
Abstract
Abstract
The use of a mouse embryonic fibroblast (MEF) feeder for culture of embryonic stem cells (ESCs) is a widely accepted method, regardless of the ESCs' origin and type. In this study, we performed the undifferentiated propagation of human ES cell lines (hESCs, H1, and HSF6) and mouse ES cell lines (mESCs, D3, and CE3), which were previously maintained on an MEF feeder, using human placenta-derived fibroblast-like cell (HPC) feeders originated from chorionic villi of women who had undergone therapeutic abortion due to known maternal disease that is aggravated by pregnancy. Moreover, we tried to introduce the HPC feeder for the establishment of inducible pluripotent stem cells (iPSCs) from human placental mesenchymal stem cells (MSCs). On the HPC feeder we were able to propagate ESCs and iPSCs colonies as an undifferentiated state up to the 50th passage and 20th passage, respectively. Maintenance of undifferentiated ESCs was identified by the expression of ALP, SSEA-1, SSEA-4, TRA-81, TRA-60, Oct-4, Nanog, or Rex-1. Also, addition of leukemia inhibitory factor was not required for undifferentiated propagation of mESCs on the HPC feeder. The efficiency and expression of three germ layer markers of embryoid bodies (EBs) from ESCs were satisfactory in both the MEF and HPC group. EBs formed from iPSCs were scant, and differentiation to the three germ layers was identifiable by reverst transcription-polymerase chain reactio (RT-PCR) only in the HPC group. In conclusion, the HPC feeder can efficiently support the undifferentiated propagation of hESCs, mESCs, and iPSCs, suggesting that human placenta may be a useful source of universal feeder cells for hESC, mESC, and iPSC culture.
Introduction
To perform studies using ESCs, indefinite propagation and maintenance of ESCs in an undifferentiated state is required. Coculture of ESCs on the feeder layer is the most widely accepted method, regardless of ESCs' origin and type. Mouse embryonic fibroblast (MEF) cells are the most widely used feeder cells for both hESCs and mESCs. However, there are certain drawbacks in the culture system using MEF as a feeder. First, MEF is usually made of fibroblasts from mouse embryos at embryonic day 13.5, and can only be propagated for approximately 7–12 passages before they senesce (Kim et al., 2002); only cells at early passages (p4–p6) are used as feeders (Conley et al., 2005). Because continuous sacrifice of many mouse embryos is required in the culture system using the MEF feeder it is very costly, and involves other ethical issues. Next, in respect to the hESC culture, there is a risk of transmission of animal pathogens, such as mouse retroviruses; therefore, new zoonosis can develop in the hESC culture system using the MEF feeder. Moreover, a product contaminated with animal contents could be immunologically unstable, and therefore, lead to an immunogenic response in the host during cell replacement therapy. Recent study has demonstrated that hES cells cultured on MEF incorporated a sialic acid originated from a nonhuman source (Martin et al., 2005). Therefore, tremendous effort toward minimizing exposure to animal products is required in the culture of hES cells.
Some issues associated with mES culture also need to be resolved. The culture system for maintaining mESCs on an MEF feeder is a full xenoenvironment. Discrepancy between the outcome from use of this system and the physiologic and immunologic features of hESCs is inevitable. Considering that much of the intellectual driving force behind the study of hESC has come from the mESC field, the situation is undesirable now, and will remain so in the future. Additionally, the role of leukemia inhibitory factor (LIF), an inhibitor of mESC differentiation (Gough et al., 1989; Williams et al., 1988), in the mESC culture using MEF is not clear. Although there is some evidence that MEF produces LIF (Burdon et al., 1999; Rathjen et al., 1990), many investigators add LIF to the media in culturing of mESC on the MEF feeder to avoid the potential risk of unwanted mESC differentiation. Culture of mESC on the MEF feeder without LIF requires more validation.
Recent reports on human placenta-derived fibroblast-like cells (HPC) have demonstrated their usefulness as a feeder in support of undifferentiated growth of primate ESCs and hESCs (Genbacev et al., 2005; Miyamoto et al., 2004; Simon et al., 2005). Based on these reports, we performed in vitro maintenance of an hESC line, SNUhES3, which was previously maintained on the MEF feeder, using HPC as the feeder layer; our findings demonstrated that HPC could support prolonged undifferentiated propagation of SNUhES3 (Kim et al., 2007). HPC has some advantages over MEF. First, it originated from a human source; therefore, concern over development of new zoonosis and a full xenoenvironment is relatively reduced compared with the MEF feeder. Next, it was known to be propagated up to the 35th passage and to support ESCs until the 12th passage (Ilic et al., 2008). Thus, the culture system using an HPC feeder can be more cost effective than an MEF feeder. Moreover, because a recent study reported on successful propagation of mESC on a human-origin feeder (human foreskin fibroblasts, human endothelial cell) (Meng et al., 2008; Zhou et al., 2008) without LIF, the attempt to culture mESC on HPC without LIF is worthwhile.
However, if HPC can be utilized as an alternative to MEF, and used as widely as MEF in culturing of ESCs, a variety of ESC lines should be maintained in an undifferentiated state on an HPC feeder. For this validation, we cultured two kinds of hESC lines (H1 and HSF6) and two kinds of mESC lines (D3 and CE3) on an HPC feeder. Moreover, we introduced the HPC feeder for the establishment of inducible pluripotent stem cells (iPSs) from placental mesenchymal stem cells (MSCs). The institutional review board at the Korea University Medical Center approved this study protocol. Placentas were used after written informed consent was obtained.
Materials and Methods
Preparation of feeder cells from placenta and MEF cells
A previously reported protocol of for preparation of feeder cells from placenta was used in this study (Genbacev et al., 2005; Kim et al., 2007). We obtained 21st-trimester human placentas at 6–8 weeks gestational age from women that had undergone therapeutic abortion due to known maternal disease that is aggravated by pregnancy. Briefly, chorionic plates of placenta were surgically isolated, minced, and incubated in 0.25% trypsin–EDTA (GIBCO–Invitrogen, Carlsbad, CA, USA) at 37 °C for 30 min. We then cultured the cells in Dulbecco's modified Eagle medium (DMEM) with 20% fetal bovine serum (FBS; HyClone Laboratories Inc., Logan, UT, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C, 5% CO2, and 95% humidity. The medium was exchanged every 3–4 days until the third passage. During culture, we removed cell debris floating in the culture medium, and found placental-derived fibroblast-like cells growing attached to the plate. At approximately 2 weeks after inoculation, colonies of fibroblast-like cells that were attached to the plates had formed (Fig. 1). Presence of MSCs was identified by flow cytometric analysis showing positive expression of CD 29 and CD 44 and negative expression of CD 34. In the primary culture of HPC from an initial 10 placentas, HPCs usually became thin and lost stability after the 20th∼25th passage, and HPCs after the 13th∼17th passage could not support undifferentiated growth of both hESCs and mESCs. Therefore, HPCs were cultured to the 12th passage, and all of these cells were stocked for the ESC culture. Frozen stock was achieved at the amount of 1 × 106 cells per cryovial after harvest with trypsinization and irradiation (1500 cGy). For ESC culture, stocked HPCs were thawed out and seeded onto 0.1% gelatin-coated 35-mm well plates at 1 × 106 cells/well for coculture with hESCs.
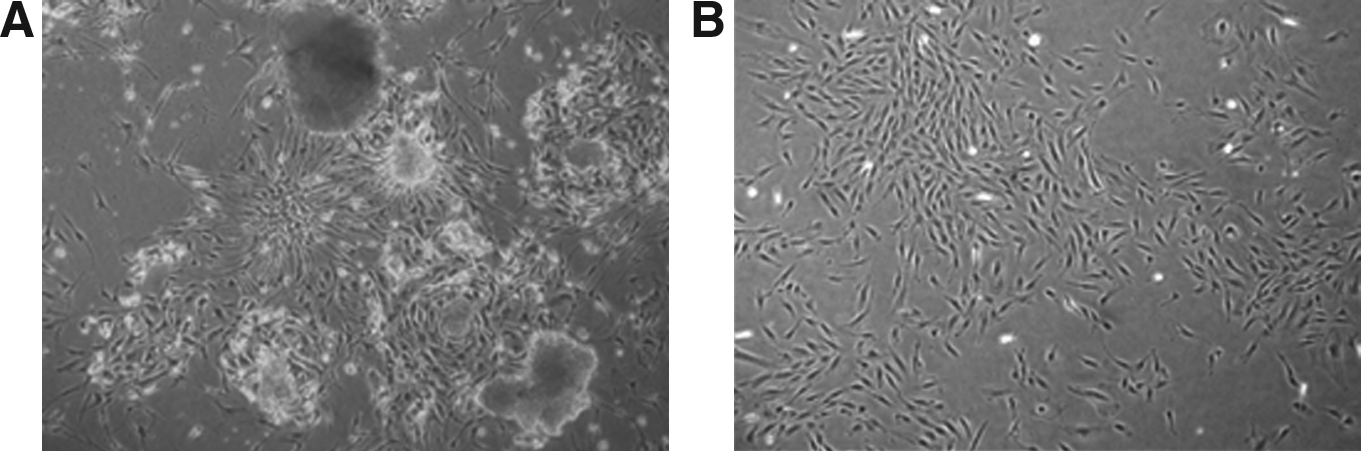
Morphology of placenta-derived cells. (
Pathogens that commonly infect the placenta were tested by reverse transcriptase-polymerase chain areaction (RT-PCR) before they were frozen as stock. These include cytomegalovirus, herpes simplex virus types 1 and 2, Chlamydia trachomatis, Chlamydia spp, Mycoplasma genitalium, Mycoplasma hominis, and Ureaplasma ureaticum. The published primer sequences were used (Genbacev et al., 2005; McDonagh et al., 2004). HPC testing positive for pathogens were abandoned.
The MEF cell line was purchased from Millipore (Bedford, MA, USA). MEF was cultured in DMEM supplemented with 10% FBS at approximately 90% confluence; typically, MEF could not be propagated after the fifth to seventh passage and could not support undifferentiated growth of both hESCs and mESCs after the fourth to fifth passage. Therefore, MEF was cultured up to the third passage and stocked for ESC culture. MEF was prepared by incubation with 10 μg/mL mitomycin for 90 min before being transferred to plates. These two feeder cells (HPC, MEF) were allowed to attach for 24 h before seeding of hESCs, mESCs, and iPSCs.
Human and mouse ESC culture
The H1 hESC line (p29) was purchased from National Stem Cell Bank and initially cultured on MEF according to the instructions of the provider. The HSF6 hESC line (p63), which was originally purchased from National Stem Cell Bank, and kindly provided by Dr. Jong Hoon Kim (Laboratory of Stem Cell Biology, College of life Sciences and Biotechnology, Korea University), was cultured on MEF. After maintenance on MEFs for 17 passages in H1 and 8 passages in HSF-6, colonies of both hESCs were transferred onto a newly prepared HPC and MEF feeder (Fig. 2). Coculture of hESC and feeder was performed in DMEM/F-12 supplemented with 20% Knockout Serum Replacer (Gibco, Gaithersburg, MD, USA), 0.1 mM β-mercaptoethanol, 1% penicillin–streptomycin (Sigma, St. Louis, MO, USA), and 4 ng/mL of recombinant human basic fibroblast growth factor (bFGF).
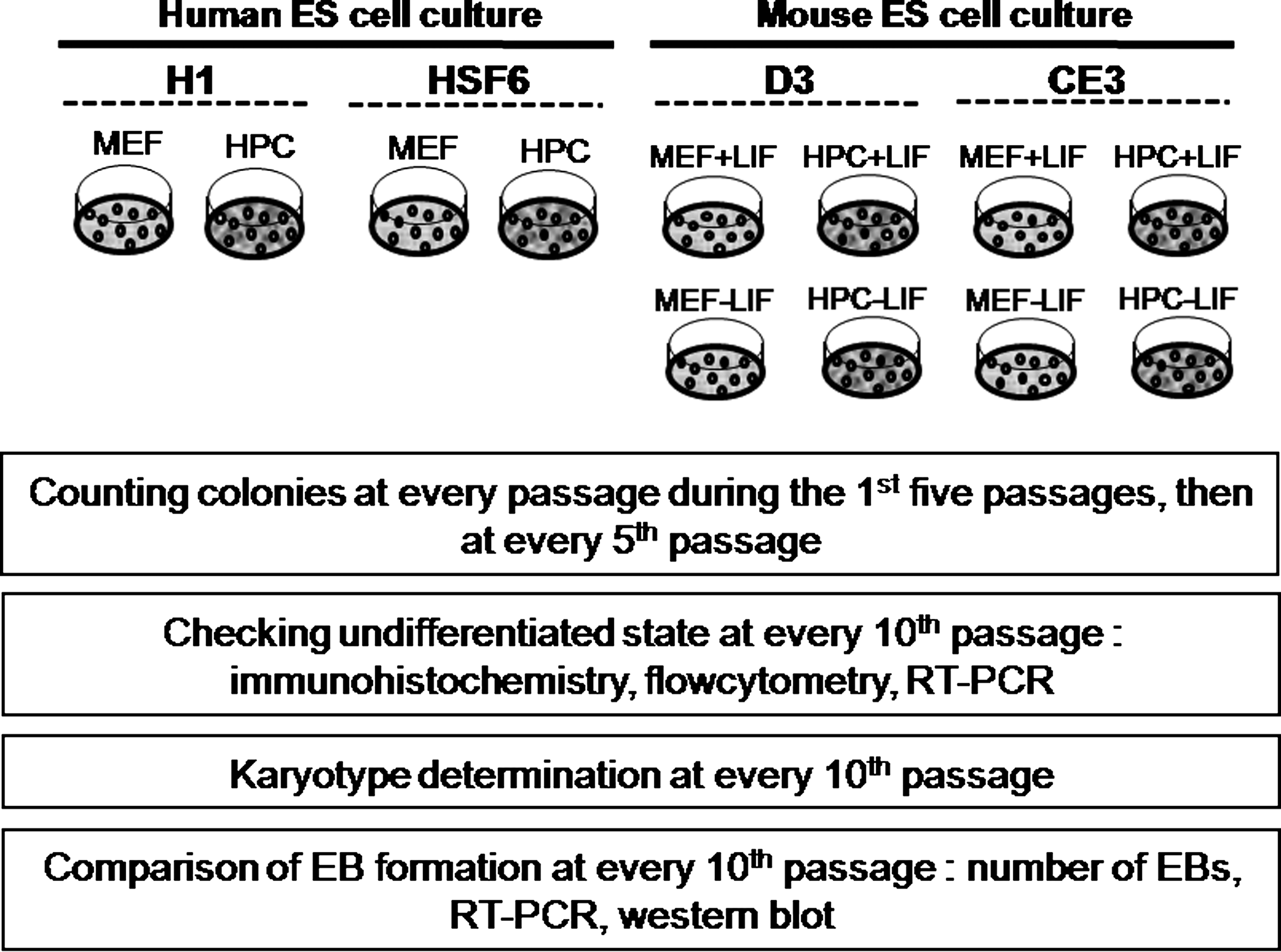
The study scheme. MEF, mouse embryonic fibroblast, HPC, Human placenta-derived fibroblast-like cell, LIF, Leukemia Inhibitory Factor, +LIF, with LIF; −LIF, without LIF.
D3 and CE3 mESC lines were purchased from ATCC (Manassas, VA, USA), and initially cultured on MEF according to the instructions of the provider. After maintenance on MEF cells for five passages, colonies of mESC were transferred onto a newly prepared HPC and MEF feeder. According to the guideline of the providers, coculture of D3 mESC and feeder cells was performed in DMEM supplemented with 20% FBS (HyClone Laboratories), 0.1 mM β-mercaptoethanol, and 1% penicillin–streptomycin (Sigma). Culture media for CE3 cell lines was the same as that of D3 cell lines, with the exception of ES-DMEM (ATCC) instead of DMEM. The two mESC lines were propagated on each feeder with or without LIF (Fig. 2).
Both hESCs and mESCs were passaged weekly by mechanical dissociation, and then cultured at 37°C, 5% CO2, and 95% humidity, and seeded at each passage at a density of 4 × 104 per plate, respectively. During culture, we counted ESC colonies at every passage during the first five passages, then at every fifth passage. We counted each colony comprising 120–300 ESCs using a grid and hemocytometer.
Production of iPSCs
iPSCs were produced according to the previously published protocol (Park et al., 2008). Our process was only different from this protocol in that we used the primary cultured HPCs as the source of the iPSCs and feeder instead of human dermal fibroblasts and MEFs. Briefly, OCT4, SOX2, KLF4, and c-MYC were introduced via the pMIG vector. SV40 large T was in pBABE-puro and hTERT was in pBABE-hygro (Addgene, Cambridge, MA, USA). These retroviral vectors, VSV-G, and Gag-Pol were transfected to 293T cells. After 48 h, retroviral transfection was identified by RT-PCR. Retrovirus-containing medium was collected and centrifuged at 70,000 × g at 4°C for 90 min and viral pallets were dissolved with DMEM. Retrovirus was infected to the primary cultured HPCs and incubated for 48 h. Culture of infected HPCs was maintained over 2∼3 weeks with the method-like hESCs culture. After newly formed colonies developed, these colonies were transferred onto the newly prepared feeder. The study design of iPS was similar to that of ESCs.
Characterization of hESCs, mESCs and iPSCs
To identify the undifferentiated state of hESCs and mESCs, morphology, expression of stemness marker, and differentiation capacity of both ESCs were examined. Cell morphology was observed each day under an inverted microscope, and expression of stemness markers were tested by immunostaining, flow cytometry, and RT-PCR. For hESCs, stemness was identified by immunostaining for alkaline phosphatase (ALP), flow cytometry for stage-specific embryonic antigen (SSEA)-1, SSEA-4, tumor rejection antigen (TRA)-60, TRA-81, and RT-PCR for Oct-4, Nanog, and Rex-1. For mESCs, stemness was identified by immunostaining for ALP, Oct-4, Nanog, SSEA-1, SSEA-4, and RT-PCR for Oct-4. Characterization of both cultured hESCs and mESCs were performed at every 10th passage (Fig. 2). Cocultures used for immunocytochemistry were established in six-well plates. Prior to analysis, adherent cell layers were fixed by addition of 10% formalin (15 min) at room temperature, permeabilized with 0.1% Triton X-100/phosphate-buffered saline (PBS) for 10 min, and incubated with primary antibodies overnight at 4°C. Primary antibodies for SSEA-1 and SSEA-4 were purchased from Hybridoma Bank, and other antibodies were purchased from Chemicon. ALP activity was detected using a commercial kit (Sigma). RT-PCR for stemness markers was also performed. Total RNA was prepared with the Qiagen RNeasy kit (Qiagen, Hilden, Germany), and reverse transcription was performed with 500 ng of total RNA primed with random hexamers using AMVreverse transcriptase (Roche Molecular Biochemicals, Mannheim, Germany). Primers for markers used in RT-PCR are described in Table 1. After PCR was performed, products were analyzed on a 1.5% agarose gel, and shown with ethium bromide. For iPSCs, stemness was identified by Nanog with immunostaining and Oct-4 and Rex-1 with RT-PCR because the colony size and amount was smaller and fewer than ESCs.
Karyotype analysis
To evaluate chromosomal stability, we performed karyotype analysis of cultured hESCs and mESCs at every 10th passage (Fig. 2). ESCs were incubated with 0.1 μg/mL colcemid for 3–4 h, trypsinized, and then incubated in 0.075 M KCl for 20 min at 37°C. After fixation with 3:1 methanol/acetic acid, the karyotypes of ESCs were analyzed at the 550-band level of resolution.
Formation of embryoid bodies (EBs) and study to confirm three lineage differentiations
EB formation was induced from cultured ESCs at every 10th passage to determine the differentiation potential of during long-term in vitro maintenance (Fig. 2). ESC colonies were harvested at each designated point in time to induce EB formation, and then transferred to each EB culture medium for human and mouse. For detection of the presence of three germ layers within the formed EB, we performed RT-PCR and Western blot analysis of EBs on day 21. Primers for this analysis are described in Table 1.
For Western blot analysis, EBs from both hESCs and mESCs were harvested, washed with PBS, and lysed in 100 μL of sodium dodecyl sulfate (SDS) sample buffer (12 mM Tris-HCl, pH 6.8, 0.4% SDS, 5% glycerol, 0.02% bromophenol blue, 0.288 mM 2-mercaptoethanol). Samples were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and electrotransferred onto polyvinylidene fluoride (PVDF) membranes (Millipore Corp). The membranes were soaked for 2 h at room temperature in blocking buffer (PBS, 0.5% Tween-20 containing 5% nonfat dry milk), and then incubated overnight at 4°C with appropriate primary antibody. Proteins were detected by horseradish peroxidase (HRP)-conjugated secondary antibody and an enhanced chemiluminescence (ECL) reagent (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Antibodies to Sox-1, brachyury, and AFP of both human and mouse were purchased from Santa Cruz Inc. (Santa Cruz, CA, USA).
In the iPSCs study, small and scanty EB formation was observed only in the HPC group, not in the MEF group, so only RT-PCR was possible and Western blot analysis cold not be done.
Statistical analysis
In each group, hESCs (H1/MEF, HSF6/MEF, H1/HPC, HSF6/HPC) and mESCs (D3/MEF/LIF+, D3/MEF/LIF−, D3/HPC/LIF+, D3/HPC/LIF−, and CE3/MEF/LIF+, CE3/MEF/LIF−, CE3/HPC/LIF+, CE3/HPC/LIF−) had five culture dishes; values for each group were the mean value of the five dishes. In each feeder group, the whole study was repeated five times, and the results were verified by two other specialists. All quantitative data were presented as mean ± SEM, and statistical significance was determined using a one-way analysis of variance (ANOVA) test. Results were considered significant when the p value was less than 0.08. The study scheme of ESCs is shown in Figure 2. The study of iPSCs was repeated only for two times as a pilot trial to focus on the formation of colonies having stem cell markers, so statistical analysis was not done.
Results
Comparison of colony counts
In hESCs, the mean numbers of colonies for the H1/MEF, H1/HPC, HSF6/MEF, and HSF6/HPC groups at the fifth passage were 154 ± 33, 151 ± 23, 144 ± 35, and 141 ± 37, respectively. There were no significant differences between H1 groups (H1/MEF and H1/HPC) and HSF6 groups (HSF6/MEF and HSF6/HPC). When we assessed colony counts serially at every fifth passage, we were able to propagate the culture of hESCs (H1 and HSF6) on both the HPC and MEF feeder up to the 50th passage (Fig 3A). Analysis of D3 mESCs was performed in the same manner as for hESCs. The mean number of colonies for the D3/MEF/LIF+, D3/MEF/LIF−, D3/HPC/LIF+, and D3/HPC/LIF− groups at the fifth passage was 210 ± 15, 205 ± 21, 223 ± 23, and 221 ± 15, respectively. There was no significant difference among all D3 groups.
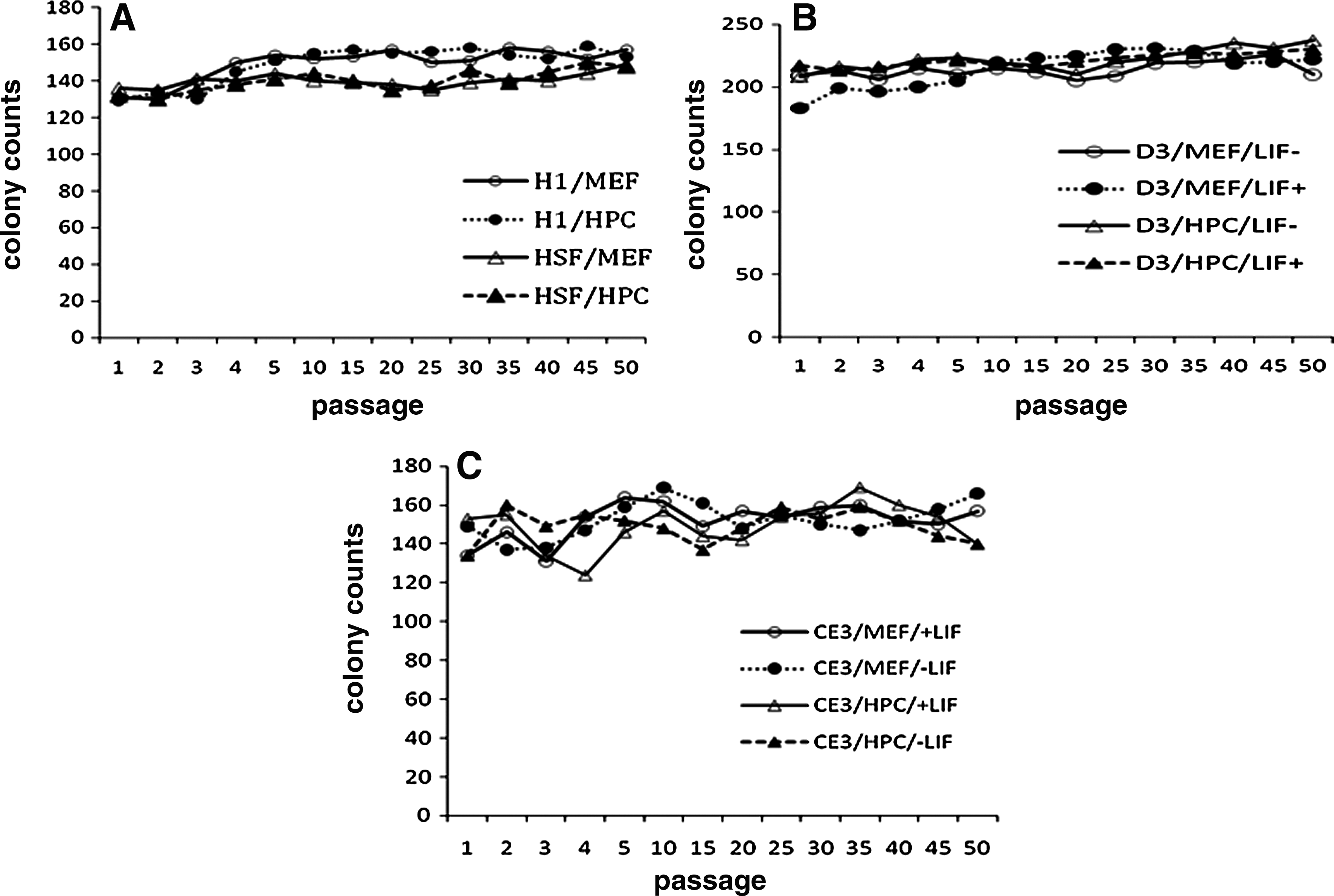
The resuts of colony counts of hESC and mESC over the passage. (
Propagation of the D3 mESC culture under four different conditions was also possible up to the 50th passage (Fig. 3B). Results from the CE3 mESC lines showed the same trend as those of the D3 mESC lines. There was no significant difference among all CE3 groups, and propagation of the CE3 mESC culture under four different conditions was also possible up to the 50th passage (Fig. 3C). The number of colonies at each measurement was similar to the results of the fifth passage in D3 and CE3 groups, respectively.
All HPCs cultured up to the 12th passage were able to support undifferentiated growth of both the hESC and mESC lines. It was the same for all MEFs cultured up to the third passage.
In the iPSC study, the morphology of colonies was relatively small compared with that of hESCs and mESCs (Fig. 7A) and the mean numbers of colonies for the MEF and HPC at the fifth passage were 18 ± 9 and 25 ± 11, respectively. The current passage number of colonies having stem cell markers was about 20 passages on the HPC feeder and 10 passages on the MEF feeder.
Undifferentiated state and chromosomal stability
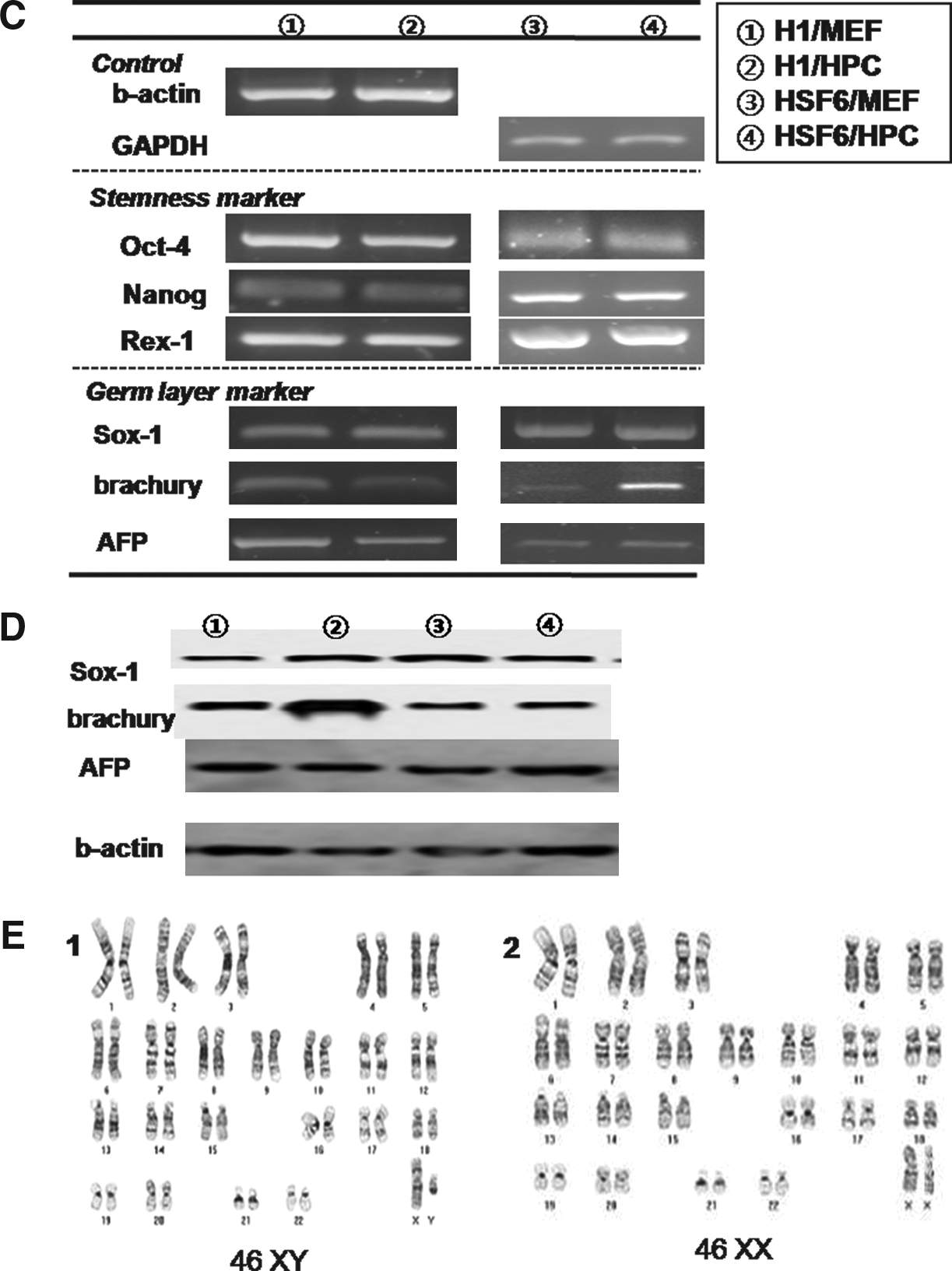
We were able to propagate cultures of two types of hESCs, for example, H1 and HSF6, up to the 50th passage on both the MEF and HPC feeders. Colonies of H1 and HSF6 hESCs were well maintained in long-time in vitro coculture with each type of feeder cell. Morphologically, there was no difference between the MEF and HPC groups in H1 and HSF6 hESC colonies (Fig. 4, A1–4). ALP staining was strongly positive in H1 and HSF colonies of both the MEF and HPC groups (Fig. 4, A5–8). Flowcytometric analysis for stemness markers showed that stemness of H1 and HSF6 on the HPC feeder was well conserved over the 50 passages. H1 hESCs of both the MEF and HPC groups showed negative expression of SSEA-1 and positive expression of SSEA-4 at every measurement (Fig. 4, B1–4 for the MEF group, B9–12 for the HPC group). TRA-60 and TRA-81, which are also stemness markers, were positive in H1 hESCs of both the MEF and HPC groups at every measurement (Fig. 4, B5-8 for the MEF group, B13–16 for the HPC group). The result from HSF6 hESCs in both the MEF and HPC groups was the same as that of H1 hESCs (data not shown). Stemness was also evaluated at every 10th passage by RT-PCR for Oct-4, Nanog, and Rex-1, which are known to be expressed in pluripotent cell populations. Expression of these markers was identified in H1 hESCs of both the MEF and HPC groups (Fig. 4C, ①, ②). In HSF6 hESCs of both the MEF and HPC groups, these markers were also identified by RT-PCR (Fig. 4C, ③, ④). Determination of hESC karyotype at every 10th passage found no karyotypic abnormalities in H1 and HSF-6 hESCs of both the MEF and HPC groups (Fig. 4, D1–2).
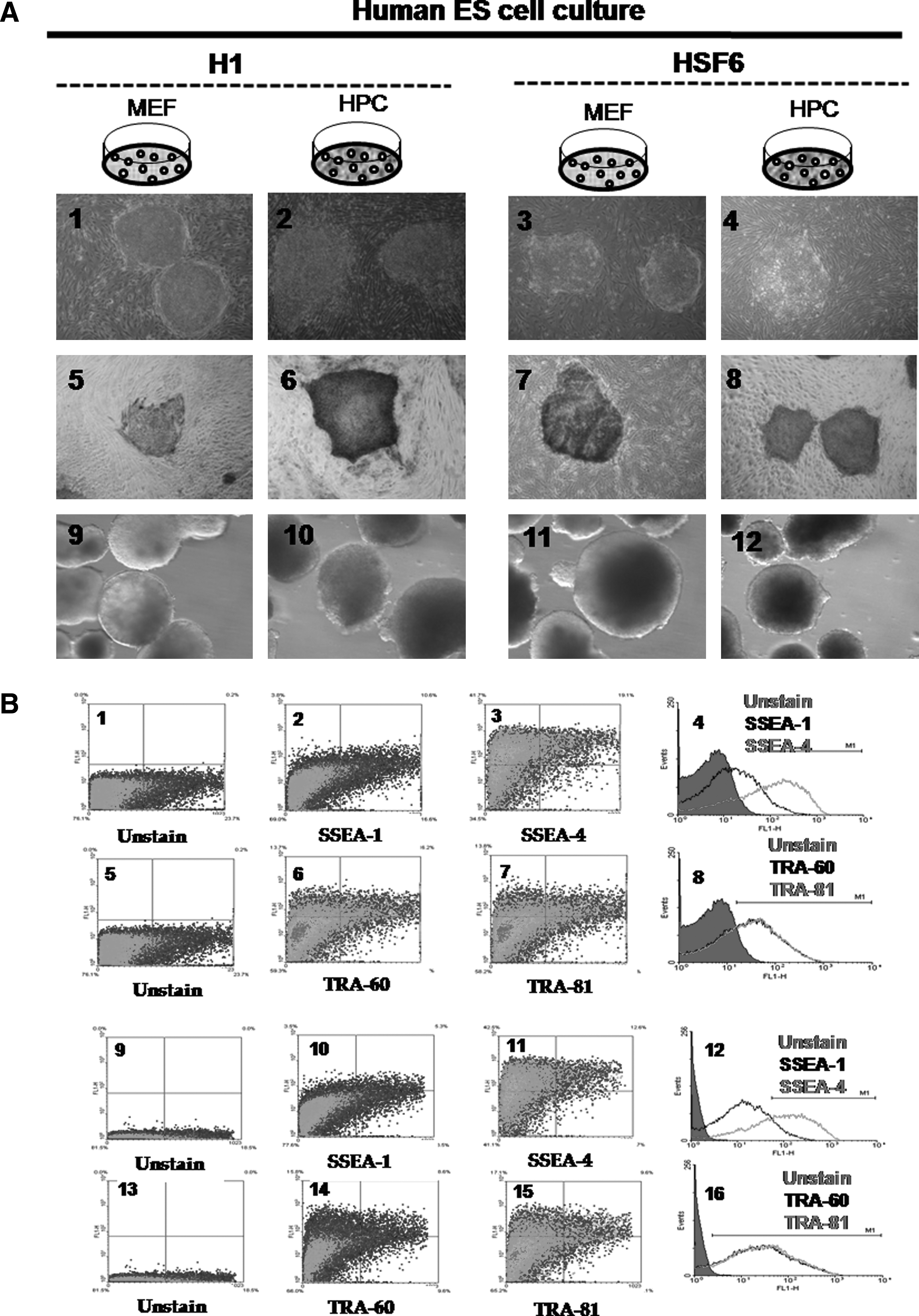
The results of hESC culture on MEF and HPC feeder at 50th passage. (
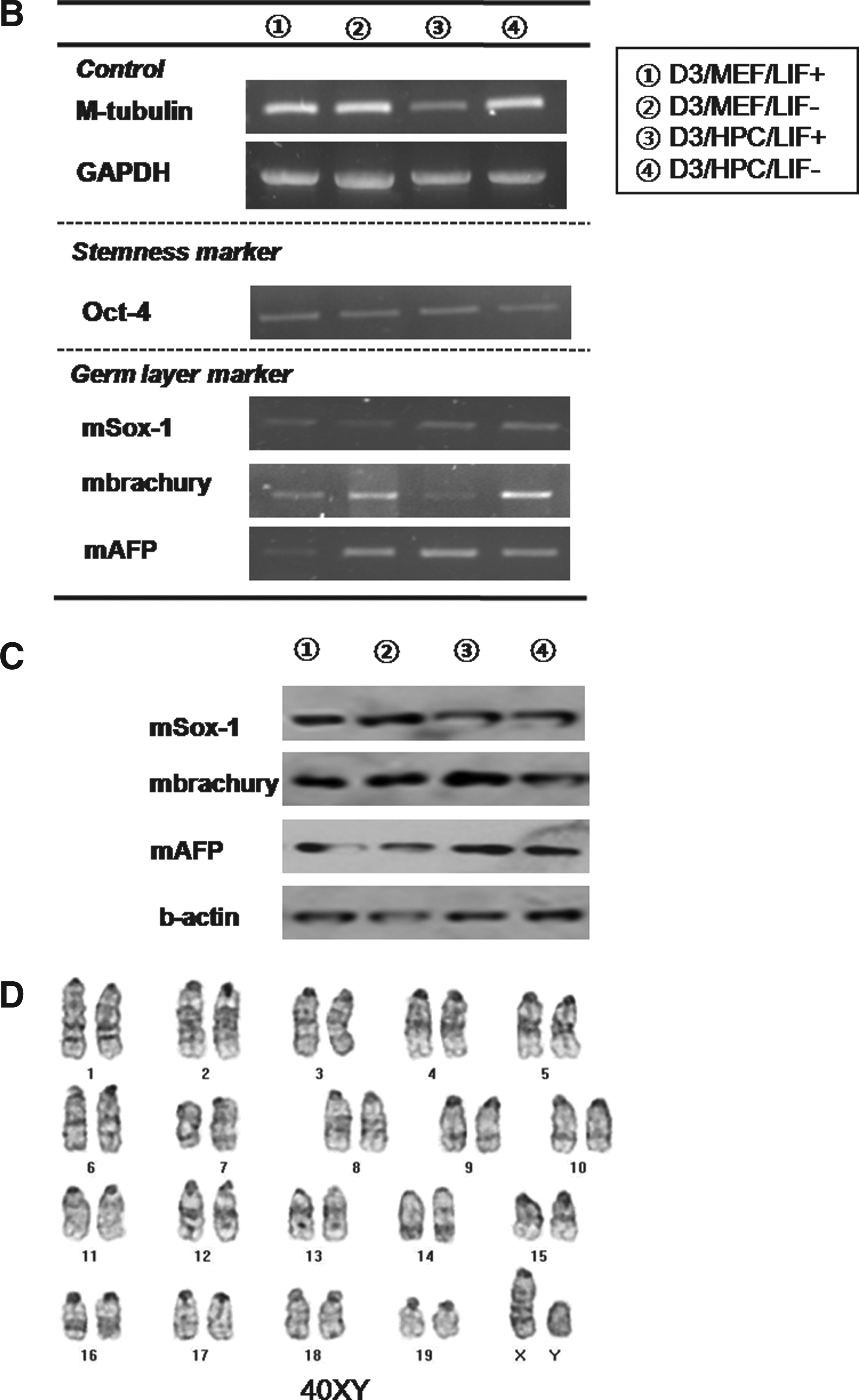
We also were also able to propagate cultures of D3 mESCs after the 50th passage under four different conditions, which included the MEF feeder with/without LIF and the HPC feeder with/without LIF. Colonies of mESCs were well maintained in long-time in vitro coculture with each feeder cell, regardless of LIF. Morphologically, there was no difference in mESC colonies among four conditions (Fig. 5, A1–4). ALP staining was strongly positive in all groups (Fig. 5, A5–8). Immunocytochemical staining for stemness markers showed well-conserved stemness of D3 mESCs in all groups over 50 passages, regardless of feeder type or LIF. Oct-4 (Fig. 5, A9–16), Nanog (Fig. 5. A17–24), and SSEA-1 (Fig. 5, A25–32) were positively expressed, and SSEA-4 was not expressed in D3 mESCs from all groups. Stemness was also evaluated at every 10th passage by RT-PCR for Oct-4. Expression of Oct-4 was identified in D3 mESCs from all groups, that is, regardless of feeder type or LIF, and at every measurement (Fig. 5B). Determination of mESC karyotype at every 10th passage found no karyotypic abnormalities in mESCs from all groups (Fig. 5C, for the D3/HPC/LIF− group). Results from CE3 mESCs were the same as those of D3 mES cells. Stemness was also well conserved in CE3 mESCs, regardless of feeder type or LIF (Fig. 6).
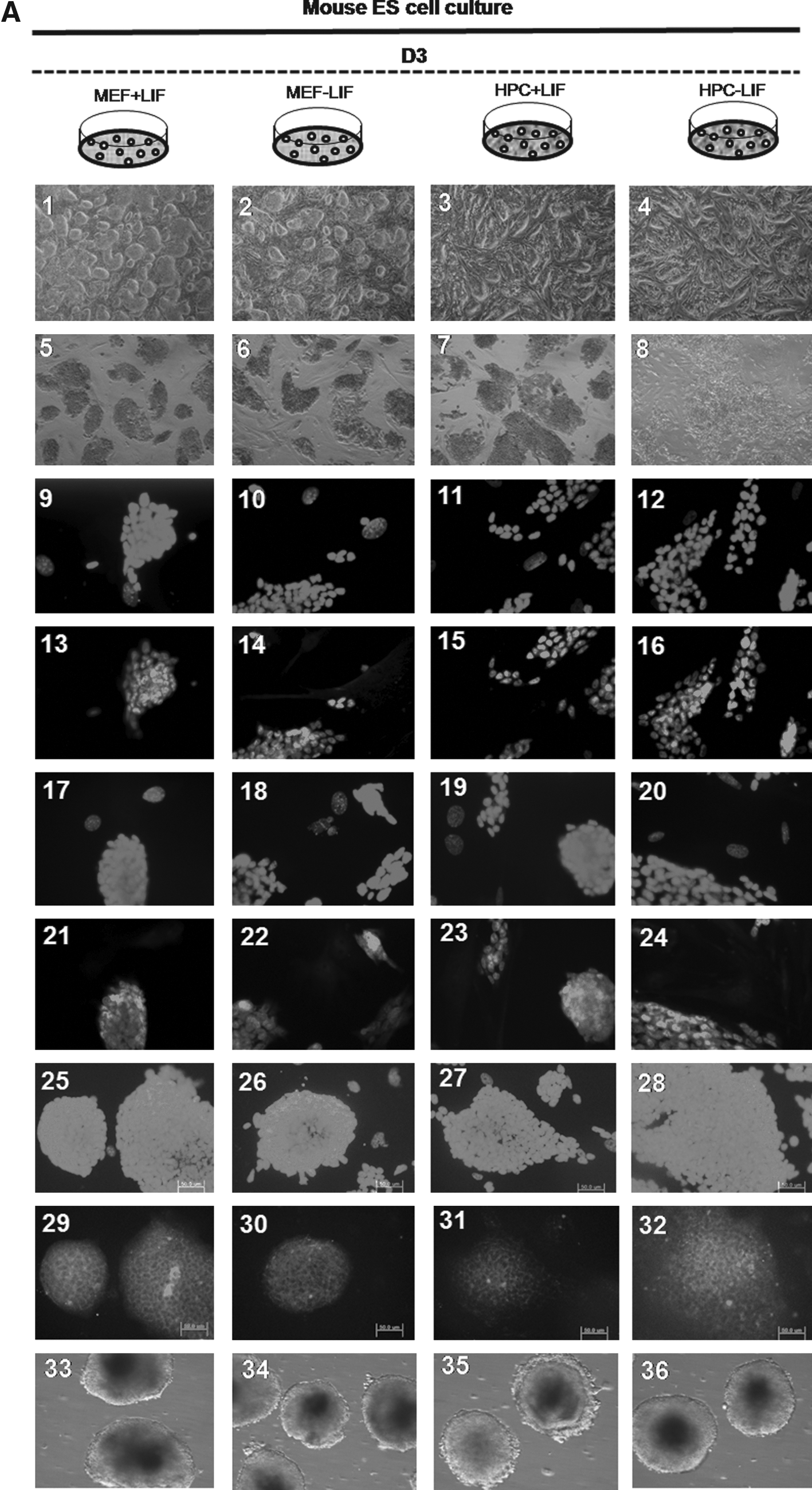
The result of D3 mES cell culture of each group at the 50th passage. (
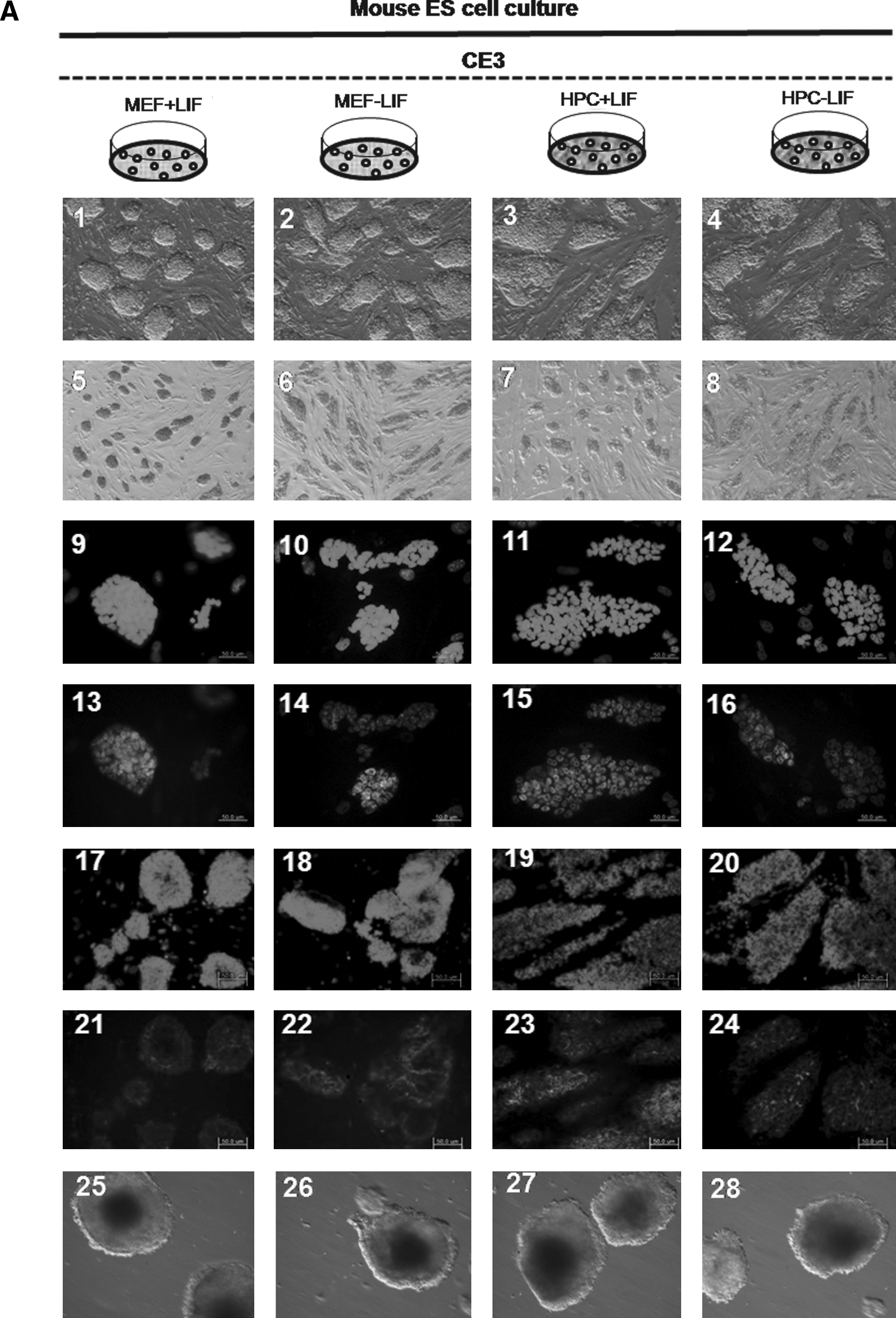
The result of CE3 mESC culture of each group at the 50th passage. (
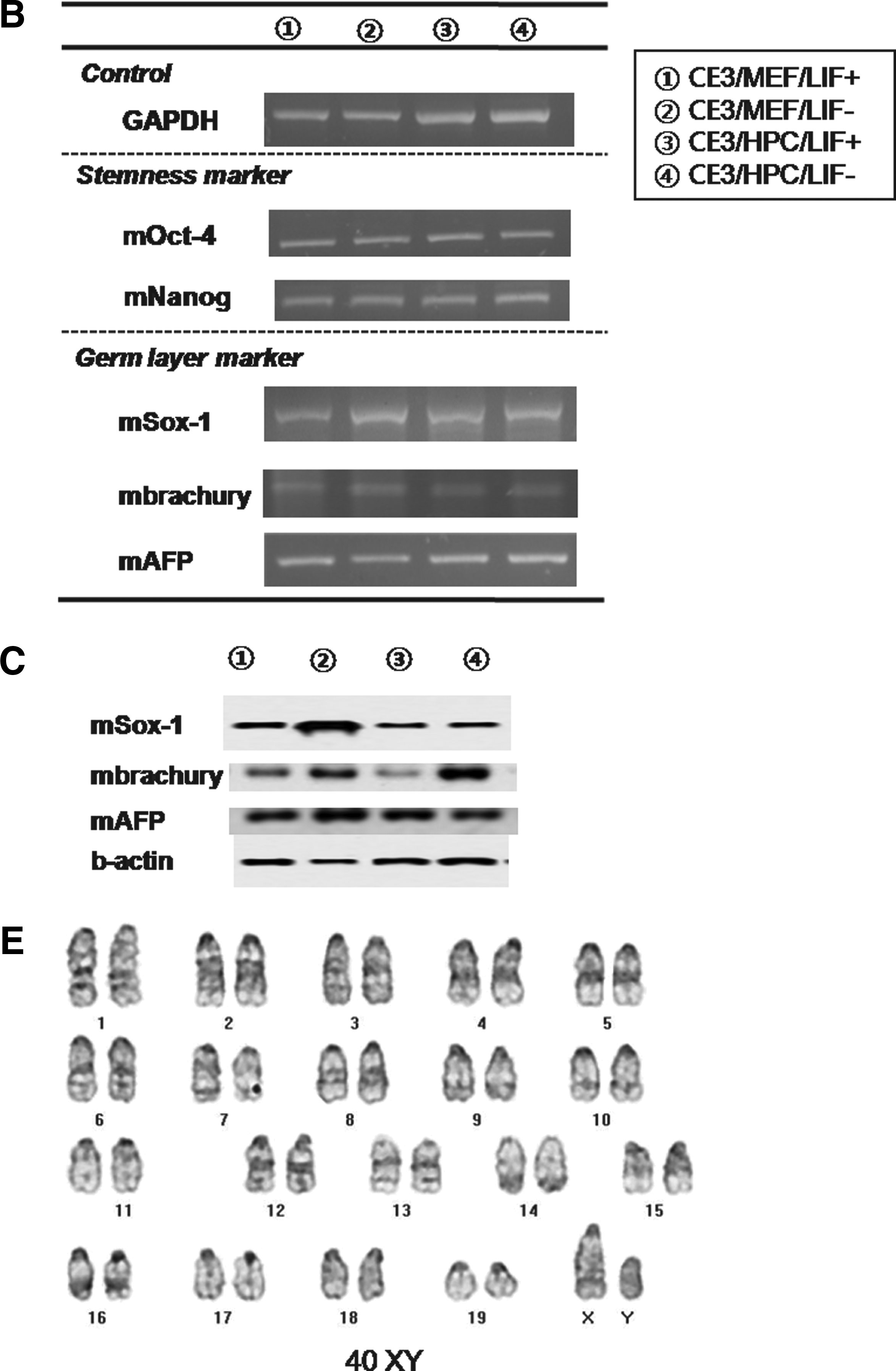
For iPSCs, stemness was identifiable by Nanog with immunostaining and Oct-4 and Rex-1 with RT-PCR, and karyotype at the 10th passage was normal (Fig. 7B–D).
EB formation and identification of lineage-specific markers
Efficiency of EB formation was calculated by the percentage of formed EBs per seeded ESC colony. In hESCs, the mean percentage of formed EBs in the H1/MEF, H1/HPC, HSF6/MEF, and HSF6/HPC groups at the fifth passage was 77.6 ± 1.6%, 78.7 ± 3.7%, 60.3 ± 5.5%, and 62.9 ± 2.8%, respectively. There were no significant differences between H1 groups (H1/MEF and H1/HPC) and between HSF groups (HSF/MEF and HSF/HPC). Evaluation of the efficiency of EB formation at every 10 passages found no significant differences between H1 groups and HSF groups. Presence of three germ layers within formed EBs was identified by RT-PCR and Western blot analysis. Expression of Sox-1, Brachyury, and AFP was demonstrated within formed EBs from all groups, indicating the presence of ectoderm, mesoderm, and endoderm (Fig. 4C and D).
In D3 mESCs, the mean percentage of formed EBs at the fifth passage in D3/MEF/LIF+, D3/MEF/LIF−, D3/HPC/LIF+, and D3/HPC/LIF− at the fifth passage was 79.1 ± 2.7%, 80.5 ± 1.5%, 77.3 ± 3.2%, and 78.7 ± 0.7 %, respectively. There was no significant difference among all mESC groups. This trend was maintained at each measurement, at every 10 passages. The presence of three germ layers within the formed EBs was identified by RT-PCR and Western blot analysis. Expression of ectodermal marker (Sox-1), mesodermal marker (Brachyury), and endodermal marker (AFP) was identified within the formed EBs from all groups, regardless of feeder type or LIF (Fig. 5B and C). These trends were also shown in CE3 mESCs. The mean percentage of formed EBs at the fifth passage in CE3/MEF/LIF+, CE3/MEF/LIF−, CE3/HPC/LIF+, and CE3/HPC/LIF− was 82.1 ± 1.9%, 81.6 ± 2.2%, 80.8 ± 3.1%, and 81.4 ± 2.4%, respectively. All lineage-specific markers were also identified, regardless of feeder type or LIF (Fig. 6B and C).
For the iPSCs study, EBs formed from iPSCs was scantly, and differentiation to three germ layers was identified by RT-PCR only in the HPC group (Fig. 7D).
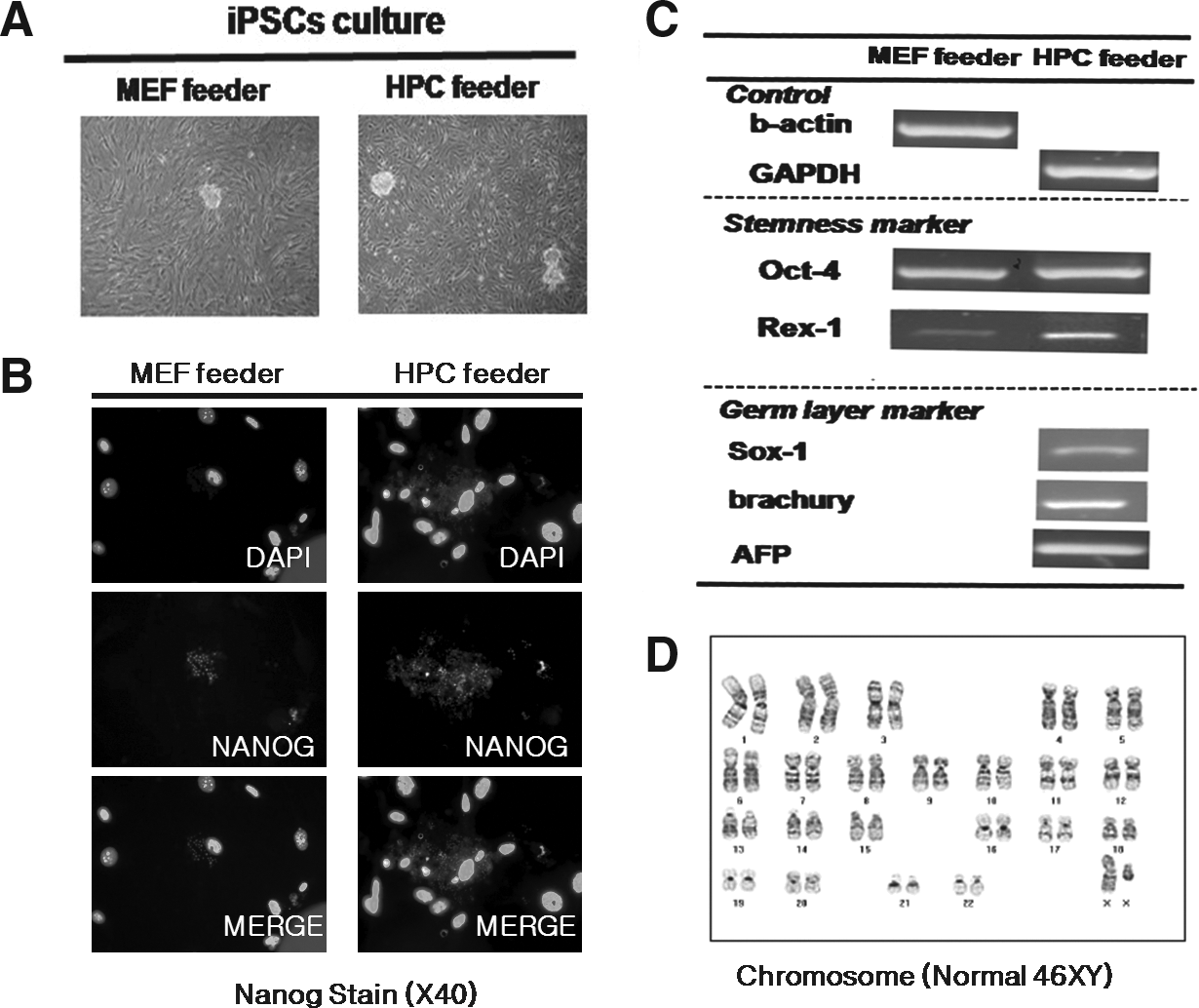
The result of iPSCs culture at the 10th passage on the MEF feeder and the 20th passage on the HPC feeder. (
Discussion
Genbacev et al. (2005) produced human ESC lines using an HPC feeder; this was the first report showing that HPC could support undifferentiated proliferation of hESCs. However, this study did not prove the validity of HPC for proliferation of hESCs previously maintained on an MEF feeder. Therefore, we first attempted to propagate the SNUhES3 hESC line on an HPC feeder that had been previously maintained on an MEF feeder, and reported successful undifferentiated proliferation of SNUhES3 hESCs (Kim et al., 2007). On the basis of our previous report, we designed this study to determine whether or not HPC could be a universal feeder, like MEF, for undifferentiated propagation of a mammalian ESC. Therefore, this study was applied to the widely used hESC lines, H1 and HSF6, as well as to the widely used mESC lines, D3 and CE3, which were previously propagated on an MEF feeder. Recently it was reported that a feeder derived from human amnion epithelial cells was useful in culturing of mESCs (Lai et al., 2009). The HPC feeder in this study was derived from the chorionic villi of human placenta. To the best of our knowledge, this study appears to be the first report demonstrating the feasibility of an HPC feeder to support undifferentiated proliferation of mESCs previously maintained on an MEF feeder and the possibility to establish iPSCs from placenta. Because HPC was previously demonstrated to support undifferentiated propagation of monkey ESCs (Miyamoto er al., 2004), HPC might have a role as a feeder for all species of established mammalian ESC lines: mice, monkeys, and humans. Therefore, we determined that HPC can be a suitable alternative to MEF as a universal feeder in mammalian ESC culture.
The rationale for choosing HPC as a feeder was that HPC feeders might provide a natural environment for ESCs similar to that of early in vivo development, which enhances growth in an undifferentiated state (Kim et al., 2007). Our study results support this theory. Although it has been suggested that placenta-derived cells might secrete some proteins in culture medium, or that they may maintain some membrane proteins for undifferentiated growth of ESCs (Miyamoto et al., 2004), further studies are still required. Presence of MSC in HPC helps to explain the underlying mechanism. Fukuchi et al. (2004) first demonstrated that human placenta-derived fibroblast-like cells were composed mainly of MSC. We also identified MSC in HPC cells by flow cytometric analysis for surface markers. Considering that bone marrow (BM) MSC provides hematopoietic stem cells with a niche for maintaining the multipotency of the cell population, placenta MSC may also provide ESCs with a niche for maintaining pluripotency of cells. We previously tried to utilize hESCs on a BM-derived MSC feeder; however, we failed to propagate undifferentiated hESCs beyond the ninth passage. Therefore, we concluded that BM-derived MSC was not appropriate for long-standing culture of undifferentiated hESCs (Kim et al., 2007). Thus, differences between placenta-derived MSC and BM-derived MSC may be helpful in determination of the mechanism of ESC support of HPC.
In addition, compared with MEF, HPC provides additional benefits as well. For hESC cultures, contamination of xenoproduct can be reduced, and these concerns can be minimized using an HPC feeder. Moreover, unlike MEF, which requires additional sacrifice of mouse embryos, and can be used as feeders only during early passages (p4–6) (Kim et al., 2002), HPC feeders can be easily obtained after delivery or therapeutic abortion with no any additional risk to donors, and can be propagated up to the 35th passage, and cells up to the 12th passage are known to be used as feeders (Ilic et al., 2008); we were also able to confirm these features. This finding can suggest the relative advantage of HPC over MEF, which can reduce overall cost of ESC culture.
This study has implications for some aspects of mESC culture. We first demonstrated successful propagation of undifferentiated mESCs on an HPC feeder without LIF. It has recently been demonstrated that addition of exogenous LIF was not required in the mESC culture when using a human-originated cell feeder, such as human foreskin fibroblasts and human endothelial cells (Meng et al., 2008; Zhou et al., 2008). Considering that HPC originate from mesoderm, it may be possible that human cells originating from mesoderm might secrete LIF or LIF equivalent, which inhibits mESC differentiation. However, this hypothesis will need further investigation in future studies. Considering the cost of LIF, the culture system using an HPC feeder can save a considerable amount of maintenance cost during mESC culture. Second, there were no differences in outcome of mESC culture on MEF feeders, regardless of LIF (D3/MEF/LIF− vs. D3/MEF/LIF+, CE3/MEF/LIF− vs. CE3/MEF/LIF+) in this study. Although it was known that no exogenous LIF was required in the mESC culture system using an MEF feeder, many investigators have usually added LIF to the medium using an MEF feeder to avoid accidental differentiation. This has been considered due to an absence of sufficient data on quantification of LIF secreted by MEF and maintenance of undifferentiated mESC growth without LIF on MEF feeder. A recent study demonstrated that the amount of LIF secretion by MEF was enough to maintain mESCs in an undifferentiated state (Lee et al., 2009), and we also confirmed that LIF was not required during culture of mESCs using an MEF feeder in this study. Therefore, addition of LIF to the culture medium for maintenance of mESCs using an MEF feeder can be regarded as an unnecessary extravagance. Finally, by using an HPC feeder, rather than MEF, in culturing of mESCs, concerns over a full xenoenvironment might be partially allayed. Thus, it can be expected that discrepancy between outcomes of research on mESCs and hESCs will be reduced with use of a human-originated feeder, such as HPC.
For the aspect of iPSCs, a recent report for the usefulness of autologous feeders for the undifferentiated propagation of human iPSCs appeared (Takahashi et al., 2009). We also had an idea that iPSCs from placental MSCs would be easily established and well propagated on HPC feeders, and found the usefulness of HPC to support the colonies having stemness markers. The current passage number in our iPSCS study was about 20, and this is similar with the report of Takahashi et al. Our study showed that colony counts, passage numbers, and EB formation efficacy of iPSCs on HPC feeders could be more favorable than those on MEF feeders. This finding might be compatible with the report (Chen et al., 2009), suggesting that MEF feeder cells did not accelerate reprogramming or increase the frequency of iPSC colonies. So, this study has a meaning to establish and propagate iPSCs from placental MSCs. But this was a pilot trial. To suspect the fact that many parts of incurable diseases are genetic or inherited disease, the establishment of iPSCs from fetal part of the placenta would be important to investigate the tools to overcome incurable genetic or inherited diseases. Therefore, we will do further study, as soon as possible.
This study validate efficacy of an HPC feeder during hESCs culture previously propagated on the MEF feeder and is the first to demonstrate usefulness of the HPC feeder for mESCs culture previously propagated on the MEF feeder and iPSCs establishment and propagation from placenta. There are some limitations to this study. First, because animal products, such as FBS, were used for culture, this culture system is not completely animal free for undifferentiated proliferation of hESCs. Although some previous studies have reported on the safety of FBS (Koc et al., 2000; Lazarus et al., 1995), where in vitro cultured MSCs with FBS were infused into patients, safety concerns associated with contact with animal products might be of concern. Second, we used only placenta from early gestation as the source of the HPC feeder, as described in earlier studies (Genbacev et al., 2005; Simon et al., 2005). For a steady supply of placental tissue, full-term placenta seems to be more practical. But the viability of placenta of early gestation is better than full-term placenta. So, banking system of early gestational placenta collected from inevitable abortion after informed consent acquisition must be considered. Placentas from mothers with healthy babies who should have therapeutic abortion due to known maternal disease that is aggravated by pregnancy might be used for the production of HPC feeders or normal iPSCs, and those from fetal diseases might be applied to establish disease-specific iPSC lines. Of course, the study with an HPC feeder originating from a full-term placenta is also required.
In conclusion, the HPC feeder can efficiently support the undifferentiated propagation of hESCs, mESCs, and iPSCs, suggesting that human placenta may be a useful source of universal feeder cells for hESCs, mESCs, and iPSCs culture.
Footnotes
Acknowledgments
This research was supported by a grant (SC-2240) from the Stem Cell Research Center of the 21st Century Frontier Research Program, funded by the Ministry of Science and Technology, Republic of Korea.
Author Disclosure Statement
All authors declared no conflict of interest in this study.