
Abstract
Abstract
The potential of induced pluripotent stem (iPS) cells, which have self-renewal ability and can differentiate into three germ layers, led us to hypothesize that iPS cells in pigs can be useful and suitable source for producing transgenic pigs. In this study, we generated iPS-like cells using doxycycline-inducible piggyBac (PB) expression vectors encoding porcine 4 transcription factors. After transfection, transfected cells were cultured until the formation of outgrowing colonies taking least of 7–10 days. The iPS-like cells demonstrated pluripotent characteristics such as self-renewal, high proliferation, expression of pluripotent markers, and aggregation ability. The embryo development through somatic cell nuclear transfer (SCNT), cleavage rate, and blastocyst formation rate did not show any significant differences. However, the total cell number of blastocysts was significantly increased with the established cell line. In conclusion, the iPS-like cell line, generated from porcine transcriptional factors using the PB transposon system, demonstrated pluripotency with the capacity for unlimited self-renewal, and could be used as donor cells to produce cloned embryos by SCNT. These cells will be suitable for gene modification and would contribute to the stability or safety of pig models in biomedical research.
Introduction
Transgenic pigs are important for studying human diseases and cell therapy models (Aigner et al., 2010; Ekser et al., 2011, 2012). To produce highly efficient transgenic pigs, the somatic cell nuclear transfer (SCNT) technique has been widely used with genetically modified donor cells. In SCNT, somatic cells are needed for the donor cells to produce embryos. Nevertheless, the production efficiency of cloned embryos or animals using SCNT is lower than that observed using other in vitro production methods (Cibelli et al., 2002; Yang et al., 2007).
Many studies have been conducted to overcome this problem. Some of these studies have focused on donor cells to enhance the efficiency of SCNT (Tian et al., 2003). The strategies used in these studies include synchrony of the cell cycle stage (Campbell et al., 1994), age (Jeon et al., 2012; Wang et al., 2015), origin (Li et al., 2014; Richter et al., 2012), passage (Li et al., 2014), and type (Kim and Hyun, 2014). However, the conditions for donor cells have not been optimized for high-efficiency production, the G0 or G1 phase of the donor cell cycle, long telomere lengths, or early passage, which would reduce damage or mutations in the chromosome of origin and cells (Wilmut et al., 2007).
These conditions facilitate nuclear reprogramming during the developmental events in SCNT-cloned embryos and coordinate the interaction of the donor nucleus with the recipient cytoplasm (Campbell et al., 1996; Wells et al., 2003). In particular, synchronizing the G1 and/or G0 phase of donor cells would generally be more suitable for reprogramming SCNT-derived embryos, and several studies have suggested using serum starvation or high-density cultured cells to synchronize the donor cell cycle stage to G1 and/or G0 for SCNT (Baguisi et al., 1999; Kasinathan et al., 2001; Polejaeva et al., 2000; Wakayama et al., 1998).
In contrast, since well-proliferated stem cells are highly activated stage to M phage, the stage is not suitable as SCNT donor cells to generate cloned embryos and animals (Oback and Wells, 2002). However, other studies that have recently used stem cells as donor cells for SCNT demonstrated that cloned embryos reconstructed with stem cells at the M phase develop more efficiently (Kou et al., 2010; Wakayama et al., 1999).
These studies reported the successful production of cloned embryos and animals from several stem cell types, and the rates of these cells appear to be higher than those of fully differentiated cells (Kou et al., 2010; Mizutani et al., 2016; Rideout et al., 2000; Wakayama et al., 1999; Zheng et al., 2009). These results suggest that undifferentiated donor cells may increase the developmental rate of embryos and the birth rate of cloned animals.
Although embryonic stem cells (ESCs) are well known, porcine ESCs have limited germline transmission (West et al., 2011). Induced pluripotent stem cells (iPSCs) have self-renewal ability and the potential for differentiation into the three germ layers, which may be useful as a novel tool for producing transgenic pigs (Hanna et al., 2007; Wu et al., 2011). iPSCs are artificial reverse-differentiated pluripotent cells produced through transcription of specific genes (Oct4, Sox2, Myc and Klf4) from fully differentiated cells. The iPSC technology was developed by Shinya Yamanaka (Takahashi and Yamanaka, 2006). Induced cell lines have various pluripotent characteristics such as self-renewal ability, expression of several pluripotent markers, teratoma formation, and differentiation into three germ layers.
Until recently, this innovative technology has been applied worldwide and in several species (Cao et al., 2012; Esteban et al., 2009; Ezashi et al., 2009; Han et al., 2011; Honda et al., 2010; Khodadadi et al., 2012; Kues et al., 2013; Liu et al., 2012; Song et al., 2013; Talluri et al., 2015; Whitworth et al., 2014; Wu et al., 2009), including humans (Park et al., 2008; Takahashi et al., 2007; Yu et al., 2007). Therefore, iPSCs have become an alternative to ESCs. Many studies have employed viral-mediated vectors to transfer exogenous transcription factors into adult cells for successful induction and regeneration (Ezashi et al., 2011; Fusaki et al., 2009; Imamura et al., 2012; Montserrat et al., 2011).
However, the use of viral-mediated vectors to induce pluripotent cells restricts their therapeutic usage because of mutation risks and the potential activation of oncogenes (Nakagawa et al., 2008). The applications of iPSCs have been evaluated in several species using adenoviral vectors (Zhou and Freed, 2009), episomal DNAs (Lorenzo et al., 2013), and small molecules (Esteban et al., 2009). Those were evaded into the host's chromosomal DNA. Using piggyBac (PB) transposition reduces the risk of insertional mutagenesis, and is safer to deliver exogenous DNA than other gene delivery systems (Wu et al., 2006).
In addition to the absence of certain pig ESCs, there is no clear evidence that piPSCs are suitable as donor cells in SCNT. In this study, we used PB transposon-inducible vector, which was constructed with four porcine transcriptional factors controlled by doxycycline, to induce undifferentiated cells. In addition, induced pluripotent stem (iPS)-like cells were established, demonstrating pluripotency with self-renewal ability. Furthermore, SCNT-derived cloned embryos were evaluated for developmental competence using the iPS-like cells as donors.
Materials and Methods
Chemicals and materials
All chemicals were obtained from Sigma-Aldrich Co. LLC (Missouri) unless otherwise stated.
Primary culture of porcine fetal fibroblasts
Primary culture was performed using a 35-day fetus. Lumps of tissues from the backside of the fetus were minutely homogenized and cultured overnight in collagenase IV in an incubator at 37°C. The homogenized tissues were washed more than three times with phosphate-buffered saline (PBS; Life Technologies, Carlsbad, CA) and collected by centrifugation at 1500 rpm for 2 minutes. The collected clusters of cells were cultured in 60-mm culture dishes with Dulbecco's Modified Eagle's medium (DMEM; Life Technologies), supplemented with 10% fetal bovine serum (FBS; Life Technologies) and 1% Pen Strep (Life Technologies).
Transcriptional factors cloning and expression vector construction
The coding domain regions of porcine Oct4 (Kim et al., 2015), Sox2, Myc, and Klf4 were amplified from pig ovarian cDNA by PCR and cloned to TA-vectors using designed cloning primers (Table 1). Among the attained porcine transcription factors, Oct, Sox2, and Myc (pOSM) were linked by 2A peptides (F2A and E2A) except for porcine Klf4 (pKlf4), which was individually constructed.
Primer Sequences of Pig Transcription Factors
To design PB expression vectors, targeted PCR DNA fragments were used for the Gateway Cloning System (Invitrogen, Carlsbad, CA) according to protocol. Targeted exogenous DNAs were amplified using PCR primer-containing Gateway sequence (F: attB1+kozak sequence and R: attB2, Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/cell). Amplified fragments were recombined with BP and LR clonase (Invitrogen). In this study, pDonor vector (Invitrogen) was used as an entry vector and PB-TET, which has an inducible expression system containing the tetracycline-dependent mini-CMV promoter from Addgene (www.addgene.org), was used as a final destination expression vector.
Transfection of constructed PB vectors
One day before transfection, fibroblasts were plated at a density of 1 × 105 cells/mL in a well of six-well plate and cultured overnight to achieve 50%–70% confluence. Next, in total 2 μg of constructed expression vector (Fig. 1A, a: PB-TET-pOSM, b: PB-TET-pKlf4, c: PB-CA-rtTA, and d: pCy43 [Sanger Institute, Hinxton, United Kingdom]), 6 μL of the transfection reagent (Fugene HD; Roche, Inc., Switzerland), and 92 μL of DMEM were incubated for ∼20 minutes at room temperature (RT) in a 1.5-mL tube, and were then overlaid on the prepared porcine fibroblasts. After 24 hours of transfection, 2 μg/mL doxycycline was added to DMEM/F12 (Life Technologies) culture media containing 15% FBS and 10 ng/mL basic fibroblast growth factor (bFGF), and transfected cells were cultured until the formation of cell colonies.
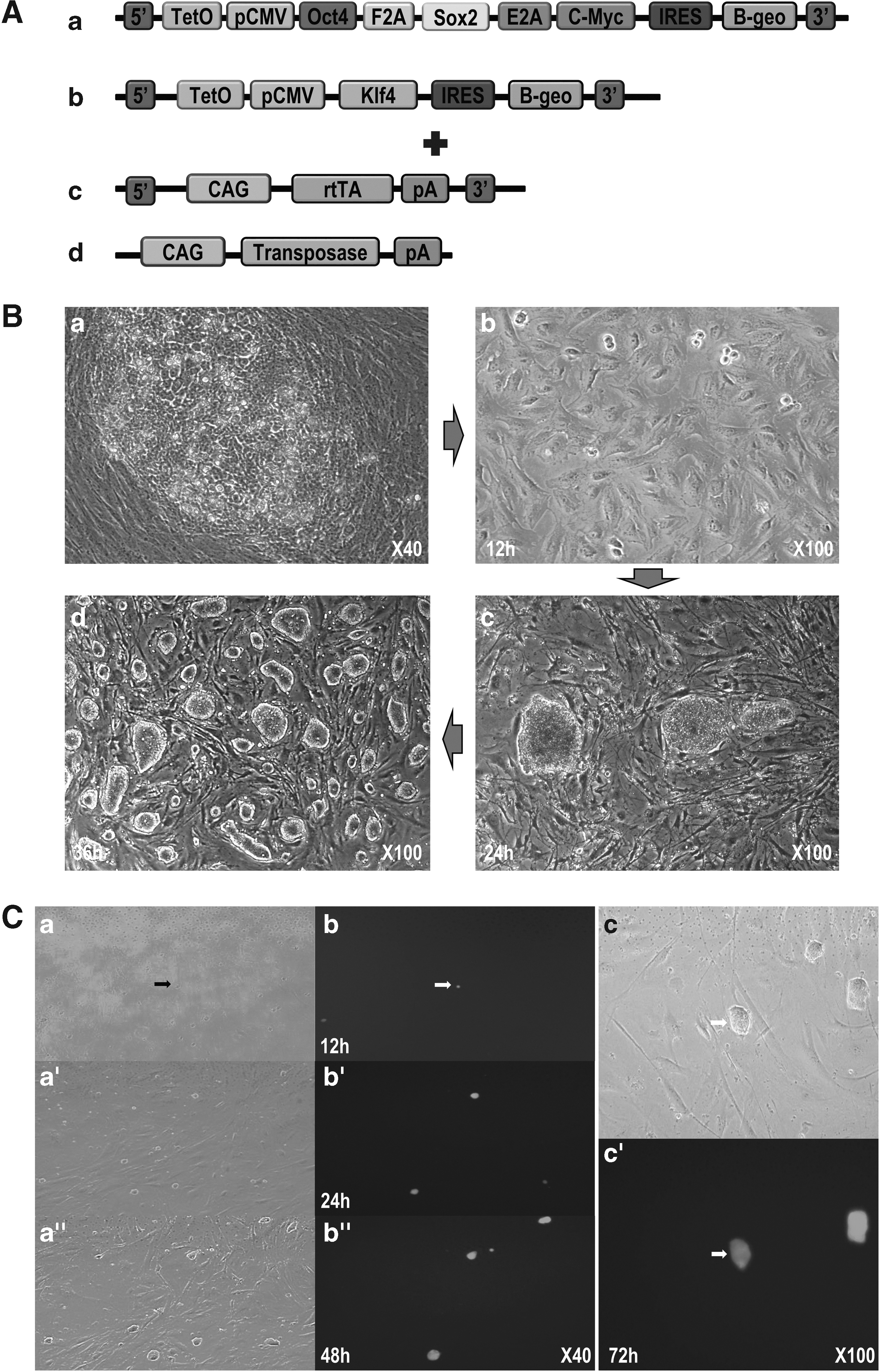
Transcription factors containing piggyBac vector constructs [
Formation of iPS-like cells and culture
iPS-like cells were cultured in stem cell culture media (DMEM/F12) containing 15% FBS, 10 ng/mL bFGF, 1% Pen Strep, and 2 μg/mL doxycycline, and maintained with mitomycin C (MMC)-treated CF1 feeder cells. In subculture process, iPS-like colonies were manually picked using a fine capillary pipette and collected in a 1.5-mL tube containing 1 mL Tryple (Invitrogen). The collected colonies were digested for 2–3 minutes in an incubator at 37°C. After digestion, cells were collected by centrifugation at 1500 rpm for 2 minutes, resuspended in stem cell culture media, and subcultured in a four-well dish on a feeder cell layer every 2 days. Cryopreservation was performed under liquid nitrogen using 70% culture medium with 20% FBS and 10% dimethyl sulfoxide.
Colony formation on different culture media conditions
To determine the optimal culture media conditions for maintaining pluripotency, generated iPS-like cells were cultured in different culture media. Well-growing iPS-like cells in control media (DMEM/F12 + 10 ng bFGF + Doxy+) were transferred to (1) media with leukemia inhibitory factor (LIF) (DMEM/F12 + 10 ng humanLIF + Doxy+) and (2) media without doxycycline (DMEM/F12 + 10 ng bFGF + Doxy-), and then cultured for 24 hours. We observed the morphology change of the iPS-like colonies at 12 and 24 hours.
Additional transfection of reporter gene to iPS-like cells
To confirm that the colonies were formed from single cells, PB-CA-GFP [a transposon vector that expresses green fluorescence protein (GFP) from a CMV early enhancer/chicken β actin (CAG) promoter] (Kim et al., 2011) was transfected into cells of the colonies using transfection reagent (Fugene HD). One day before transfection, the colonies were subcultured on CF1 feeder cells in a new four-well dish and cultured overnight. On the following day, 1 μg of the constructed vector was mixed with 3 μL of Fugene HD and 96 μL of DMEM, incubated for ∼20 minutes at RT in a 1.5-mL tube, and overlaid on the colonies.
After 24 hours of transfection, the colonies were subcultured in stem cell culture media. To establish stable GFP-expressing cells, only GFP-expressing colonies were selected and subcultured manually. Moreover, red fluorescence protein (RFP)-expressing cells were established following the same procedures using the PB-CA-DsRed II vector, which expresses RFP from the CAG promoter.
Alkaline phosphatase staining and CDy1 staining
To determine the existence of pluripotent cell surface marker of transfected cells, the cells were stained with alkaline phosphatase (AP), using AP staining kit (Vector Laboratories, Burlingame, CA) and CDy1 (Active motif, Carlsbad, CA) staining according to manufacture's protocol (Kang et al., 2011).
Immunocytochemistry of pluripotent markers
To determine the location of Oct4 and Sox2 protein expression, the cells were immunostained. The cells were fixed in 4% paraformaldehyde for 20 minutes, permeabilized with 0.1% Triton X-100 (Sigma) for 10 minutes, and blocked with 10% normal goat serum for 1 hour. The fixed cells were incubated in a refrigerator at 4°C overnight with primary antibodies (Oct3/4; Santa Cruz Biotechnology, Inc., CA, Sox2; R&D System, Minneapolis, MN, SSEA-1, 4 and Nanog; Millipore, 1:2000), followed by secondary antibodies (Cy3-AffiniPure Goat Anti-Mouse IgG, Jackson ImmunoResearch Laboratories, Inc., PA, 1:5000) for 2 hours. In addition, DAPI was used as a counterstain. The stained cells were examined under ultraviolet light using a fluorescence microscope (Nikon, Japan).
Generation of chimeric parthenotes by iPS-like cells aggregation
To determine the pluripotency of the cells, chimeric embryos were generated through iPSCs aggregation with parthenote blastomeres (Nakano et al., 2013). Established RFP-expressing iPS-like cells were used to generate chimeric embryos by injecting 15–20 RFP-expressing iPS-like cells to a 4–8 cell electro-activated parthenote using a fine injection needle attached to a micromanipulator.
The parthenotes were prepared 2 days in advance by electronic activation following the same protocol as the activation process described for SCNT (described below). The cell-injected parthenotes were cultured in porcine zygote medium-5 (PZM-5; Funakoshi Corporation, Tokyo, Japan) for 7 days and observed under ultraviolet light using a fluorescent microscope (Nikon, Japan). In addition, the aggregated and hatched blastocysts were seed cultured on a MMC-treated feeder cell layer. The obtained trophoblasts were stained using a β-gal staining kit (Invitrogen) according to the manufacturer's protocol.
SCNT and embryo culture
To produce cloned porcine embryos, SCNT was performed as previously described (Park et al., 2014). Initially, porcine ovaries were collected from a slaughterhouse in 0.9% (w/v) NaCl solution at 30–37°C. Antral follicles (3–6 mm) were aspirated using an 18-gauge needle attached to a 10-mL syringe and collected in a conical tube at 39°C. A few minutes later, the sediment was washed with Dulbecco's PBS (Invitrogen) containing 1% Pen Strep. Cumulus-oocyte complexes (COCs) with intact compact cumulus cell layers were selected and washed three times in TCM-Hepes before being transferred to a modified TCM-199 supplemented with 10 ng/mL EGF, 0.57 mM cysteine, 0.91 mM sodium pyruvate, 5 μg/mL insulin, 1% (v/v) Pen Strep, 0.5 μg/mL follicle-stimulating hormone, 0.5 μg/mL luteinizing hormone, and 10% porcine follicular fluid.
After 22 hours of culture, the cultured COCs were moved to in vitro maturation (IVM) medium without gonadotropin for additional 22 hours. The COCs were cultured at 38°C with 5% CO2. After 44 hours of maturation, the cumulus cells of the COCs were removed by pipetting with 0.1% hyaluronidase in TCM-Hepes.
To prepare donor cells, iPS-like cells were subcultured or thawed on MMC-treated CF1 feeder cells 2 days before SCNT. Subsequently, colonies were manually picked using a fine capillary pipette and collected in 1.5-mL tubes containing 1 mL Tryple (Invitrogen) solution. The colonies were enzymatically digested for 2–3 minutes in an incubator at 37°C. The digested cells were collected by brief centrifugation, resuspended in modified Tyrode's albumin lactate pyruvate (TALP)-Hepes, and stored at RT. The solution contained a mix of prepared cells and some CF1 feeder cells; however, both types of cells were easily distinguished by their morphology and size.
Matured oocyte stained with 5 μg/mL bisbenzimide (Hoechst 33342) for 10 minutes and observed under an inverted microscope equipped with epifluorescence at 200 × magnification. Each matured oocyte was held with holding micropipette (150 μm inner diameter), and then enucleated with a micromanipulator (Nikon-Narishige, Tokyo, Japan) in TALP with 5 μg/mL cytochalasin B. The first polar body and adjacent cytoplasm, containing the metaphase-II chromosomes, were aspirated using a fine micropipette. The enucleated oocytes were placed in TALP-Hepes and used for SCNT. Transfected cells were used as donor cells for SCNT. The donor cells were injected into the perivitelline space of the enucleated oocytes and electrically fused using an electro cell fusion generator (LF101; Nepa Gene Co., Japan).
After 40 minutes of fusion, the fused embryos were artificially activated with a single direct current pulse of 1.5 kV/cm for 60 μsec using BTX Electro Cell Manipulator 2001 (BTX, Inc., San Diego). The activated SCNT embryos were cultured in PZM-5 (Yoshioka et al., 2002) covered with mineral oil at 39°C in 5% CO2, 5% O2, and 90% N2. The embryos were evaluated for cleavage on day 2 and blastocyst formation from days 5 to 7, and the total cell number of cloned blastocysts was counted on day 7. In addition, some of the cloned blastocysts were stained using a β-gal staining kit (Invitrogen). To compare within the established iPS-like cell line and normal porcine fetal fibroblasts (PFFs), a general SCNT procedure was performed with the PFFs.
Statistical analysis
All experiments were replicated at least three times and statistically analyzed using Prism 5 software (GraphPad, La Jolla, CA). Student's t-test was used to determine any differences between two groups. A p-value of <0.05 was considered statistically significant.
Results
Colony formation and single cell-driven culture
From day 10, iPS-like colonies were formed in different spots (Fig. 1B-a), then after culturing on MMC-treated CF1 feeder cells. After 12 hours, the subcultured cells adhered onto feeder cells as tiny colonies (Fig. 1B-b). After 24 hours of subculture, the colonies grown had a clear margin and compact dome-like shape (Fig. 1B-c). Over time, the iPS-like colonies grew bigger having previous form at 36 hours (Fig. 1B-d). The colonies were routinely single cell passaged every 2–3 days by Tryple (Invitrogen) and single cell grew up to a new colony (Supplementary Video S1). The generated colonies were cultured up to 37th passages by single cell culture.
To determine whether the colonies were formed from single cells, the colonies were additionally transfected with a reporter gene (GFP); only GFP-expressing colonies were selectively cultured (Fig. 1C-a′). The GFP-expressing single cells grew and divided on the CF1 feeder cell layer (Fig. 1C-b′), and cells formed dome-like colonies after 48 hours of culture. The colonies expressed GFP homogeneously (Fig. 1C-c′). The results clearly demonstrated that a colony could be generated from a single cell, and that the transgenic cell had self-renewal ability.
Optimization of culture media condition
The iPS-like cells retained their morphology and had a dome-like shape, which is a typical stem cell characteristic that can be observed in control media (Fig. 2a). However, the colonies regressed to a fibroblast-like shape, losing their dome-like shape in both media (LIF-dependent medium and absence of doxycycline). In LIF-containing medium, the iPS-like cells gradually differentiated to normal fibroblast cells as before transfection (Fig. 2-b, b′). In addition, in the absence of doxycycline, the iPS-like cells had deformed morphology during colony formation and eventually lost their pluripotent ability (Fig. 2c, c′).
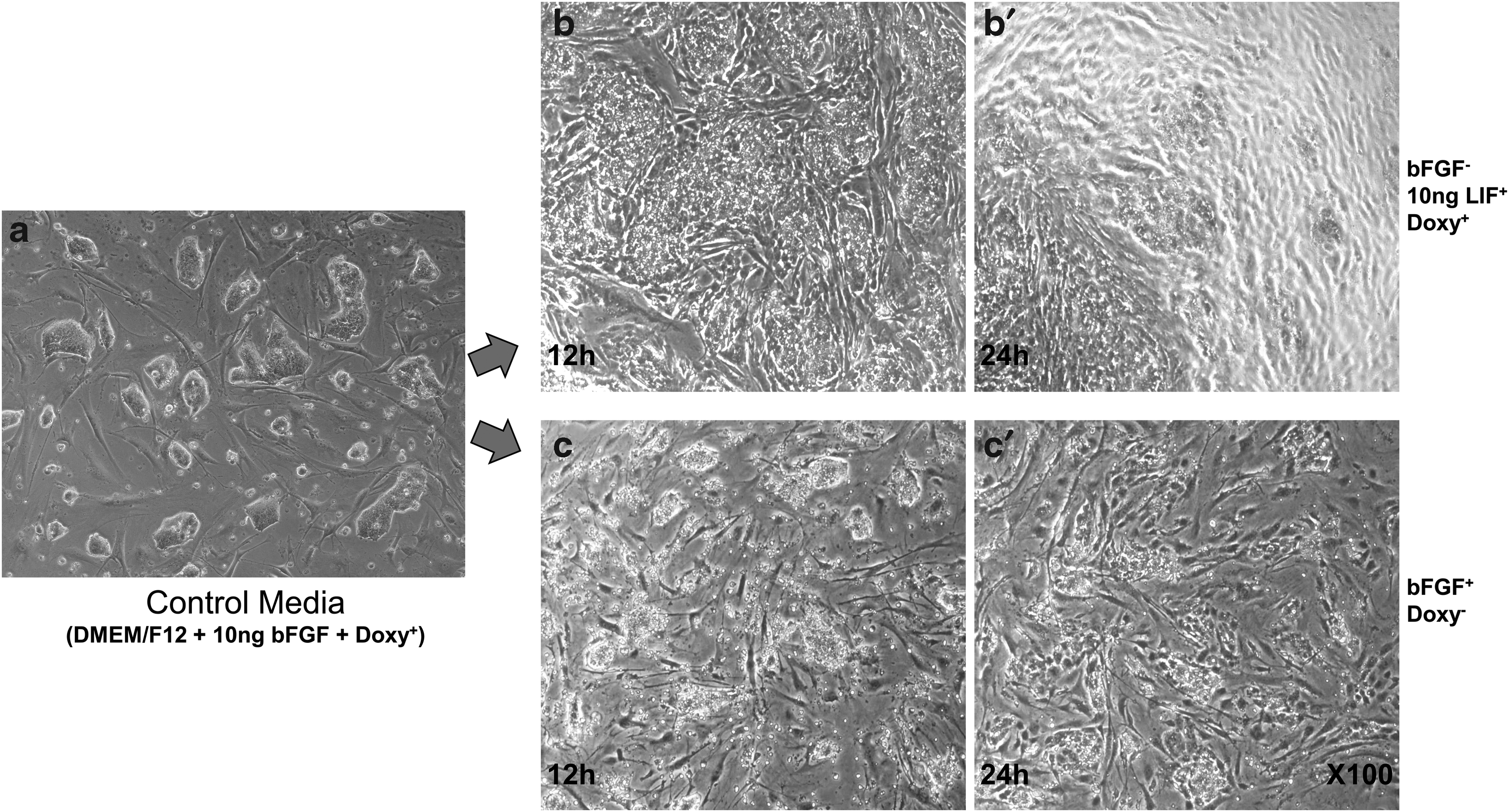
The colony deformation on different media conditions. To prove the optimized culture media conditions, well-growing iPS-like cells in control media
Characterization of iPS-like cells
To identify the characteristics of iPS-like cells, the expressions of certain pluripotent markers were evaluated by specific staining. AP and CDy1 staining showed positive results (Fig. 3A, B-c). AP and CDy1 only stained the surface of iPS-like cells but not CF1 feeder cells. The CDy1-stained parts corresponded to the DAPI-stained cell nucleus (Fig. 3B-d).
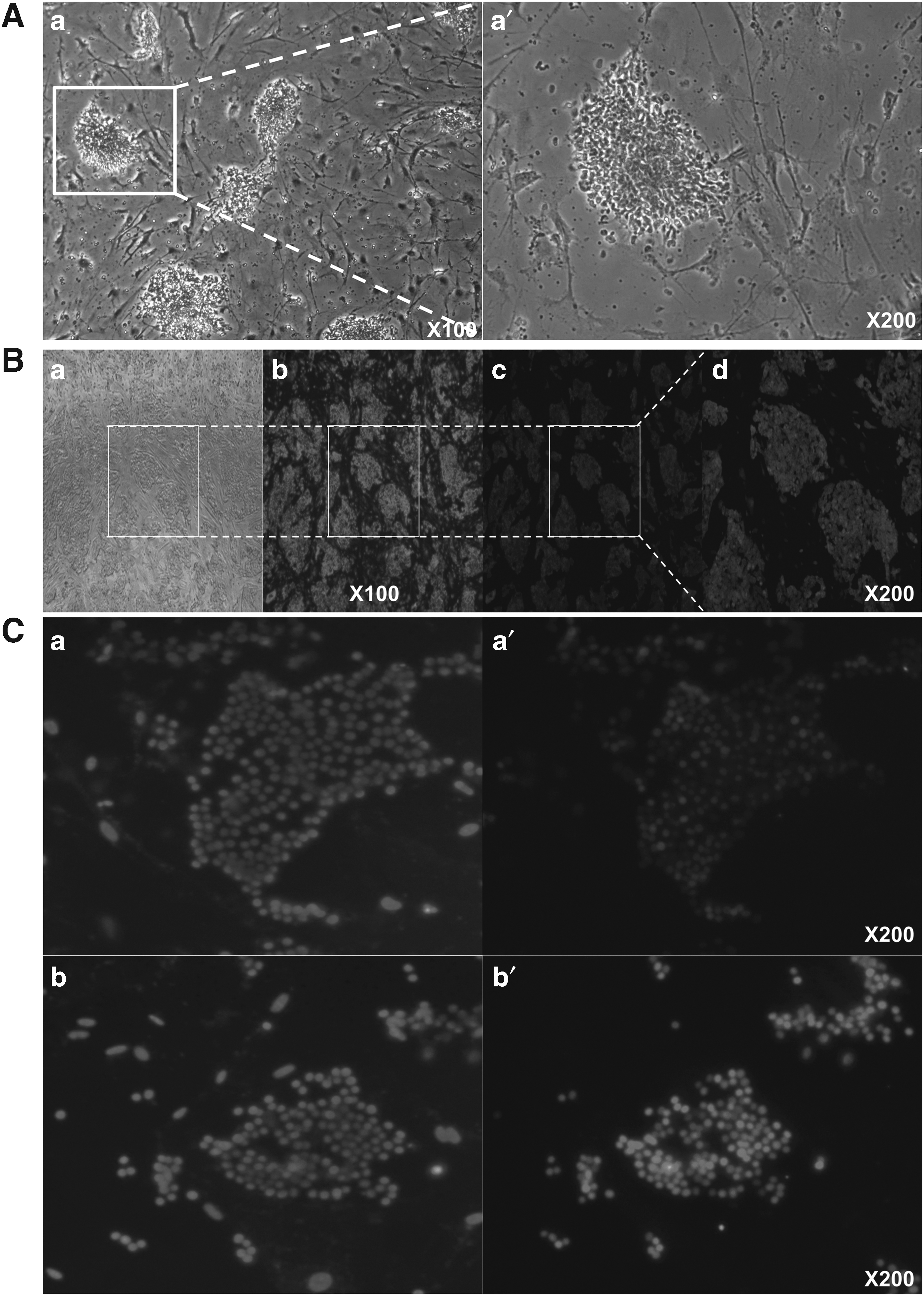
Fluorescence staining pluripotency cell markers of iPS-like cells. AP was weakly stained on the surface of iPS-like cells, but it showed positive activity
Based on immunocytochemistry results, Oct4 and Sox2 were localized and strongly expressed in the nucleus of the established cell lines (Fig. 3C). However, other pluripotent markers including SSEA-1, SSEA-4, and Nanog were not detected.
In addition, the iPS-like stem cell colonies change the morphology into several cell types such as cuboidal, spherical, and fibroblast-like cells (Supplementary Fig. S1).
Aggregation with parthenogenetic blastomeres
The iPS-like cells demonstrated aggregation with the blastomeres of parthenogenetically activated embryos and generated chimeric blastocysts. The blastocysts, derived from the transgenic iPS-like colonies with RFP expression (Supplementary Fig. S1), were mosaically expressed in both the inner cell mass and epiblast regions (Fig. 4A-a′). However, RFP expression was not observed in normal parthenogenetically activated blastocysts (not injected cell, Fig. 4A-b′). The β-gal staining results clearly indicated that the cells were successfully aggregated with parthenogenetic blastomeres. Indigo-blue-stained parts were observed in the blastocysts and the cultured trophoblasts, which were established by seeding the blastocysts onto CF1 feeder cells (Fig. 4B-a, a′).
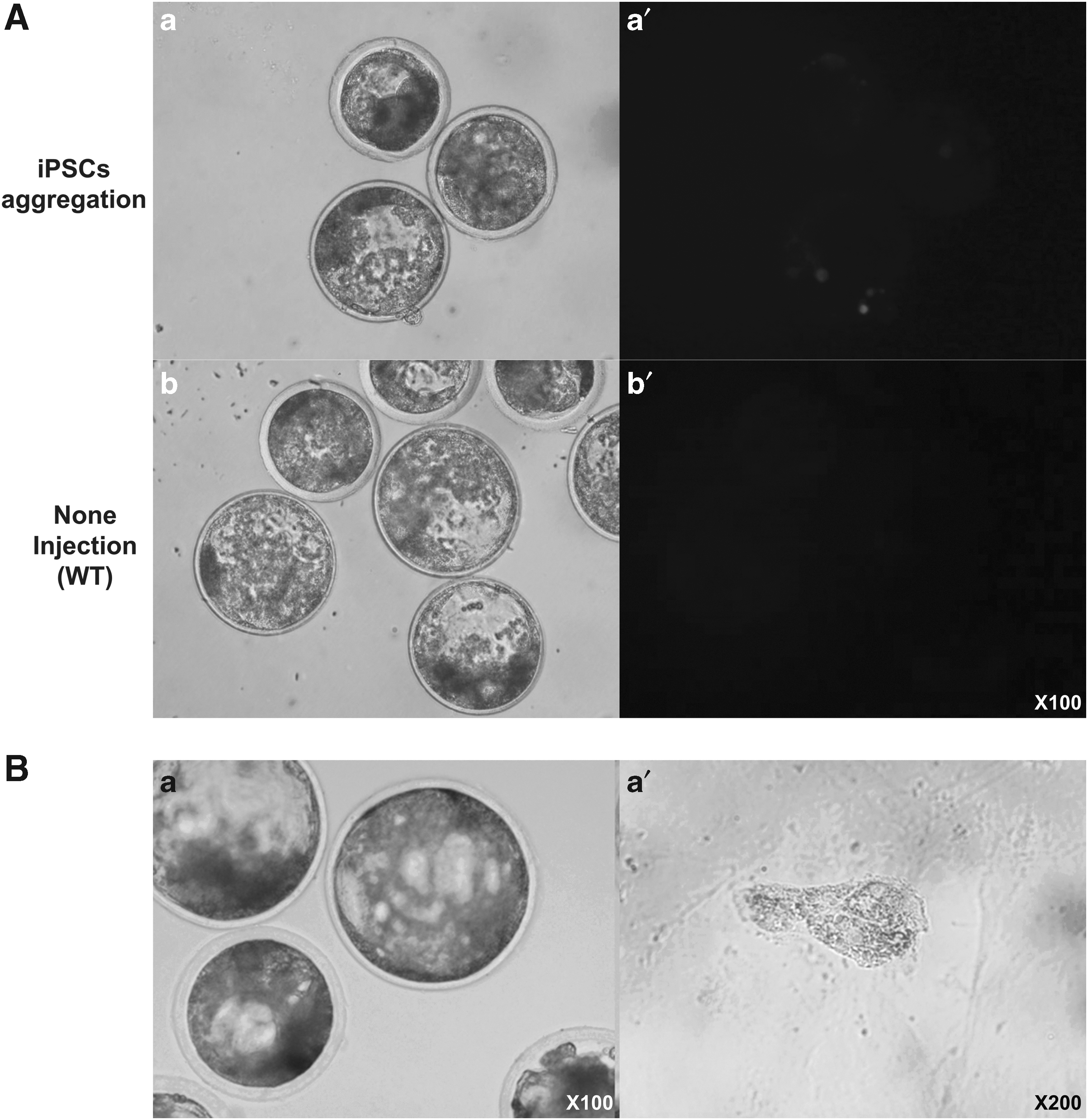
Generation of chimeric parthenotes by transgenic cells aggregation. After injection of iPS-like cells into parthenotes
Developmental competence of SCNT-derived embryos
To assess the developmental potential of using iPS-like cells as donors for nuclear transfer, single iPS-like cells were reconstructed with enucleated oocytes and developed into the preimplantation stage. Of 306 cloned embryos, 145 (47.3% ± 2.7%) embryos developed into the two-cell stage, and 48 (15.7% ± 2.3%) blastocysts were generated (Table 2). The cleavage rate and blastocyst formation rate were not significantly different between fetal fibroblasts and iPS-like cells.
Comparison Developmental Rate of Cloned Embryos Derived from Fibroblasts or iPS-Like Cells
Within the same column, values with different superscripts are significantly different (p < 0.05).
iPS, induced pluripotent stem.
However, the cell number of blastocysts was significantly higher compared with that of fetal fibroblasts (47.4 ± 3.8 vs. 36.5 ± 2.5). Furthermore, the origin of blastocysts was identified by β-gal staining and the reporter gene expression of the SCNT donor cells. The generated cloned blastocysts were stained by β-gal (Fig. 5A-b, b′). In addition, GFP-expressing iPS-like cells were successfully reprogrammed to preimplantation embryos, which expressed GFP without mosaicism (Fig. 5B-b, b′).
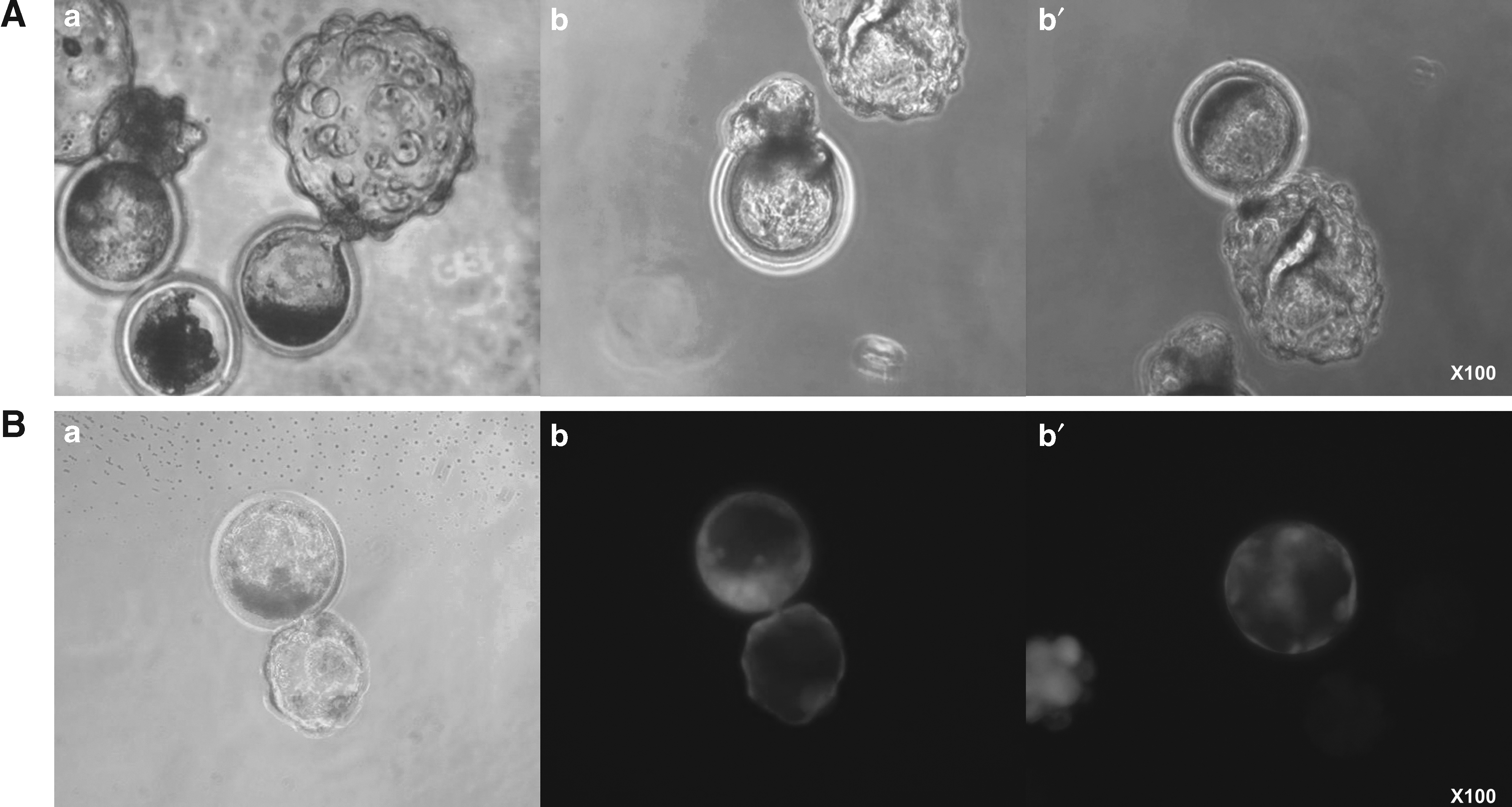
The blastocysts derived from porcine iPS-like cells by SCNT. Blastocyst development was normally observed
Discussion
The data demonstrated that the established iPS-like cells from fetal fibroblasts using PB and porcine transcription factors could propagate without senescence from a single cell with self-renewal ability. In this study, pluripotent characteristics, such as self-renewal ability, dome colony formation, and rapid proliferation, were preserved during growth in the stem cell media. Pluripotent cells are categorized into primed and naïve types. Primed pluripotent cells, including mouse epiblast stem cells, human ESCs, and human iPSCs, appear as flattened colonies, have low clonogenicity, and depend on bFGF/activin for self-renewal.
Naïve stem cells, such as mouse ESCs, are tight domed colonies, are highly clonogenic from single cells, and are LIF/Stat3 dependent, with self-renewal ability (Nichols and Smith, 2009). Ambiguously, the present established iPS-like cells were not classified into these cell types as they had a compact, domed colony morphology and high clonogenicity from single cells but progress through a bFGF/activin-dependent self-renewal pathway.
The stem cell markers AP, Oct4, and Sox2 were detectable on the iPS-like cell surface or in the nucleus. However, the sensitivity of AP staining was low to determine whether iPS-like cells were pluripotent (Fig. 3A). Another staining substance, CDy1, was strongly detected in present iPS-like cells only, not feeder cells. On the contrary, SSEA-1, SSEA-4, and Nanog were not detected. Oct4 and Sox2 are the main key factors in stem cell whether the cell type is naïve or primed status cells. These factors should be present in the nucleus of stem cells.
However, in the case of SSEA-1 and 4, there is still controversy on their expression and role as stem cell markers in pigs. The expressions of these markers can vary according to several studies (Esteban et al., 2009; Ezashi et al., 2009; Wu et al., 2009). Studies on the expression and levels of pluripotent markers have indicated that the pluripotency of stem cells may be limited in the porcine inner cell mass. Therefore, the role of transcription factors and pluripotent marker expression in pigs remains unclear, and further studies are needed for a complete characterization.
The iPS-like cell colonies occasionally changed morphology into several cell types such as cuboidal, spherical, and fibroblast-like cells during the cell culture, as the cells have differentiation ability. Thus, we performed embryonic body (EB) formation to evaluate differentiating into three germ layers, but the iPS-like cells failed the EB formation. Although the iPS-like cells showed important features such as self-renewal and stem cell markers, they showed partial rather than full pluripotency.
Cell aggregation is another feasible method to determine pluripotency (Nakano et al., 2013). iPS-like cells (15–20 cells) were injected into an electrically activated parthenote, and the embryos were developed into a chimeric blastocyst. The chimeric blastocysts were identified using RFP-expressing cells and β-gal staining. The iPS-like cells aggregated completely with the blastomere of the parthenote. As a result, the chimeric blastocysts expressed RFP and had indigo blue spots after β-gal staining. The established cell line must retain pluripotency without exogenous inducing factors to successfully generate iPSCs. However, the iPS-like cell line in this study did not maintain pluripotency in the absence of doxycycline, which controls the transcription of exogenously targeted DNAs in the PB transposon vectors.
These results are in agreement with those of previous studies; however, the continuous expression of exogenous transcription factors is still necessary to maintain the pluripotency of presumptive iPSCs in pigs (Esteban et al., 2009; Ezashi et al., 2009; Wu et al., 2009). There have been no reports of porcine iPSCs in which the inducing factors are silenced and pluripotency is maintained by endogenous pluripotent factors (Kues et al., 2013).
The transformed cells were successfully reprogrammed into the blastocyst stage after reconstruction with pig oocytes. Furthermore, an additional gene (GFP or RFP) was transfected and expressed in preimplantation embryos without mosaicism. The iPS-like cells developed in the study can be maintained in vitro without senescence, which allows easy manipulation of another gene within the cells of large animals. Furthermore, developmental competence of the cloned embryos derived from the iPS-like cells appeared greater than that of the embryos reconstructed with fibroblasts.
In conclusion, we demonstrated that the constructed PB transposon vector associated with four porcine transcription factors could be used to generate iPS-like cells, which are stained positive with some stem cell markers. These cells could potentially be used as suitable donors for generating genetically modified embryos and animals through SCNT. The cells could also be a valuable source for generating pig models for human biomedical research.
Footnotes
Acknowledgments
This study was financially supported by NRF (No. 2011-0014941), the Research Institute of Veterinary Science, and BK21 PLUS Program for Creative Veterinary Science Research.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.