
Abstract
Purpose:
This study aims to generate a more potent oncolytic adenovirus, Ad.hTERT-E1A/CMV-CD, which combines therapeutic gene and oncolytic effect.
Methods:
A human telomerase reverse transcriptase (hTERT) gene promoter was used to regulate the expression of adenoviral immediate-early gene 1A (E1A) to induce selective replication of recombinant adenovirus in tumor cells. To further enhance antitumor effect, a cytomegalovirus (CMV) promoter-driven Escherichia coli cytosine deaminase (CD) gene expression cassette was further incorporated into E1 region and the antitumor effect of this novel adenovirus was evaluated in vitro and in vivo.
Results:
Ad.hTERT-E1A/CMV-CD was capable to selectively replicate and lyse in various human tumor cell lines, including NCIH460, SW1990, and HeLa, while causing no damage to primary fibroblasts. The combined therapy of Ad.hTERT-E1A/CMV-CD with prodrug 5-fluorocytosine (5-FC) elicited a greater killing effect on tumor cells than Ad.hTERT-E1A/CMV-CD alone, and it synergistically suppressed tumor growth in BALB/c nude mice bearing human lung tumor.
Conclusions:
As telomerase is reactivated in a broad spectrum of tumors and prodrug 5-FC is much safe than its metabolized 5-fluorouracil, a chemotherapeutic agent in the treatment of many malignancies, Ad.hTERT-E1A/CMV-CD in combination with 5-FC may be a potential strategy for the treatment of a wide range of solid tumors.
Introduction
Despite advances in surgery, radiation, and chemotherapy, the prognosis for most malignant tumors remains poor. This fact accentuates the need for a new generation of more effective therapies for cancer. Replication-competent viruses have recently emerged as a promising new platform for the treatment of cancers, as they are able to selectively replicate in tumor cells and increase local concentration of viral particles and then lyse tumor cells. In addition, the oncolytic effect will result in spread of viral vectors to adjacent tumor cells, thereby inducing bystander effect. Many of these viruses have entered clinical trials, including vaccinia virus, herpes simplex virus-1, measles virus, Newcastle disease virus, and reovirus; however, oncolytic adenovirus, such as ONYX-015, is most widely used. 1 –10
Two main strategies have been utilized to construct oncolytic adenoviruses. One involves the use of tumor- or tissue-specific promoter to control the expression of adenoviral gene or genes that are essential for its replication. Another is based on deleting adenoviral gene or genes encoding protein responsible for completing adenoviral lytic life cycle in normal cells, but not in tumor cells, thereby restricting viral replication to tumor cells. 7,8 The first tested oncolytic adenovirus was ONYX-015 (dl1520), which has a deletion in E1B region, enabling it to replicate selectively in cells deficient in the p53 pathway. 9,10 However, it is now clear that ONYX-015 replication in some tumor cell lines appears to be independent of p53 status and its antitumor efficacy remains limited. 11,12
As human telomerase reverse transcriptase (hTERT) gene is reactivated in >85% of human cancers, 13,14 and early gene 1A (E1A) is an essential gene for adenoviral replication, the authors and other groups had used hTERT promoter to drive adenoviral E1A gene expression to develop tumor-specific replication-competent adenoviral vectors such as Ad.hTERT-E1A. And such vectors have been shown to be superior to ONYX-015 in terms of selective replication and oncolytic effect. 15 –19
Escherichia coli cytosine deaminase (CD) gene encodes a nonmammalian enzyme that can convert the nontoxic antifungal agent, 5-fluorocytosine (5-FC), into the chemotherapeutic drug, 5-fluorouracil (5-FU). 20 Because 5-FU is one of the most potent as well as widely used antitumor drugs, the advantage of using the CD/5-FC (enzyme/prodrug) therapy strategy is that it can limit cytotoxic 5-FU to tumor cells and spare the normal cells, 21,22 and therefore, CD gene has been chosen as a therapeutic gene to further enhance the antitumor effect of oncolytic adenovirus Ad.hTERT-E1A. The results from this study suggested that Ad.hTERT-E1A/CMV-CD in combination with prodrug 5-FC may be a potential strategy for the treatment of a wide range of tumors.
Materials and Methods
Cell lines and cell culture
Human embryonic kidney 293 (HEK293) cells were purchased from Invitrogen (San Diego, CA). The human cancer cells NCIH460 (lung cancer cell line), A549 (lung cancer cell line), SW1990 (pancreas cancer cell line), HeLa (cervical carcinoma cell line), and SMMC-7721 (hepatoma cell line) were obtained from the Cells Bank of the Chinese Academy of Science (Shanghai, China). Primary human dermal fibroblasts derived from bioptic tissue for dermatoplasty (a written informed consent was obtained from patients) were provided by the authors' laboratory. Cells were cultured in DMEM medium supplemented with 10% fetal bovine serum. All cancer cells possess telomerase activities, whereas primary human dermal fibroblasts do not possess telomerase activity according to a previous study. 15
Construction and preparation of recombinant adenoviruses
The complete cDNA sequence of the E. coli CD gene lacking the stop codon was amplified by PCR and inserted into pcDNA3.1/HA-myc-His(−)Z (a gift from the State Laboratory for Oncogene and Related Gene Research, Shanghai Jiaotong University, Shanghai, China) to obtain an HA-tag. The HA-tagged CD gene was cut and inserted into the plasmid pEGFP-N1 to get a complete CD-HA-tag expression cassette, which has cytomegalovirus (CMV) promoter, CD-HA-tag coding sequence, and SV40 poly A. This cassette was then inserted into the plasmid pDC311-hTERT-E1A to yield pDC311-hTERT-E1A-CMV-CD-HA-tag. The plasmid, pDC311-hTERT-E1A, contained an hTERT-controlled E1A expression cassette between BglII and SalI site of multiple cloning sites in pDC311, which has a majority of adenoviral genome except E1 and E3 regions (Microbix, Toronto, Canada).
To package the recombinant adenoviral vector, the shuttle vector, pDC311-hTERT-E1A-CMV-CD-HA-tag, was then transfected by Lipofectamine2000 (Invitrogen) into HEK293 cells together with adenoviral backbone plasmid pBHGloxΔE13cre (Microbix), following the manufacturer's protocol. The viral comets appeared 7–14 days after co-transfection. The plaque assay was carried out for a total of three rounds to pick up more homogeneous adenoviral vectors, and thereafter, the large scale of preparation was performed using HEK293 cells and purified by CsCl gradient centrifugation according to the protocol described by Huang et al. 15 The presence of the CD gene was determined by PCR. Ad.GFP (a replication-defective Ad expressing the green fluorescent protein gene [GFP]), Ad.null (a replication-defective Ad expressing no extraneous gene), dl309 (a wild-type Ad having a completely intact E1 region), and Ad.hTERT-E1A (a replication-competent adenovirus with hTERT-driven E1A expression cassette) were also propagated with the same method described above. The schematic diagram of Ad.hTERT-E1A/CMV-CD is shown in Figure 1A.
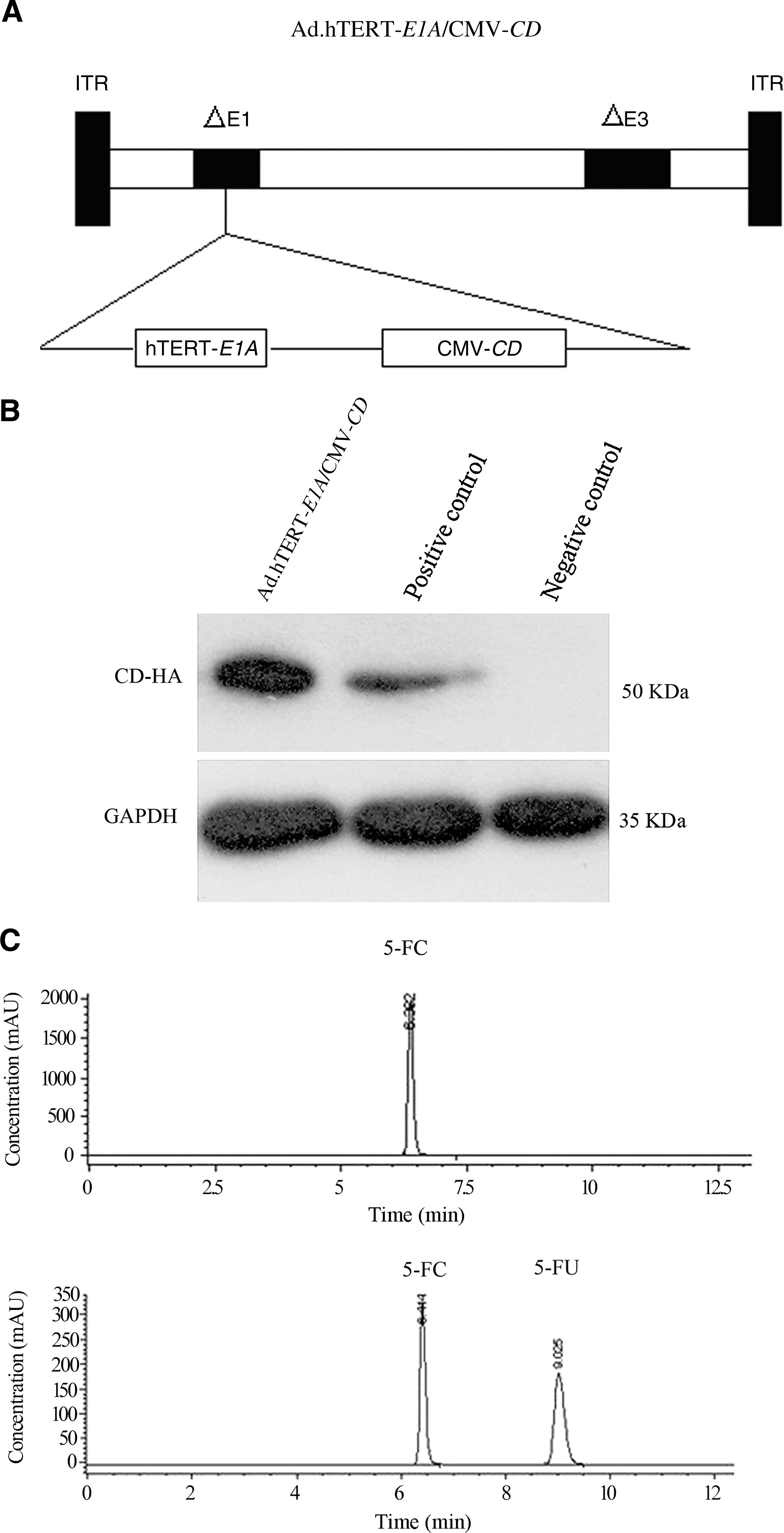
Ad.hTERT-E1A/CMV-CD adenoviral construct and its character. (
Western blot analysis
The cells were plated in six-well plates and infected with Ad.hTERT-E1A/CMV-CD at a multiplicity of infection (MOI) of 10 plaque forming units (PFU), that is, 10 MOI/cell. Forty-eight (48) hours after infection the infected cells were harvested and lysed in sodium dodecyl sulfate (SDS) sample buffer (containing 10 mM β-mercaptoethanol, 100 mM Tris-Cl [pH 6.8], 2% SDS, and 0.1% bromophenol blue). Protein samples were separated by 10% SDS polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Amersham, Buckinghamshire, United Kingdom). The blots were probed with anti-HA (Sigma, St. Louis, MO) monoclonal antibody detected for CD or anti-Ad2 E1A (Santa Cruz Biotechnology, Santa Cruz, CA) directed against adenoviral E1A, followed by a secondary horseradish peroxidase-conjugated antibody. The antigen–antibody complexes were observed using the enhanced chemiluminescence kit (Roche, New York, NY) as recommended by the manufacturer.
Analysis of 5-FC metabolites using high-performance liquid chromatography
NCIH460 cells (4 × 105) were infected with Ad.hTERT-E1A/CMV-CD or Ad.hTERT-E1A (as control) at 20 MOI. Twenty-four (24) hours later, the cells were grown in DMEM containing 5 mM 5-FC (Sigma). The cells and media were harvested after 48 hours of incubation with 5-FC. The cells were lysed by three rounds of freezing–thawing and then centrifuged for 10 minutes at 14,000g. Aliquots (20 μL) of each sample or standard were analyzed by HPLC on a Partisphere C18 column (4.6 × 250 mm; Whatman, Kent, United Kingdom), which was previously equilibrated with 0.1% trifluoroacetic acid (TFA). The elution was performed with acetonitrile containing 0.1% (v/v) TFA with a gradient of 0%–20%, 20%–70%, and 70%–100% at a flow rate of 0.7 mL/min. The UV absorbance at 254 nm was detected.
In vitro viral replication assay
To determine virus progeny production, NCIH460, HeLa, SW1990, and primary human fibroblasts plated in six-well plates were infected with Ad.hTERT-E1A/CMV-CD or dl309 at 10 MOI, respectively. After 4 hours of incubation at 37 C, cells were washed once with phosphate-buffered saline (PBS) and maintained in growth medium. Five (5) days later, the cells and medium were harvested, freeze–thawed three times, and centrifuged to collect the supernatant. The titers of vectors were determined by the plaque assay in HEK293 cells.
Cytotoxicity assay
Tumor cells (NCIH460, HeLa, and SW1990) or primary human fibroblasts were plated in six-well plates. On the next day, the cells were infected with Ad.hTERT-E1A/CMV-CD, dl309, or Ad.GFP at 10 MOI, respectively. At the time points indicated, the cytopathic effects (CPEs) were monitored by light microscopy. To further quantify the CPEs, cells (5 × 103 cells/well, 96-well plate) were either mock infected or infected with increasing amounts of Ad.hTERT-E1A/CMV-CD. The fresh medium was replaced at 24 hours after infection and the cytotoxicity was assessed with Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Gaithersburg, MD) after 5 days, according to the manufacturer's protocol.
For prodrug sensitivity or cytotoxicity assays, cells (5 × 103 cells/well, 96-well plate) were either mock infected or infected with Ad.hTERT-E1A/CMV-CD at 10 MOI. The fresh medium with or without different concentrations of 5-FC were applied at 24 hours after infection. The survived cells were quantified by CCK-8 assay after 5 days. A crystal violet assay was also performed. The cells (5 × 105 cells/well, 24-well plate) were infected with Ad.hTERT-E1A/CMV-CD at 10 MOI or mock infected. Twenty-four (24) hours later, the medium containing virus was removed and the fresh medium with or without 5-FC (50 μg/mL) was applied every 2 days. Five (5) days after 5-FC treatment, the cells were washed twice with ddH2O and stained with 1% crystal violet in 70% methanol for 30 minutes. The pictures were captured with a digital camera.
Animal experiments
Specific pathogen-free male athymic BALB/c nude mice, 4–6 weeks old (20–30 g), were obtained from the Institute of Animal Center (Chinese Academy of Sciences, Shanghai, China). Mice were housed 5 per cage and allowed free access to food and water. All animal procedures were performed according to principles of laboratory animal care (NIH publication No. 85-23, revised 1985) and the current Chinese regulations and standards on the use of laboratory animals. For tumor cell implantation, NCIH460 cells (5 × 106) were injected subcutaneously into the right dorsal lumbar region in 100 μL PBS. When the tumors grew to ∼100 mm3, the animals were randomly divided into 5 groups, each containing at least 7 animals. Animals were subjected to intratumoral injection with 1 × 109 PFU of Ad.null, Ad.hTERT-E1A/CMV-CD, or PBS. Three (3) days after viral injection, the Ad.hTERT-E1A/CMV-CD-injected and PBS-injected animals were either left untreated or administered 5-FC (500 mg/[kg day]) for 14 consecutive days. The tumor growth was assessed by measuring bidimensional diameters twice a week with calipers. The tumor volumes (V) were calculated according to the following formula: V = 1/2ab 2, where a represents the largest diameter and b is the smallest diameter. All the animals were sacrificed at 4 weeks after treatment.
Statistical analysis
The data from different groups were compared statistically by one-way analysis of variance using SPSS12.0 statistical software. A p-value of <0.05 was considered statistically significant.
Results
CD converting 5-FC into 5-FU in Ad.hTERT-E1A/CMV-CD-infected cells
A tumor-specific replication-competent adenovirus was generated by inserting an hTERT promoter-driven E1A expression cassette into adenoviral vector. To further enhance antitumor efficacy, a CD gene was engineered into the vector. CD gene expression was confirmed by western blot on NCIH460 cells infected with or without Ad.hTERT-E1A/CMV-CD (Fig. 1B). To further validate that CD gene was functional, the medium or lysate from NCIH460 cells was analyzed for 5-FU metabolites by HPLC after infection with Ad.hTERT-E1A/CMV-CD or Ad.hTERT-E1A and exposure to 5 mM 5-FC. The medium and cell lysate from Ad.hTERT-E1A/CMV-CD-infected cells showed two peaks, which were identified as 5-FC and 5-FU by comparison with the standards. In contrast, the medium and cell lysate from Ad.hTERT-E1A-infected cells showed a single peak, which was identical to 5-FC (Fig. 1C). Thus, the prodrug 5-FC could be transformed into the chemotherapeutic drug 5-FU in Ad.hTERT-E1A/CMV-CD-infected cells.
Ad.hTERT-E1A/CMV-CD selectively killed tumor cells but not normal cells
A series of experiments were conducted to assess whether Ad.hTERT-E1A/CMV-CD preferentially replicated in tumor cells but not in normal cells. At first, the different types of tumor cells including NCIH460, SW1990, and HeLa, or normal cells, that is, primary fibroblasts were infected with either 10 MOI Ad.hTERT-E1A/CMV-CD, dl309, or Ad.GFP, and thereafter, the CPEs were observed by light microscopy. The tumor cells displayed similar CPEs after infection of Ad.hTERT-E1A/CMV-CD or dl309 with the exception of Ad.GFP. In contrast, CPEs were only observed on fibroblasts infected by dl309 but not by Ad.hTERT-E1A/CMV-CD or Ad.GFP (Supplemental Fig. 1A, available online at
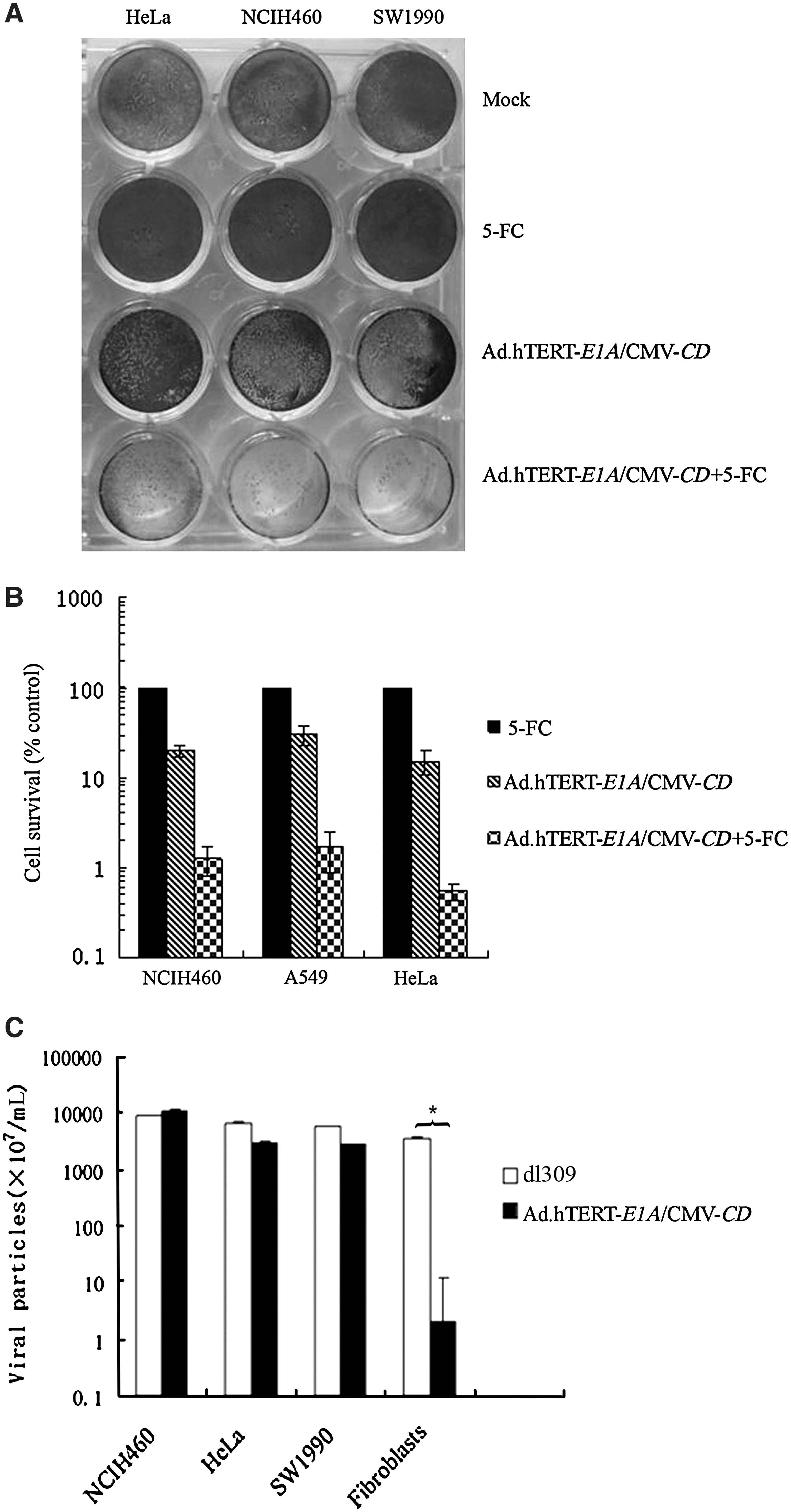
Tumor selective replication and cytotoxicity of Ad.hTERT-E1A/CMV-CD. (
Ad.hTERT-E1A/CMV-CD replicated in tumor cells but not in normal cells
The differences in production of adenoviral progenies in Ad.hTERT-E1A/CMV-CD- or dl309-infected tumor cells or primary fibroblasts were the direct evidence for recombinant adenovirus replication selectivity. The results from plaque assay indicated that the tumor cells produced a large amount of adenoviral particles after infection with Ad.hTERT-E1A/CMV-CD, which were comparable to that infected with dl309 (p > 0.05). In contrast, the primary fibroblasts produced remarkably attenuated viral particles after infection with Ad.hTERT-E1A/CMV-CD, which were about 1800-fold less than that after infection with dl309 (p < 0.01; Fig. 2C).
The E1A gene expression in Ad.hTERT-E1A/CMV-CD-infected tumor cells or primary fibroblasts indicated the transcriptional activation of viral and cellular genes needed for virus replication. 18,23 Western blot analysis was then carried out to examine E1A expression. The positive signal was observed only in HEK293 cells, a positive control, which has been stably transduced by partial adenovirus genome including E1A, and Ad.hTERT-E1A/CMV-CD-infected tumor cells but not primary fibroblasts (Supplemental Fig. 1B). These data indicated that Ad.hTERT-E1A/CMV-CD replication was restricted to tumor cells.
Growth inhibition mediated by Ad.hTERT-E1A/CMV-CD/5-FC in vitro
To quantify the growth inhibition mediated by Ad.hTERT-E1A/CMV-CD/5-FC in vitro, tumor cells (NCIH460, SW1990, and HeLa) or primary fibroblasts were infected with adenoviral vector at various MOI. All tumor cells were efficiently killed by Ad.hTERT-E1A/CMV-CD in a dose-dependent manner, but normal fibroblasts were resistant to Ad-hTERT-E1A/CMV-CD infection (p < 0.05; Fig. 3A). This growth inhibition effect could be further enhanced by adding prodrug 5-FC, although 5-FC alone showed no significant inhibition on tumor cell growth. As expected, 5-FC-increased CPE was also dose dependent (Fig. 3B).
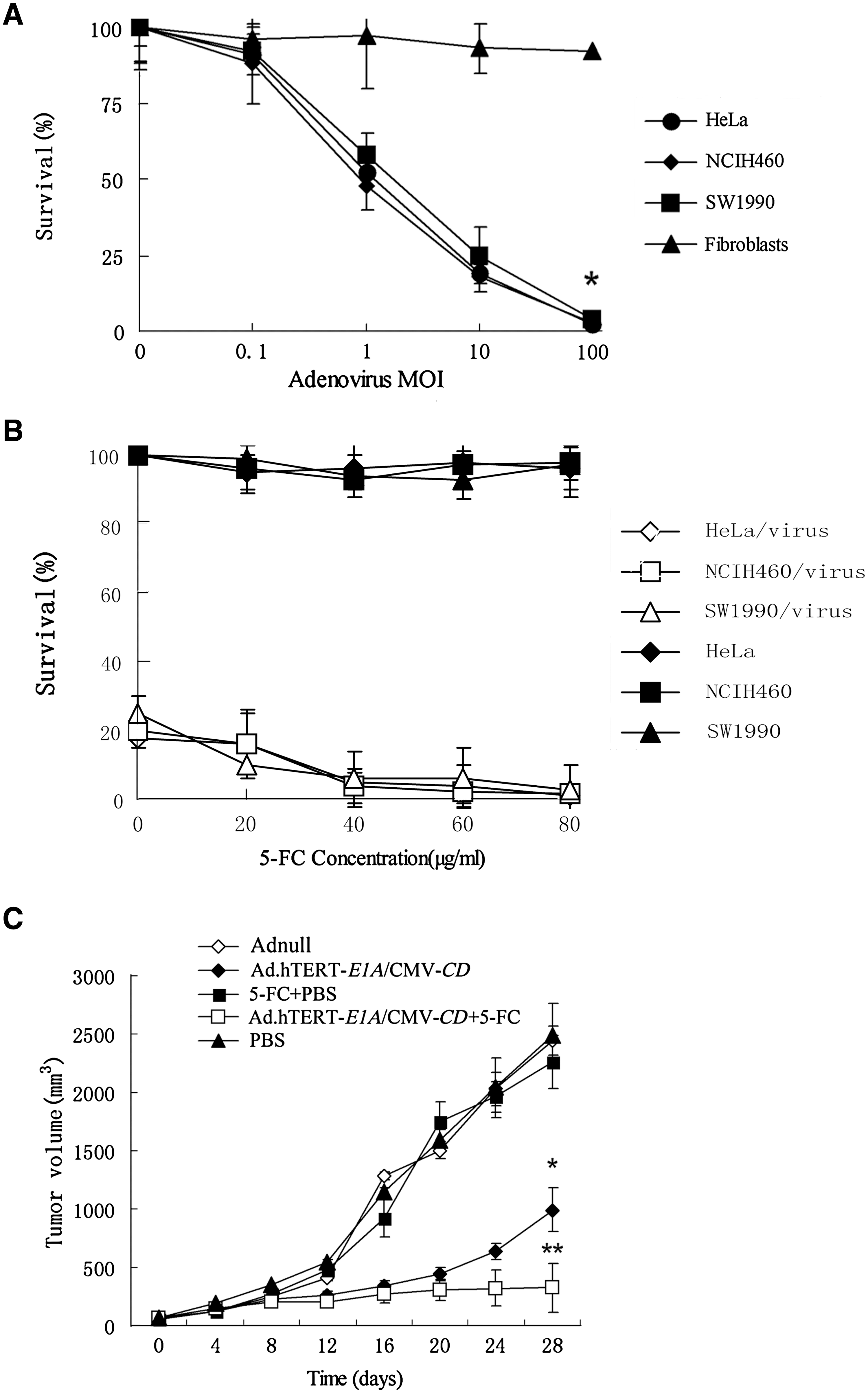
Growth inhibition mediated by Ad.hTERT-E1A/CMV-CD/5-FC in vitro and in vivo. (
Ad.hTERT-E1A/CMV-CD/5FC suppressed tumor growth in vivo
The therapeutic efficacy of Ad.hTERT-E1A/CMV-CD, either alone or in combination with prodrug 5-FC, was evaluated using nude mice bearing established NCIH460 subcutaneous xenografts. When the tumors reached a volume of ∼100 mm3, the nude mice were randomly assigned to each group and treated according to the description in the Materials and Methods section. The tumor growth was assessed by measuring bidimensional diameters twice a week with calipers, and the tumor growth curves were drawn (Fig. 3C). At the end of this experiment, the tumor sizes were 2488.80 ± 468.97, 2261.00 ± 362.87, 2440.00 ± 227.99, 993.01 ± 317.62, and 326.80 ± 236.66 mm3 for the PBS alone, PBS plus 5-FC intraperitoneal injection, Ad.null, Ad.hTERT-E1A/CMV-CD alone, Ad.hTERT-E1A/CMV-CD plus 5-FC groups, respectively. The volume of tumors treated with PBS alone or PBS plus 5-FC were identical to those of tumors treated with Ad.null, a replication-incompetent adenovirus (p = 0.995; p = 1.000). Intratumoral inoculation of Ad.hTERT-E1A/CMV-CD itself resulted in delayed tumor growth, which was significantly different from tumors treated with PBS on day 28 post-treatment (p = 0.006). More importantly, tumor growth inhibition was remarkably increased in the group of Ad.hTERT-E1A/CMV-CD plus 5-FC intraperitoneally administration, which resulted in 3.04 times smaller in tumor size at the end point in comparison with Ad.hTERT-E1A/CMV-CD treatment alone (p = 0.032).
Discussion
Oncolytic adenoviruses can selectively replicate in malignancies, and therefore, in addition to oncolytic effect, an increased viral spread and transgene expression in tumor mass can also be obtained accompanying with the replication process. 1 –10 As E1A gene product is essential for adenovirus replication and the lack of E1B has been identified to deplete adenoviral replication in normal cells but not in p53 mutated cells, which occurred in almost 85% tumors, the novel adenoviral vector constructed in this study has increased stringency for replication than adenoviral vector with E1B gene in theory. 9,10 Therefore, it could be thought to be double-controlled, that is, p53 mutant and telomerase activation associated.
Data from clinical trails indicated that oncolytic adenovirus itself was insufficient to kill tumor cells when used as a monotherapeutic agent. 9,10 A new strategy by inserting one or two therapeutic genes into the oncolytic adenoviral vector is emerging and is called as “armed oncolytic adenovirus” or “gene–viral therapy.” 23 To enhance therapeutic efficacy, a CD gene expression cassette was inserted into an oncolytic adenoviral vector. This novel oncolytic adenoviral vector has both hTERT promoter-driven E1 gene expression cassette and a constitutive expression CD gene cassette in E1 region and it efficiently replicated in tumor cells, similar to wild-type adenovirus dl309. Accordingly, this vector conferred CPE on tumor cells, but not on normal fibroblasts. The preliminary data suggest that Ad.hTERT-E1A/CMV-CD could efficiently convert 5-FC to 5-FU in infected tumor cells, as measured by HPLC, and Ad.hTERT-E1A/CMV-CD in combination with prodrug 5-FC was more efficacious and less toxic than the conventional administration of the chemotherapeutic agent 5-FU and resulted in synergistic suppression of tumor growth in vitro and in vivo.
Although oncolytic adenoviruses hold the promise to be more efficient gene delivery vehicles and powerful gene–viral therapy methods, the safety issues should be addressed before this mode of therapy can be introduced into the clinical setting. According to a previous study, adenoviral leaking into circulation and consequent infection in liver, lung, spleen, and kidney could occur after intratumoral injection. 24 As systemic administration of adenoviral vector is required for those patients with disseminated tumors or multiple metastatic lesions, the stringency of adenoviral replication is the prerequisite for developing oncolytic adenoviral therapy and the biodistribution and potential toxicity should be extensively investigated. It is worth mentioning that the application of Ad.hTERT-E1A/CMV-CD has some limitations because it contains a suicide gene, which may cause some undesirable effect in nontargeted organs or tissues. The intratumoral injection may be more preferential for this novel vector. Fortunately, the suicide gene/prodrug systems, such as CD/5-FC in this study, could be used as a fail-safe mechanism to stop adenoviral vector replication when the risk for disseminated adenovirus infection occurs, thereby providing a means to control viral runaway infections. 25
Conclusions
A novel “armed oncolytic adenovirus” (Ad.hTERT-E1A/CMV-CD) was generated. Its replicative capacity was mediated by hTERT activity and p53 gene status in host cells and its tumor cell killing effect was further enhanced by CD/5-FC therapy strategy. This novel oncolytic adenovirus showed extensive growth suppression on different types of tumor cells and xenografted human lung cancer in BALB/c nude mice. The results from this study suggested that Ad.hTERT-E1A/CMV-CD in combination with prodrug may be an effective and safe strategy for the treatment of a wide range of solid tumors.
Footnotes
Acknowledgments
This work was supported by grants from the National Basic Research Project of China (2010CB529902), National High-Tech R&D Program (2007AA021202), and National Natural Science Foundation for Outstanding Youth (30325043).
Disclosure Statement
There are no financial supports or relationships that may pose a conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.