in the recruitment of caspase-1, which processes and releases the inflammatory cytokines IL-1β and IL-18 (Roh and Sohn, 2018). IL-1β, in particular, is a hallmark cytokine of chronic inflammation. It exacerbates cellular senescence, EC dysfunction, and vascular remodeling, accelerating the progression of CVD, including atherosclerosis, plaque instability, myocardial fibrosis, myocardial infarction, and heart failure.
Impacts of Radiation on Vascular Cellular Senescence
Radiation disrupts cardiovascular homeostasis by inducing EC injuries across various organs, contributing to long-term vascular dysfunction (Allen et al., 2019; Fliedner et al., 2005; Venkatesulu et al., 2018). These injuries are associated with increased CD44 expression (Lowe and Raj, 2014) and elevated levels of pro-inflammatory cytokines such as IL-6 and IL-8 (Stojanović et al., 2020). One prominent mechanism underlying radiation-induced vascular damage is its impact on adhesion molecule expression and EC apoptosis. A 2 Gy dose of whole-body irradiation significantly increases VCAM-1 expression, particularly in atherosclerosis-prone regions. This effect is mediated by enhanced activation of p90RSK and phosphorylation of ERK5 at serine 496 (Le, 2023b; Le et al., 2013; Nguyen et al., 2023; Vu et al., 2018; Wang and Tournier, 2006). Experimental evidence shows that inhibition of p90RSK or overexpression of the ERK5 S496A mutant prevents radiation-induced NF-κB activation, VCAM-1 expression, and EC apoptosis (Vu et al., 2018). These findings highlight the pivotal role of the p90RSK-ERK5 signaling axis in mitigating RICVD in cancer survivors (Vu et al., 2018). Radiation also modulates lipid metabolism in ECs, enhancing their uptake of LDL for lysosomal degradation and reverse cholesterol transport pathways. This process reduces lipid accumulation within the intima and impacts lesion initiation and growth, while paracellular leakage remains unaffected (Ikeda et al., 2021). Furthermore, radiation-induced reductions in CHK1 expression in ECs have been linked to fibrotic changes and the formation of vulnerable plaques (Nguyen et al., 2023).
SIRT1 plays a critical protective role in maintaining vascular homeostasis by inhibiting inflammation through the NF-κB signaling pathways and preserving endothelial nitric oxide synthase (eNOS) activity (Man et al., 2019; Yang et al., 2012). However, in senescent ECs and aged arteries, the loss of SIRT1-mediated acetylation of LKB1 leads to irreversible changes in the vascular wall, contributing to increased vascular stiffness and adverse arterial remodeling (Bai et al., 2016). Postradiation, increased acetylation levels of key proteins suggest potential SIRT dysfunction, as SIRT1 activity decreases due to NAD+ depletion and elevated ROS production. This dysfunction exacerbates radiation-induced vascular injury. The essential role of SIRT1 in vascular health is further demonstrated in mouse models with EC-specific deletion of exon 4 in SIRT1. These mice exhibit impaired angiogenesis and reduced vasorelaxation capacity (Vasko et al., 2014). Similarly, SIRT6 contributes to vascular protection by preventing premature senescence and preserving cellular replicative capacity and angiogenic potential. In ECs, hydrogen peroxide-induced senescence is associated with reduced SIRT6 expression, which promotes cellular aging and functional decline. However, overexpression of SIRT6 partially reverses these effects, restoring some aspects of EC function (Cardus et al., 2013).
Radiation induces mitochondrial dysfunction and cellular senescence in immune cells, a phenomenon known as immunosenescence, which impairs both innate and adaptive immunity and contributes to the progression of CVD (Song et al., 2020). Radiation exposure alters the functions and metabolism of key immune cells, including peripheral blood mononuclear cells (PBMC) and macrophages (MCs), promoting a pro-inflammatory environment (Bauer et al., 2011; Genard et al., 2018; Loinard et al., 2023; Moertl et al., 2020). Specifically, radiation skews MC polarization toward the inflammatory M1 phenotype, characterized by elevated secretion of inflammatory factors such as TNF-α, IL-1β, and IFN-γ (Genard et al., 2018). In vitro studies using whole blood samples and PBMCs from healthy donors exposed to radiation doses of 2 and 6 Gy have demonstrated increased secretion of these pro-inflammatory cytokines, along with dysregulation of key proteins and microRNAs involved in immune function (Moertl et al., 2020). Furthermore, radiation-induced IL-1β production in cultured alveolar MCs and blood monocytes has been shown to promote cellular senescence, further amplifying inflammatory signaling (Wu et al., 2017). Senescent immune cells contribute to the SASP, releasing altered profiles of inflammatory secretomes that sustain chronic inflammation and exacerbate vascular dysfunction. This cascade of events links radiation exposure to the development and progression of CVD.
SASP Components as Communicating Factors in RICVD
In patients with thoracic cancer undergoing RT, the heart is exposed to radiation, affecting various cell types, including cardiomyocytes, ECs, and immune cells (Litviňuková et al., 2020). Although cardiomyocytes exhibit resistance to radiation-induced apoptosis, ECs are highly susceptible to senescence rather than apoptosis (Menendez et al., 1998; Venkatesulu et al., 2018), leading to myocardial injury and contributing significantly to RICVD (Boerma et al., 2016; Boerma and Hauer-Jensen, 2010; Lee and Mallik, 2005). Radiation-induced senescent ECs exhibit several pathological features, including increased ROS production, elevated expression of adhesion molecules, mitochondrial dysfunction, decreased NO production, impaired angiogenesis, and the release of SASP components (Dong et al., 2015; Kim et al., 2014; Lafargue et al., 2017; Lowe and Raj, 2014; Mendonca et al., 2011; Oh et al., 2001; Panganiban et al., 2013; Yentrapalli et al., 2013). Similarly, senescent MCs undergo metabolic reprogramming, adopt a low-energy state, experience mitochondrial dysfunction, and release SASP components (Abe et al., 2023; Kotla et al., 2021). Encapsulated within extracellular vesicles (EVs), these SASP factors serve as potent signaling molecules, propagating inflammation and dysfunction by attracting and influencing neighboring cells (Sun et al., 2022) (Fig. 9).
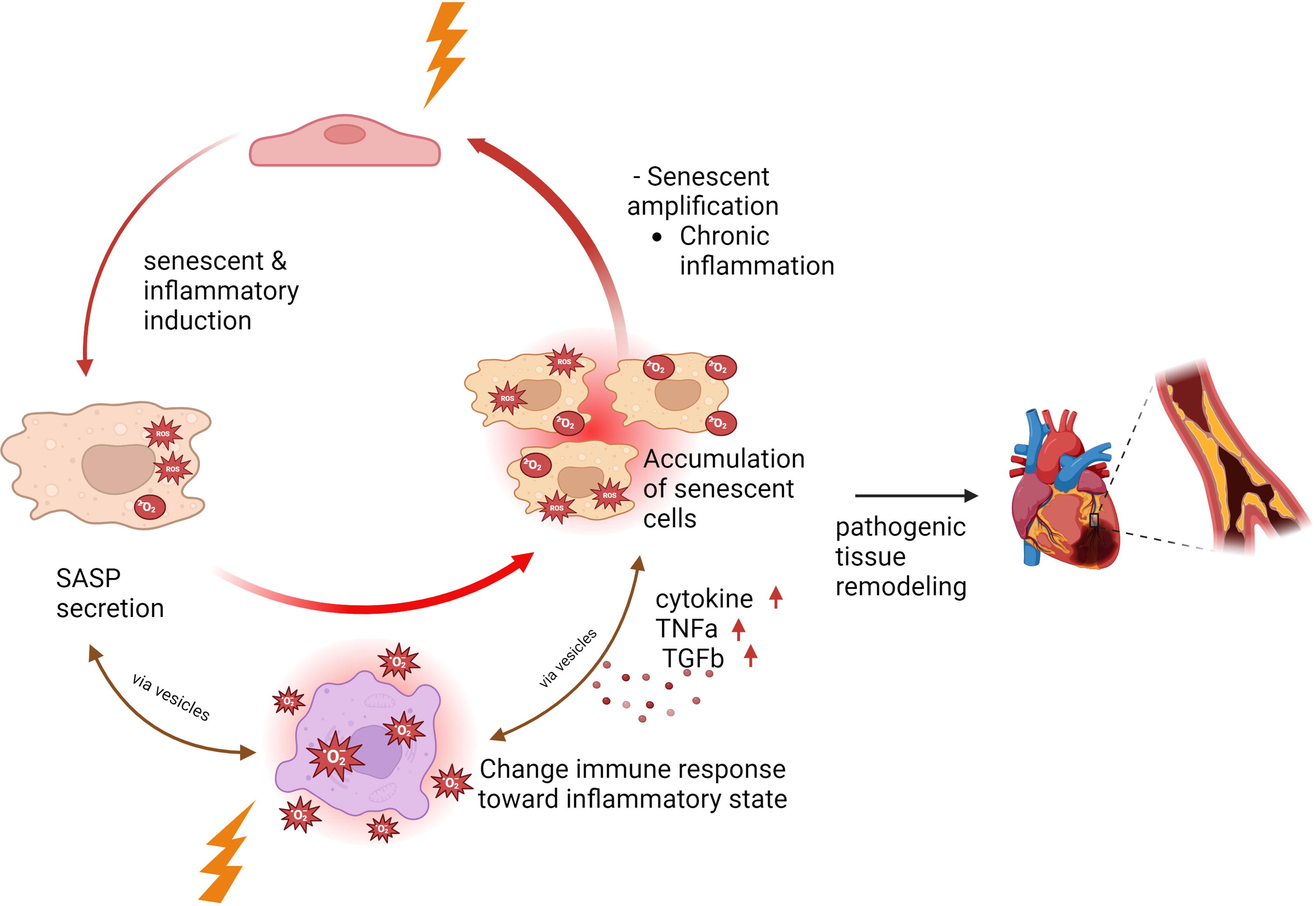
In PBMCs from patients with thoracic cancer before and after RT, a decrease in B cell subtypes and an increase in TBX21 (T-bet) expression in naive B cells were observed post-RT. These changes correlated with p90RSK activation, along with elevated CD38 expression in certain B and T cell populations. In vitro experiments demonstrated that p90RSK activation drives these alterations, contributing to immune senescence (Imanishi et al., 2022). In a hypercholesterolemia mouse model, phosphorylation of ERK5 at S496 induced senescent MCs and atherosclerosis by upregulating AHR activity while inhibiting NRF2 activity via SUMOylation (Abe et al., 2023; Le et al., 2012, 2013; Vu et al., 2018). Similarly, senescent MCs induced by chemotherapy or RT persistently release SASP components due to p90RSK-mediated ERK5 S496 phosphorylation. This process depletes NAD+, alters metabolism, and induces reversible mitochondrial dysfunction without affecting ATP levels. This feedback loop sensitizes MCs to ROS, perpetuating inflammation and tissue damage. A similar phenomenon has been observed in circulating monocytes from patients with cancer post-RT (Abe et al., 2023; Imanishi et al., 2022; Kotla et al., 2021).
ECs effectively uptake EVs derived from PBMC and MCs (Moertl et al., 2020), amplifying chronic senescence and inflammation across tissues and organs (Wang et al., 2020). This intercellular communication exacerbates atherosclerosis and cardiac fibrosis, promoting plaque instability and rupture, thereby increasing the risk of cardiovascular events (Prata et al., 2018). Radiation-induced metabolic alterations, including increased lactate biosynthesis, elevate lactate levels in the microenvironment. This accumulation inhibits immune cell-mediated clearance of senescent cells by impairing MCT1 activity, which is essential for T cell activation and immune cell proliferation (Lopez et al., 2023). Additionally, lactate disrupts dendritic cell function, hindering the presentation of senescent-associated antigens to immune cells (Brown et al., 2020), further facilitating the accumulation of senescent cells throughout tissues and organs. Our findings (Abe et al., 2023; Imanishi et al., 2022; Kotla et al., 2021) and the established role of senescent ECs in RICVD (Ashrafian et al., 2007; Banerjee et al., 2023; Jain et al., 2023; Karlstaedt et al., 2022; Le, 2023a) suggest significant interactions between MCs and ECs mediated by SASP components released by both cell types. These interactions form a feedback loop that propagates senescence and inflammation, further aggravating cardiovascular dysfunction.
Moving Forward
To advance our understanding and treatment of RICVD, innovative research approaches and therapeutic strategies must be prioritized. Microphysiological systems or organ-on-chip models provide a promising platform for investigating the complex mechanisms underlying RICVD. These systems mimic tissue organization and function using primary human cells in controlled microenvironments, offering an ethical and efficient alternative to animal studies. The use of patient-specific cells within these 3D models has the potential to accelerate the translation of findings into therapeutic applications. However, it is essential to validate the relevance of these findings through human samples and clinical research to ensure their applicability to RICVD.
To mitigate RICVD, several strategies have been proposed to address the harmful effects of IR-induced metabolic shifts and inflammasome activation. Enhancing AMPK activity through activators such as metformin or salicylates or increasing cellular NAD+ levels via the administration of precursors like nicotinamide riboside or nicotinamide mononucleotide, has the potential to protect cells against oxidative stress, restore metabolic balance, and reduce the accumulation of senescent cells. Additionally, targeting EVs or lactate metabolism could limit the dissemination of senescence and immune evasion within the vascular microenvironment. Another promising approach involves enhancing NRF2/antioxidant response element (ARE) signaling or eNOS activity to strengthen defenses against excessive IR-induced oxidative stress. Activation of NRF2 promotes the transcription of critical antioxidant and redox-regulating genes, enhancing the cellular capacity to counteract inflammatory cascades (Abe et al., 2023; Carlson et al., 2020), NO, produced by eNOS, plays a pivotal role in maintaining vascular function and modulating NRF2 signaling. Evidence underscores the importance of NO and NRF2 in neuronal and cardiovascular protection. For instance, Calabrese et al. (2010) demonstrated that NRF2 activation induces the expression of “vita-genes,” which safeguard neurons from cellular stress by enhancing mitochondrial function and reducing oxidative stress (Calabrese et al., 2010). Similarly, Calabrese et al. (2007) highlighted the neuroprotective effects of NO through the activation of intracellular signaling pathways that regulate cardiovascular, immune, and nervous system functions (Calabrese et al., 2007, 2008). La Rosa et al. (2020) further demonstrated that NRF2 signaling ameliorates pathological defects in Friedreich’s ataxia, a neurodegenerative disorder, showcasing the potential therapeutic benefits of this pathway. These findings collectively suggest that targeting NRF2/ARE signaling and eNOS activity could serve as viable therapeutic strategies for mitigating RICVD and enhancing resilience to IR-induced vascular damage (La Rosa et al., 2020).
The inhibition of inflammasome activation and improvement of mitochondrial function represent additional therapeutic targets for RICVD. Administering NLRP3 inhibitors, ROS scavengers, or clinically approved agents such as anakinra or canakinumab may reduce IL-1β/IL-18-driven inflammation and vascular damage. Mitochondrial dysfunction caused by radiation could be addressed with mitochondrial-targeted antioxidants, such as MitoQ, to restore mitochondrial function and prevent further damage to the vasculature.
FLASH radiotherapy represents an innovative approach to minimizing the toxicity of conventional RT while maintaining high tumor control efficacy. This technique involves the delivery of radiation at ultra-high dose rates (≥40 Gy/s), which achieves effective tumor control with minimal or no damage to normal cells and tissues (Matuszak et al., 2022). For example, Favaudon et al. (2014) demonstrated that applying FLASH at 60 Gy/s to orthotopic and xenograft tumors in mice mitigated pulmonary fibrosis compared with conventional RT (Favaudon et al., 2014). Recent studies have extended these findings across various tumor types and species, including zebrafish, mice, pigs, cats, and humans, providing compelling evidence of its broad applicability (Beyreuther et al., 2019; Konradsson et al., 2021; Vozenin et al., 2019). Specific investigations have further validated the therapeutic potential of FLASH. Diffenderfer et al. (2020) found that whole-abdominal FLASH treatment in mice with pancreatic tumors preserved the proliferative capacity of normal cells without exhibiting tissue-sparing effects (Diffenderfer et al., 2020). Similarly, Zhang et al. (2020) reported improved survival rates in mice exposed to 16 Gy FLASH (100%) compared with conventional RT (40%) at 21 days postirradiation (Zhang et al., 2020). The protective effect of FLASH on normal cells is attributed to its ultrafast delivery of radiation doses, which creates transient hypoxia in the tumor microenvironment. This hypoxic state offers resistance to radiation-induced damage in normal cells but not in cancer cells due to their differing oxygenation statuses (Chow and Ruda, 2024).
FLASH radiotherapy has also been shown to preserve vascular integrity and function. In a preclinical subcutaneous Lewis lung carcinoma model, FLASH at 15 Gy demonstrated effective tumor control while maintaining normal vasculature, unlike the critical vascular collapse observed with conventional RT (Kim et al., 2021). This vascular protection is attributed to the FLASH-induced reduction in myosin light chain phosphorylation (p-MLC), which mitigates EC contraction and reduces immune cell infiltration. Despite these promising findings, the clinical adoption of FLASH therapy necessitates further investigation. The accumulating data underscore its efficacy in cancer treatment; however, its long-term safety and effectiveness in broader patient populations remain to be confirmed. Rigorous validation in clinical trials is essential to ensure its feasibility and optimize its use in radiation oncology. Once fully established, FLASH radiotherapy could revolutionize cancer treatment by significantly reducing adverse effects while maintaining therapeutic efficacy.
Footnotes
Authors’ Contributions
N.-T.L. structured and wrote the article. J.A. and K.C. contributed to writing, editing, and creating the figures. All authors reviewed, provided critical feedback, and approved the final version.
Availability of Data and Materials
All figures were created using BioRender.
Author Disclosure Statement
A.D. has served as a consultant to Bayer. Other authors have nothing to disclose. No competing interests.
Funding Information
This study received partial support from the National Institutes of Health (NIH) for N.-T.L. (HL157790, HL149303, HL163857, HL134740), J.P.C. (HL148338, HL157790), and J.A. (HL149303, AI156921). Additional funding was provided by NASA, BARDA, and the USFDA (Contract No. 80ARC023CA002). J.A. and Schadler were also supported by the Cancer Prevention and Research Institute of Texas (CPRIT, RP190256). The study was further supported by the NIH NHLBI (Award No. R01HL157790), an NSF CAREER Award (No. 1944322) to A.J., and an Institutional Research Grant (IRG) Program from the University of Texas MD Anderson Cancer Center to S.K. G.W. received support from the National Institute of General Medical Sciences (NIGMS, R35GM150460).