
Abstract
Abstract
Trauma or degenerative diseases of the central nervous system (CNS) cause the loss of neurons or glial cells. Stem cell transplantation has become a vital strategy for CNS regeneration. It is necessary to effectively induce nonneurogenic stem cells to differentiate into neurogenic cell lineages because of the limited source of neurogenic stem cells, relatively difficult cultivation, and ethical issues. Previous studies have found that dental stem cells can be used for transplantation therapy. The aim of this study was to explore a better inductive mode and time point for dental stem cells to differentiate into neural-like cells and evaluate a better candidate cell. In this study, dental follicle stem cells (DFSCs), dental papilla stem cells (DPSCs), and stem cells from apical papilla (SCAPs) were cultivated in five different modes. The proliferation ability, morphology, and expression of neural marker genes were analyzed. Results showed that DFSCs showed a higher proliferation potential. The proliferation was decreased after cultivation in chemical inductive medium as cultivation modes 3 and 5. The cells could present neural-like cell morphology after cultivation with human epidermal growth factor (EGF) and fibroblast growth factor-basic (bFGF) as cultivation modes 4 and 5. The vast majority of DFSCs gene expression levels in mode 4 on the third day was upregulated significantly. In conclusion, our data suggested that different dental stem cells exhibited different neural differentiation potentials. DFSCs might be the better candidate cell type. Furthermore, cultivation mode 4 and timing of the third day may promote differentiation into neurogenic cell lineages more effectively before transplantation to treat neurological diseases.
Introduction
T
Stem cells are self-renewing cells with multidifferentiation potential. Several types of stem cells have been used for the research of treatments for neurological diseases, including embryonic stem cells (ESCs), neural stem cells, Schwann cells, olfactory ensheathing cells, bone marrow mesenchymal stem cells (MSCs), and so forth (Carletti et al., 2011; Chiu et al., 2009; Sadan et al., 2009; Tomaskovic-Crook and Crook, 2011). However, most of these cells have limited applications in clinical treatment because of ethical issues, limited resource, and high cultivation requirements in vitro (Nunes et al., 2003; Palmer et al., 2001; Schwartz et al., 2003). Dental stem cells originate from the neural crest, and the dental cell organizations often contain cells that are still in an undifferentiated state during the process of tooth development. For instance, dental follicle stem cells (DFSCs) and dental papilla stem cells (DPSCs) exist in the developing tooth germ prior to eruption (Morsczeck et al., 2005); the stem cells from apical papilla (SCAPs) are contained in root apical papilla tissue on the exterior of the root foramen area (Sonoyama et al., 2006). All of these cells are easily collected from teeth removed surgically for clinical reasons and the cells cultivated in vitro. Cellular therapies have the objective of opening up new methods and mechanisms to cure neurological diseases via differentiation and replacement of the pathological cells (Honmou et al., 2012; Kim et al., 2012; Sakai et al., 2012; Ziavra et al., 2012). However, the differentiation mechanisms of stem cells are not clear, and the differentiation of these cells is difficult to control in vivo. It is interesting to determine how to make stem cells differentiate into neurogenic cell lineages more effectively after transplantation.
Previous studies have suggested that MSCs have the potential to be induced to neural differentiation using conditional medium in vitro, such as chemical inductive medium (CIM), cytokines, and co-culture with nerve cells (Hermann et al., 2004; Wislet-Gendebien et al., 2003; Woodbury et al., 2000). But our study and previous reports have shown that cells cultivated with CIM for a long time cause cellular apoptosis. The ethical issues and limited resource also must be considered in determining the method of co-culture with nerve cells. Therefore, it is important in neural differentiation to compare the pattern of induction, for instance, CIM for a short time, cytokines, or CIM for a short time plus cytokines.
This study investigated the capacities of human dental stem cells that were isolated from the teeth germs and teeth to differentiate into different neurogenic cell lineages after cultivation using different modes in vitro. Ultimately, this work evaluated which type of dental stem cells is the better candidate for cellular therapy and which cultivation mode and time point are beneficial for the stem cells differentiating into neurogenic cell lineage before transplantation.
Materials and Methods
Participants
Three (2 female and 1 male) donors (age 18–22 years) participated in this study. All of the teeth or teeth germs used in this study were surgically removed for clinical reasons. This study was compatible with the Code of Ethical Principles for Medical Research Involving Human Subjects of the World Medical Association (Declaration of Helsinki) and was approved by the Institutional Review Board (IRB) of Sichuan University. All participants provided written informed consent.
Isolation of dental stem cells from individual human teeth germs and teeth samples
Normal human impacted third molars were surgically removed and collected at the West China Hospital of Stomatology. Teeth germs and teeth surfaces were cleaned and then washed with phosphate-buffered solution (PBS). Attached dental follicles and dental papillar tissues were isolated from the mineralized tooth; root apical papilla tissues were gently separated from the surface of the root. Human DFSCs, DPSCs, and SCAPs were isolated and cultivated as described previously (Gronthos et al., 2000; Morsczeck et al., 2005; Sonoyama et al., 2006). Briefly, the dental stem cell isolated from the tissues were cultivated in α-Modified Eagle's Medium (α-MEM; Hyclone, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone, USA) in a humidified atmosphere at 37° and 5% CO2. The cultivation medium was replaced every 2 days.
Identification of dental stem cells
For cellular immunofluorescence, 3×103 DFSCs, DPSCs, and SCAPs were seeded in each well of 24-well plate overnight. The cells were fixed with 4% polyoxymethylene for 10 min and permeabilized with 0.5% Triton for 15 min at room temperature. After washing twice in PBS for 5 min each, the cells were blocked with 10% goat serum for 30 min at room temperature. The cells were incubated with primary antibodies for 2 h in a humidified chamber at 37°C. After washing twice in PBS for 5 min each, the cells were incubated with secondary antibodies for another 2 h in a humidified chamber at 37°C. Following three rinses in PBS for 5 min each, the nuclei were stained with 100 ng/mL of 4′,6-diamidino-2-phenylindole (DAPI) for 3 min under the conditions of protection from light. All samples were examined under a fluorescence microscope (Leica Optical, Germany). Primary andtibodies were anti-vimentin [monoclonal mouse immunoglobulin G (IgG), 1:500, Santa Cruz, CA, USA] and anti-CK14 (monoclonal mouse IgM, 1:200, Abcam, UK).
For flow cytometry, 3×106 of DFSCs, DPSCs, and SCAPs were incubated with fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-conjugated primary monoclonal antibodies against CD34 (BD Biosciences, USA), CD44 (BD Biosciences, USA), CD90 (BD Biosciences, USA), and CD105 (BD Biosciences, USA). Cells were incubated with antibodies in the dark at 4°C for 30 min and then washed twice with PBS containing 0.1% bovine serum albumin (BSA). Flow cytometry was performed with a Beckman Coulter Cytomics FC 500 MPL system (Beckman Coulter, USA). The data were analyzed by CXP Software (Beckman Coulter, USA).
Cultivation mode formulations for neural differentiation
Human dental stem cells at passage 4 were used for experiments. Four types of cultivation medium were selected to cultivate cells: (1) α-MEM with 10% FBS; (2) neural CIM; (3) neural medium (NM) consisting of Neurobasal-A Medium (Invitrogen, USA) with B27 (Invitrogen, USA) and GlutaMAX Supplement (Gibco, USA); (4) NM with recombinant human epidermal growth factor (EGF; Peprotech, USA) and recombinant human basic fibroblast growth factor (bFGF; Peprotech, USA). The cultivation mode formulations with four types of medium are listed in Table 1, and α-MEM with 10% FBS was the control mode. The components of CIM are α-MEM, 2% dimethyl sulfoxide (DMSO; Solarbio, China), 200 μM butylated hydroxyanisole (Sigma, USA), 25 mM KCl (Bodi, China), 2 mM valproic acid sodium salt (Sigma, USA), 10 mM forskolin (Sigma, USA), 1 mM hydroxycortisone (Aladdin, China), and 5 μg/mL insulin (Novo Nordisk, Denmark) (Li et al., 2011). The final concentration of EGF and bFGF in this study was 20 ng/mL. The cultivation medium was replaced every 2 days.
α-MEM, α-Modified Eagle's Medium; FBS, fetal bovine serum; NM, neural medium; CIM, chemical inductive medium; EGF, epidermal growth factor; bFGF, basic fibroblast growth factor.
Cell proliferation assay
For the evaluation of cell proliferation, 1×103 cells were cultivated on 96-well plates with Nunclon Delta surface modification (Nunc, Wiesbaden, Germany). A Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Japan) was used to evaluate the proliferation ability of the dental stem cells cultivated in five different modes for 7 consecutive days. Briefly, after the cells in different cultivation modes were grown for 8 h to adhere at the bottom of the plates, the cultivation media of mode 2 and mode 4 were changed to NM and NM with EGF and bFGF, respectively. Especially, the cultivation media of mode 3 and mode 5 were changed to CIM for 4 h initially on the first day; the cultivation media of mode 3 and mode 5 were changed to NM and NM with EGF and bFGF, respectively, as for mode 2 and mode 4. On the third and seventh day, the original cultivation medium was replaced by 150 μL α-MEM with 10% FBS containing 15 μL CCK-8 for each well of a 96-well plate. After incubation at 37°C for 4 h, 100 μL of solution was taken from each sample and added to one well of a new 96-well plate. Three parallel replicates were prepared, and the absorbance at 450 nm was detected using a spectrophotometer (Thermo, USA). This assay was repeated at least three times.
Reverse transcription-PCR and quantitative RT-PCR
Total RNA was extracted from cells using RNAiso Plus (Takara, Japan). The complementary DNA (cDNA) synthesis was performed using a RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, USA). Briefly, 2 μL of total RNA with Oligo(dT)18 primer, Reaction Buffer, RiboLock RNase Inhibitor, dNTP mix, and RevertAid-MuLV Reverse Transcriptase were placed in an MJ Research PTC-200 Peltier Thermal Cycler (Bio-Rad, USA). For quantitative reverse transcription polymerase chain reaction (qRT-PCR), 1 μL of cDNA and SYBR Premix Ex Taq II (perfect real time) (Takara, Japan) were placed in an Eco Real-Time PCR System (Illumina, USA). All of the operating procedures were according to the manufacturer's protocols and described previously (Wu et al., 2009). We monitored the expression of the following genes: 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase), doublecortin (DCX), microtubule-associated protein-2 (MAP2), neurofilament medium polypeptide (NEFM), nestin (NES), chondroitin sulfate proteoglycan-4 (CSPG4), nerve growth factor receptor (NGFR), tubulin beta 3 class III (TUBB3), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). PCR primer sequences and PCR product sizes are listed in Table 2. Sequences for primers were obtained from Primer3web (http://primer3.wi.mit.edu/). All of the PCRs were run in triplicate. The relative expression of the housekeeping gene GAPDH was used as reference for normalization. For quantification, relative expression levels were calculated using the delta/delta calculation method, as described previously (Livak and Schmittgen, 2001).
NCBI, National Center for Biotechnology Information.
Statistical analysis
All data are expressed as the mean±standard deviation (SD). Statistical significance was analyzed using IBM SPSS Statistics 21.0 software (IBM SPSS, USA). Analysis of variance followed by the nonparametric test of multiple independent samples was used to assess the significant differences of multiple samples, and a value of p<0.05 was considered statistically significant.
Results
Identification and characterization of DFSCs, DPSCs, and SCAPs
The primary DFSCs, DPSCs, and SCAPs were obtained after 3–7 days of culture. After two trysinization passages and subculture, the spindle-shaped DFSCs, DPSCs, and SCAPs were purified (Fig. 1A–C). All three types of dental stem cells were positive for vimentin but negative for cytokeratin-14 (Fig. 1D–I). Flow cytometric analysis showed that the DFSCs, DPSCs, and SCAPs stained positively for CD44, CD90, and CD105, but did not show expression of CD34 (Fig. 2).
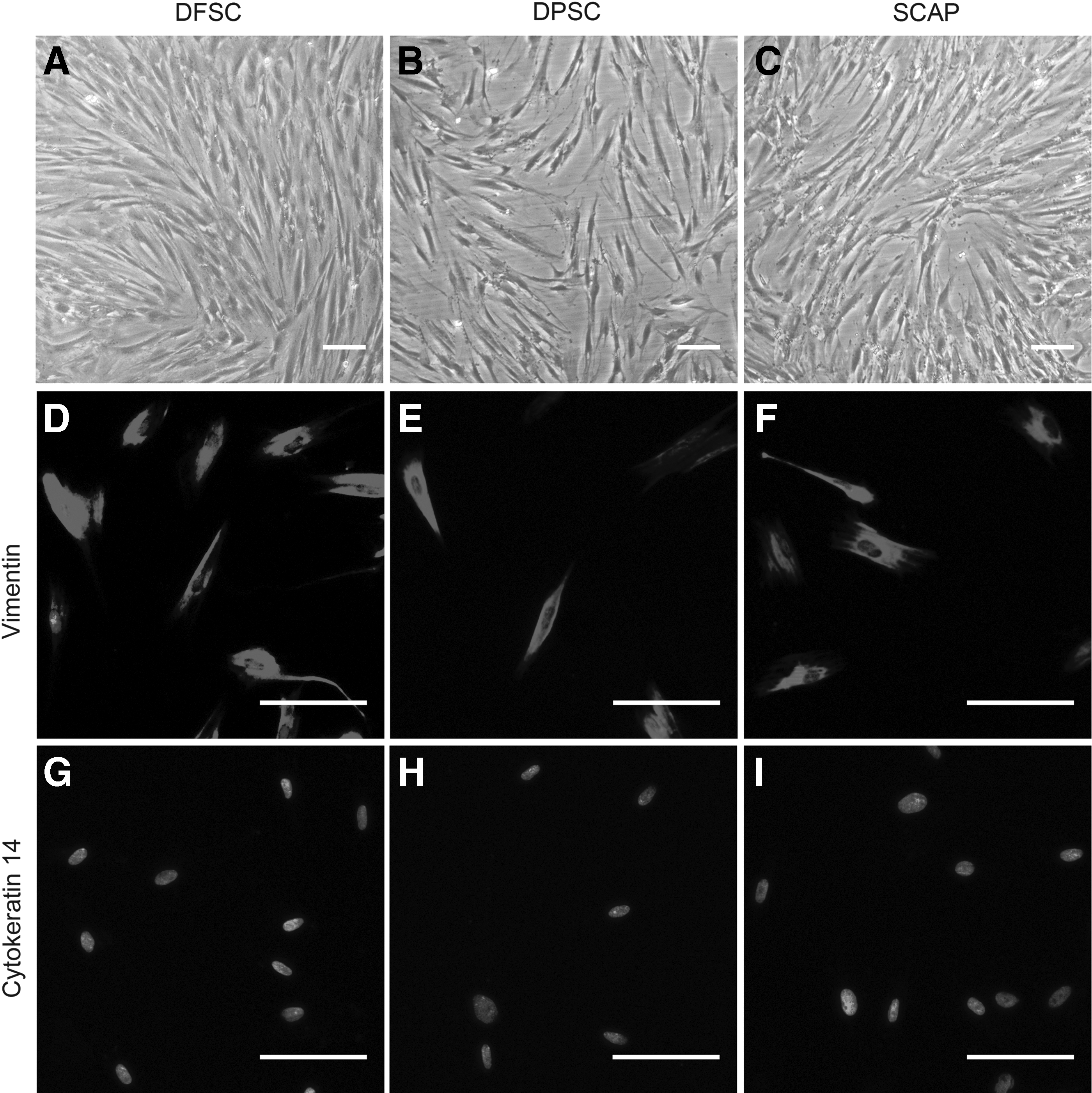
Evaluation of the biological characteristics of human DFSCs, DPSCs, and SCAPs. (
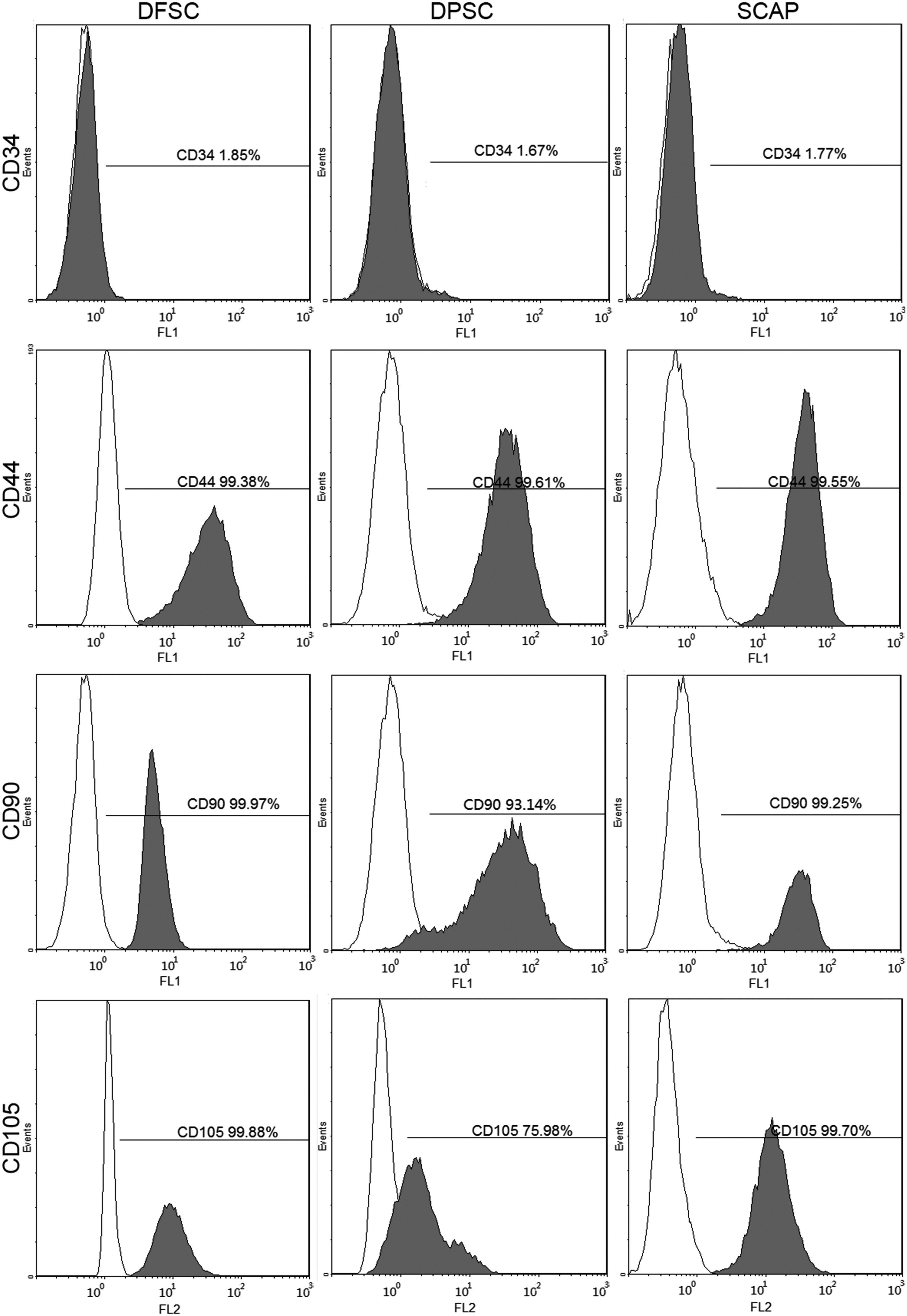
Flow cytometry detected the antigens and analyzed immunophenotypic characteristics of human DFSCs, DPSCs, and SCAPs.
Cell proliferation in five cultivation modes in vitro
After cultivation for 3 days, compared with other groups in mode 1, DFSCs showed a higher proliferation potential. The cell proliferation of DFSCs and DPSCs in mode 2 was reduced slightly after cultivation, but the proliferation ability of SCAPs was increased compared with cultivation in α-MEM (mode 1). Cell proliferation was decreased slightly in all types of dental stem cells after cultivation in NM with EGF and bFGF (mode 2 vs. mode 4). Compared with the cells cultivated in mode 2 and mode 4 (without CIM), the cell proliferation in mode 3 and mode 5 was reduced about 50% (Fig. 3A).
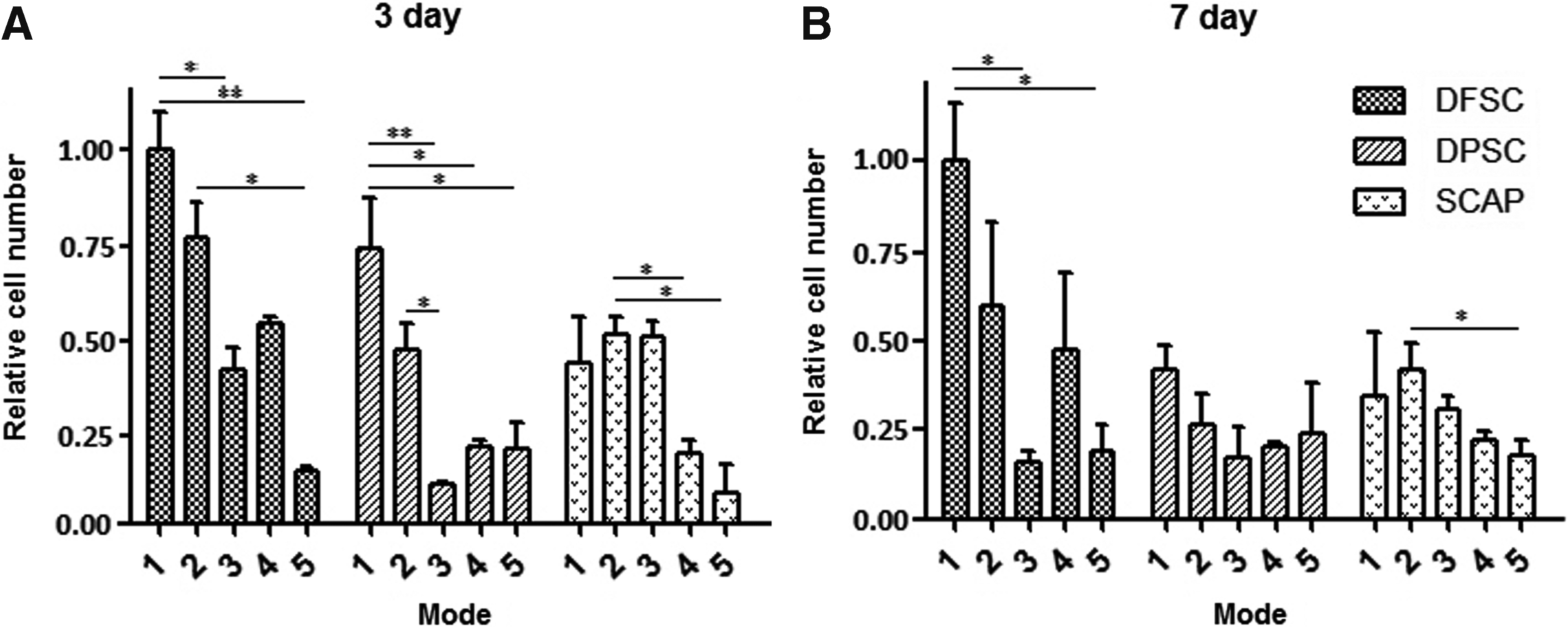
Cell proliferation of dental stem cells after cultivation in five different modes. (
After 7 days of cultivation, DFSCs in mode 1 continued to maintain their high proliferation status. As on the third day, the cell proliferation of DFSCs and DPSCs in mode 2 was reduced slightly; in contrast, the proliferation ability of SCAPs was increased compared with cultivation in mode 1. The distinct decrease of the proliferation ability in the DFSCs group was observed in mode 3 and mode 5, which included the CIM inductive step (Fig. 3B).
Cellular morphology of the dental cells after neural differentiation
After an initial cultivation for 4 h, the cellular morphology of dental stem cells that were induced by CIM (mode 3 and mode 5) presented significant changes, for instance, neural-like cells with protruding cell bodies and many long neurite-like extensions. As shown in Figure 4A, compared with other cells, the numbers of neurite-like extensions and morphology of DFSCs induced by CIM were the most obvious. Compared with mode 1, the cells in other modes not mentioned above had little differences, were spindle-like in shape, and adhered to the plastic tissue culture dishes (Fig. 4A).
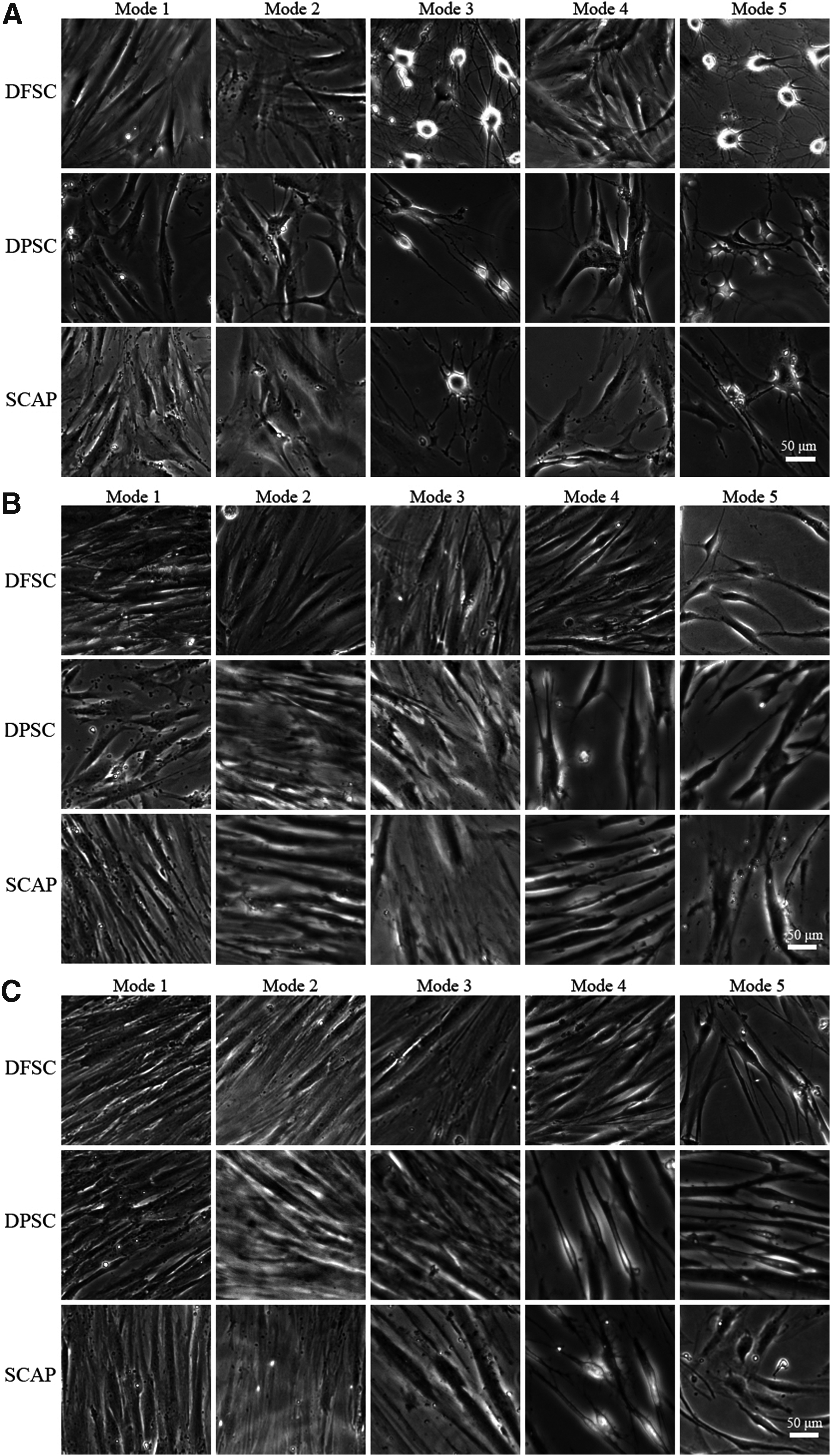
The cellular morphology of DFSCs, DPSCs, and SCAPs after cultivation in five different modes. (
The original neural-like shape of the stem cells induced with CIM for 4 h initially disappeared when they were cultivated with NM for 3 days. In addition, the cells were close to the bottom of culture dishes and there were no neurite-like extensions. The neurite-like extensions of DFSCs were well maintained in mode 5; moreover, this phenomenon in this type cells could also be observed in mode 4. DPSCs and SCAPs in mode 4 and mode 5 maintained a certain neurite-like structure, although not as obviously as before. The other cells not mentioned still maintained the spindle-like in shape (Fig. 4B).
After 7 days of cultivation, all types of cells in modes 1, 2, and 3 were closely arranged and adhered to the plastic tissue culture dishes. Especially the cellular morphology in mode 2 was most representative. DFSCs in mode 5 continued to maintain their neural-like cell morphology, and neurite-like extensions were also evident. DPSCs and SCAPs cultivated in mode 4 did not present obvious neural-like cellular morphology (Fig. 4C).
Expression levels of neural-related genes after cultivation in five modes
The data of quantitative RT-PCR revealed the expression levels of neural-related cell markers were upregulated or downregulated after cultivation in four types of conditional modes compared with control mode 1. In the DFSCs group, most of the neural cell markers except MAP2 were upregulated after cultivation in conditional modes (Fig. 5). In the DPSCs group, most of the neural cell markers were downregulated with the exception of DCX and NGFR in some modes (Fig. 6). In the SCAPs group, the expression levels of CSPG, DCX, and NEFM were upregulated in some modes but also downregulated in other modes; the MAP2 levels were downregulated as in the DFSCs group (Fig. 7).
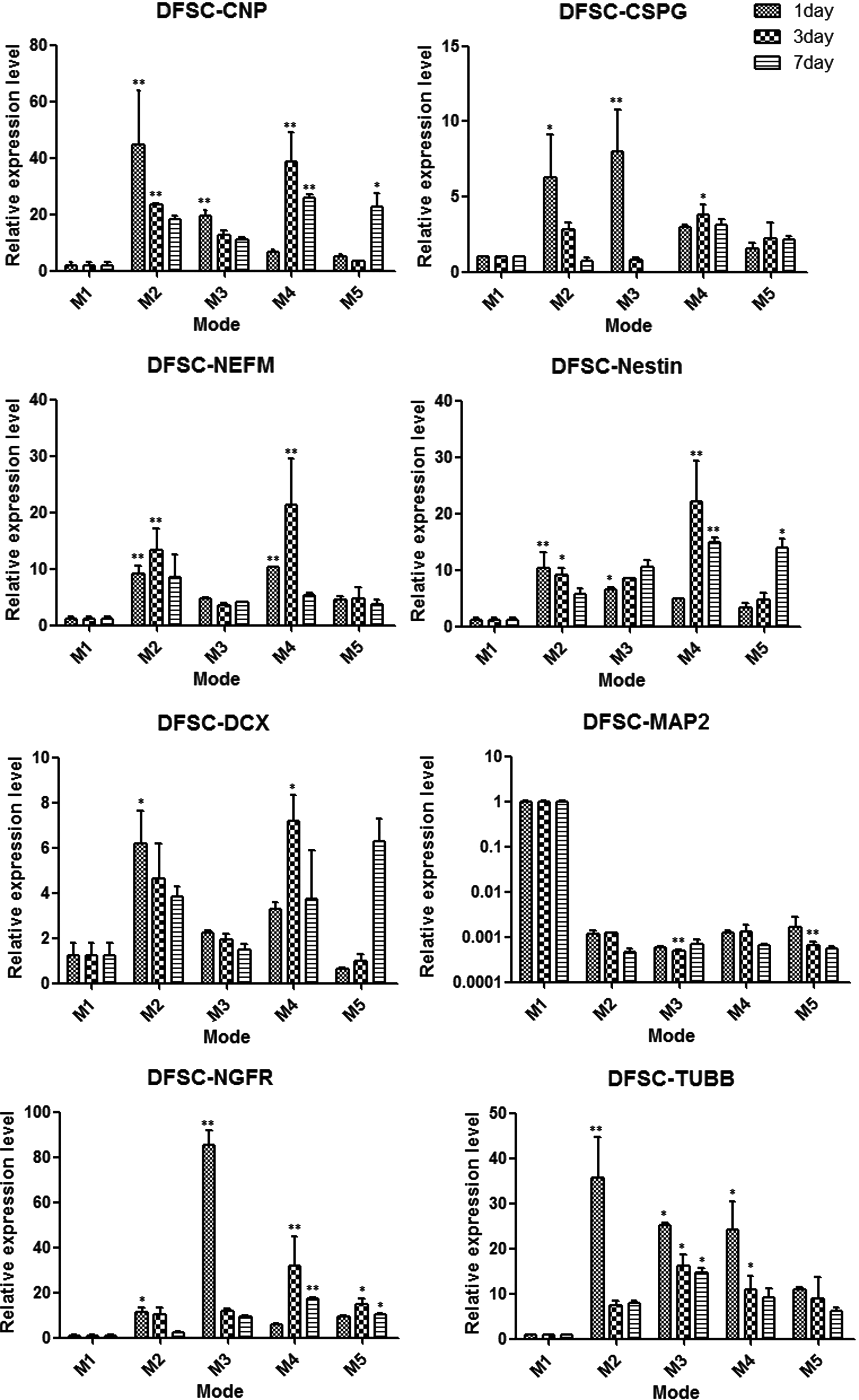
Comparison the expression changes of neural-related genes in DFSCs after cultivation in five different modes for 1, 3, and 7 days. (*) p<0.05, (**) p<0.01.
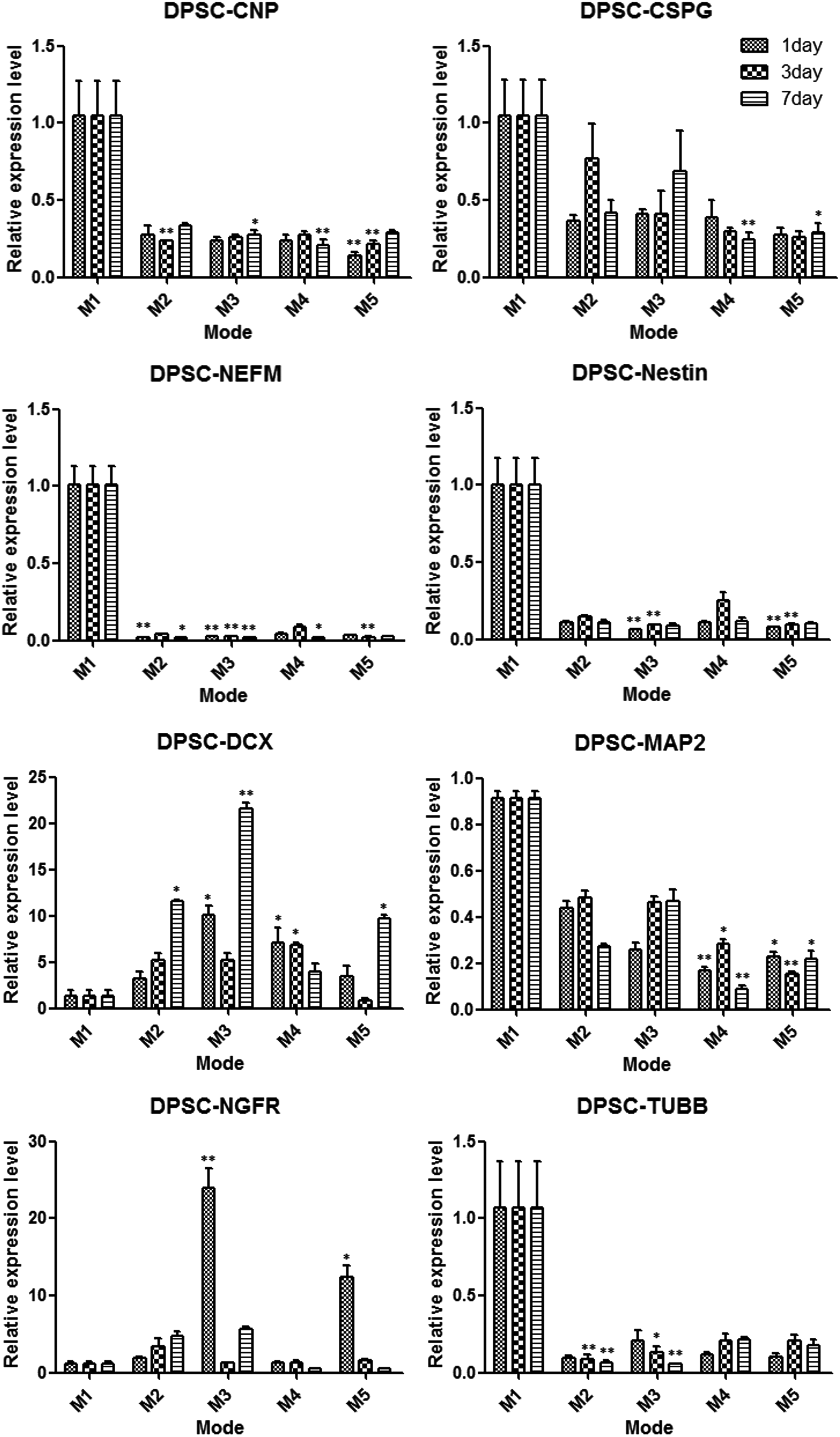
Comparison the expression changes of neural-related genes in DPSCs after cultivation in five different modes for 1, 3, and 7 days. (*) p<0.05, (**) p<0.01.
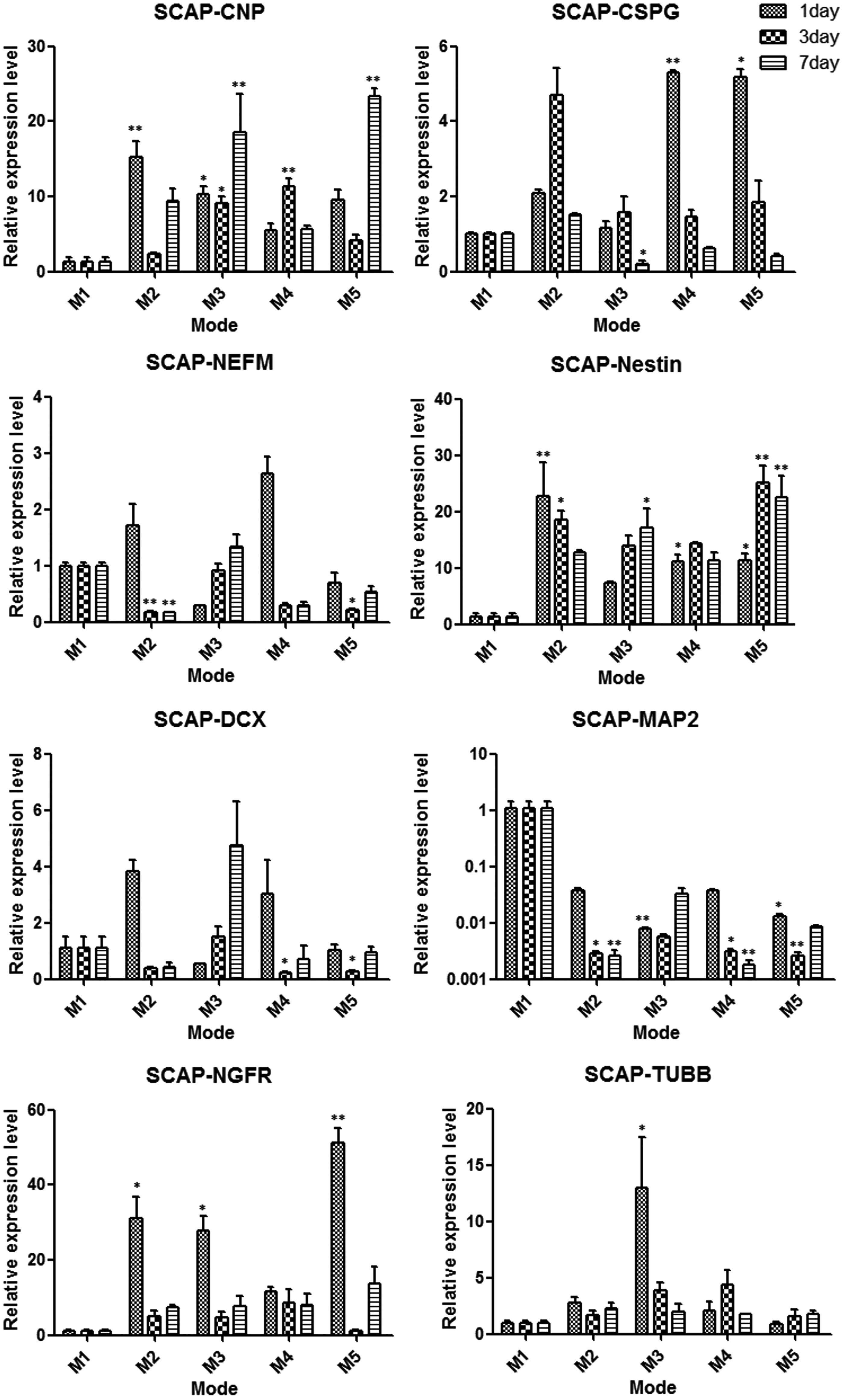
Comparison the expression changes of neural-related genes in SCAPs after cultivation in five different modes for 1, 3, and 7 days. (*) p<0.05, (**) p<0.01.
Discussion
The central nervous system of mammals has a limited capacity to replace cells that have been lost as a consequence injury or degenerative diseases (Kang et al., 2010). Because of the complexity of pathogenesis, it is still difficult to cure neurological diseases using traditional clinical treatments, such as microsurgical operation for spinal cold injury, drug delivery for degenerative diseases, and rehabilitation training (Juknis et al., 2012; Poewe et al., 2012). Therefore, stem cell transplantation might be a potential method for curing neurological diseases via differentiation and replacement of the pathological cells (Honmou et al., 2012; Kim et al., 2012; Sakai et al., 2012; Ziavra et al., 2012).
Stem cells have the ability to be a potential candidate for replacement therapy for neurological diseases, for instance, ESCs, neural stem cells, Schwann cells, olfactory ensheathing cells, and bone marrow MSCs (Carletti et al., 2011; Chiu et al., 2009; Sadan et al., 2009; Tomaskovic-Crook and Crook, 2011). But these candidate cells are facing multitudes of problems, such as the ethical issue, limited resource, and so on. Dental stem cells originate from the neural crest, and thus these cells have the possibility of differentiating into neural-like cells. Meanwhile, dental stem cells, such as DFSCs, DPSCs, and SCAPs, are easily collected from teeth or teeth germs that are surgically removed for clinical reasons. In this study, many primary cells were obtained using enzyme digestion and tissue culture methods. The miscellaneous epithelial cells could be removed, and purified MSCs could be obtained with subculture. The immunofluorescence results showed that vimentin staining was positive and cytokeratin-14 staining was negative; thus, DFSCs, DPSCs, and SCAPs had an ecto-mesenchymal origin. Flow cytometric data showed that DFSCs, DPSCs, and SCAPs did not express the surface antigen of CD34 hematopoietic stem cells but expressed the antigens of CD44, CD90, and CD105 MSCs, indicating that three types of cells had phenotypic characteristics of MSCs (Dominici et al., 2006). Thus, these dental stem cells might be a better candidate cells for the treatment of neurological diseases.
Survival, proliferation, and neural differentiation of nonneurogenic stem cells are important for cell replacement therapy to treat neurological diseases. Dental stem cells transplanted into a host would suffer many challenges, such as immunological rejection, nutritional supplies, and so on. This study found DFSCs in α-MEM with 10% FBS (basic medium) as cultivation mode 1 showed a higher proliferation ability compared with DPSCs and SCAPs. Therefore, the DFSCs would have a high proliferation potential to increase cell availability and integration of transplanted cells into the host tissue (Daviaud et al., 2013). For mode 2 and mode 4, because the medium microenvironment was changed, the proliferation ability of stem cells after cultivation in NM and NM with EGF and bFGF was decreased slightly, but not considered statistically significant. It has been reported that EGF and bFGF could stimulate cell proliferation, so this slight decrease might be caused by the NM (Yang et al., 2014). For mode 3 and mode 5, data showed that after the cells were induced with CIM for 4 h initially, their proliferation abilities were inhibited seriously, although cellular morphology presented significant changes to neural-like cells and extensions (Fig. 4A). It has been reported that rapid changes of cellular morphology after treatment with CIM might be the result of rapid disruptions of the cytoskeleton (Corti et al., 2004; Lei et al., 2007). In this study, the cells cultivated in CIM for only 7 days suffered apoptosis or death (data not shown), which might have been caused by the cytotoxicity of the CIM components and lack of FBS for too long. Therefore, chemical treatment protocols for neural differentiation should be used prudently (Morsczeck et al., 2010). Purely in terms of cellular morphology, the cells of mode 4 and 5 in 7 days were the most like neurons, with protruding cell bodies and long neurite-like extensions. It is interesting that the neuron-like morphology of mode 3 appeared after induction by CIM but disappeared after the cells were cultivated in NM (without bFGF and EGF) for the remaining few days, indicating the important role of these two growth factors for forming and maintaining the neural-like morphology (Hu et al., 2013; Yang et al., 2014).
The gene expression data suggested that these dental stem cells were moving toward neural genotypes and demonstrated some genetic changes in different degrees (Figs. 5–7). The expression levels of CNPase and CSPG were upregulated both in DFSCs and SCAPs after induction. CNPase is the third most abundant myelin protein in the central nervous system and is expressed in both oligodendrocytes and Schwann cells, indicating that the myelin production potential of DFSCs and SCAPs was enhanced after induction (Radtke et al., 2011; Yuan et al., 2002). Moreover, CSPG is expressed on the surface of oligodendrocyte precursor cells but not on astrocytes or neurons in the central nervous system. It seemed that our neural induction treatment drives these two type cells to become oligodendrocyte progenitors and promotes oligodendrocyte production and the development of myelinating processes (Busch et al., 2010; Chatterjee et al., 2008). The DFSCs and DPSCs differentially expressed DCX, a protein that is specifically expressed in neuronal cells for microtubule reorganization and synaptogenesis and frequently used as a neurogenesis marker, especially in mode 4 on the third day of DFSCs (Dehmelt and Halpain, 2007; Saaltink et al., 2012; Toriyama et al., 2012). This study also found DCX expression levels were increased more significantly in mode 2 and mode 4 (without CIM induction) than in mode 3 and mode 5. Therefore, it seemed that CIM did not promote synaptogenesis, but rapid disruptions of the cytoskeleton presented neural-like morphology (Lei et al., 2007; Morsczeck et al., 2010).
The two genetic markers that increased significantly were NEFM and NES in DFSCs and SCAPs after induction, especially in mode 4 on the third day of DFSCs. NEFM influenced a many-fold growth in axonal diameter and volume during myelination, so the DFSCs in mode 4 on the third day may have advantages to form large axonal extensions (Jacomy et al., 1999; Rao et al., 2003). Moreover, Nestin is a critical protein for embryonic neural development and is accepted as a marker of neural progenitor cells, thus implying that the neural induction mode drives DFSCs and SCAPs to become neural progenitors (Frederiksen and McKay, 1988; Wiese et al., 2004). In addition, NGFR (also called p75NTR), which was actually recognized as a pleiotropic factor for stimulating differentiation and maintenance of neurons during development, was increased in most of cultivation modes. This result indicated that the inductive modes stimulated neuronal differentiation of dental stem cells (Micera et al., 2007).
Interestingly, the expression levels of MAP2, which play a key role in dendritic outgrowth, branching, and synaptogenesis, were downregulated in all dental stem cells after induction. It was speculated that synaptogenesis might be involved in many different types of signaling, including the intercoordination of DCX and MAP2 (Mundy et al., 2008; Yamaguchi et al., 2008). Taken together, DFSCs, DPSCs, and SCAPs did not exhibit the same neural differentiation potential, and the vast majority of the DFSCs gene expression levels in mode 4 on the third day were upregulated significantly.
Conclusions
This study showed that different dental stem cells exhibited different neural differentiation potentials. DFSCs might be the better candidate cell type for neural differentiation. Furthermore, the cultivation mode 4 and timing of the third day might promote differentiation into the neurogenic cell lineage more effectively before transplantation and provide therapeutic benefits for cell transplantation therapy to treat neurological diseases.
Footnotes
Acknowledgments
This study was supported by National Basic Research Program (China, 2010CB944800), National High-Technology Research and Development Program (China, 2011AA030107), Nature Science Foundation of China (China, 81271095, 8127111981200792, and 30973348), International Cooperation Program of China (China, 2013DFG32770 and 2011DFA51970), Doctoral Foundation of Ministry of Education of China (20110181120067 and 20110181110089), China Postdoctoral Science Foundation (China, 2012M511934), Key Technology R&D Program of Sichuan Province (2012SZ0013, 12ZC0493, 13ZC0971, 2013GZX0158, and 13ZC0979), and Basic Research Program of Sichuan Province (2011JY0125, 12JC0212, and 2013JY0019).
Author Disclosure Statement
The authors declare that no competing financial interests exist.