
Abstract
Abstract
The development of next-generation sequencing platforms increased substantially the capacity of data generation. In addition, in the past years, the costs for whole genome sequencing have been reduced that made it easier to access this technology. As a result, the storage and analysis of the data generated became a challenge, ushering in the development of bioinformatic tools, such as programs and programming languages, able to store, process, and analyze this huge amount of information. In this article, we present MELC genomics, a framework for genome assembly in a simple and fast workflow.
1. Introduction
T
Bioinformatics refers to computational and analytical methods to biological problems, that is, the search and use of patterns in biological data and the development of methods for database access and queries.
In the past decades, the amount of biological data has grown exponentially and researches leading to the development of sophisticated bioinformatics resources made possible the rapidly and cost-effectively data mining and analyses, approaching the generation of genomics data and its analysis by conventional biological methods (Cantacessi et al., 2012).
The bioinformatics area has developed rapidly, making available many tools, aiding to the understanding of genome-related issues, such as programs that do not require extensive computational knowledge. For instance, for genome assembly strategies, programs such as SOAPdenovo (Luo et al., 2012) and SPAdes (Bankevich et al., 2012) have been widely used.
Although easy to use, different programs are necessary to perform the whole pipeline for data analysis. For example, to perform the assembly of an entire genome, some programs are required from the checking of the sequenced data quality to the checking of the assembly quality itself.
Some frameworks and web servers have been developed and made publicly available, such as the Genome Analysis Toolkit, a structured programming framework designed to offer a wide variety of tools with a primary focus on variant discovery and genotyping (Ghoneimy and El-Seoud, 2016), as well as the public servers BioExtract (Lushbough et al., 2010) and Galaxy (Giardine et al., 2005).
BioExtract Server is an open, web-based system wherein researchers are able to construct their own pipeline, by recording tasks performed. These tasks may include querying multiple, distributed data sources, saving query results as searchable data extracts, and executing local and web-accessible analytic and computational tools. Furthermore, this server includes integrated data interfaces, such as National Center for Biotechnology Information nucleotide and protein databases, the European Molecular Biology Laboratory (EMBL-Bank) nonredundant nucleotide database, the Universal Protein Resource (UniProt), and the UniProt Reference Clusters (UniRef) database.
Galaxy is a scientific workflow system and a data integration platform for biological data, providing a graphical user interface for specifying which data to operate on, which steps to take, and which order to do them. It supports data uploads from the user's computer and directly from many online resources, such as USCS Genome Browser, BioMart, and InterMine. Originally Galaxy was developed for genomics analysis, but currently it is also used for gene expression, proteomics, transcriptomics, and other areas in life sciences.
Here we present MELC genomics, a framework that integrates some of the most used programs for a complete assembly pipeline, ranging from small to large data sets. Through a simple, intuitive, and user-friendly web interface, MELC genomics can be processed on any browser, besides it is simple and fast to learn by the user.
In brief, MELC genomics allows users to perform the checking of the quality before and after processing of the raw sequenced data files; data processing, for example, trimming of adapters and low-quality bases; genome de novo assembly; quality assembly checking; and the comparison among different assembly approaches.
2. Framework Structure and Usage
The first version of MELC genomics was developed to be executed only in Linux distribution of 64-bit. Programmed in PHP (Hypertext Preprocessor) language, which is a widely used open source general purpose scripting language, MELC genomics is also suited for web development and can be embedded into HTML. Moreover, our framework can be used in any web browser, which means that specific plugin is required.
The MELC genomic source code is very simple and can be downloaded from the link (https://github.com/evaldocosta/melc). The user can install the MELC program in a local server and it can then be accessed from the network. The first version allows only one user to submit a job, user default admin.
The MELC install is very simple, when uncompress the files has one filename called README.md with install instructions. All tasks submitted to MELC create logs that can be accessed only by the program. In future MELC genomics version, the logs will be sent by email to users.
MELC genomics enables users to perform data treatment and genome assembly in a user friendly web-based interface (Fig. 1) and, since it is structured in modules independently, pipeline can be performed in a workflow or separately, according to user needs (Fig. 2).
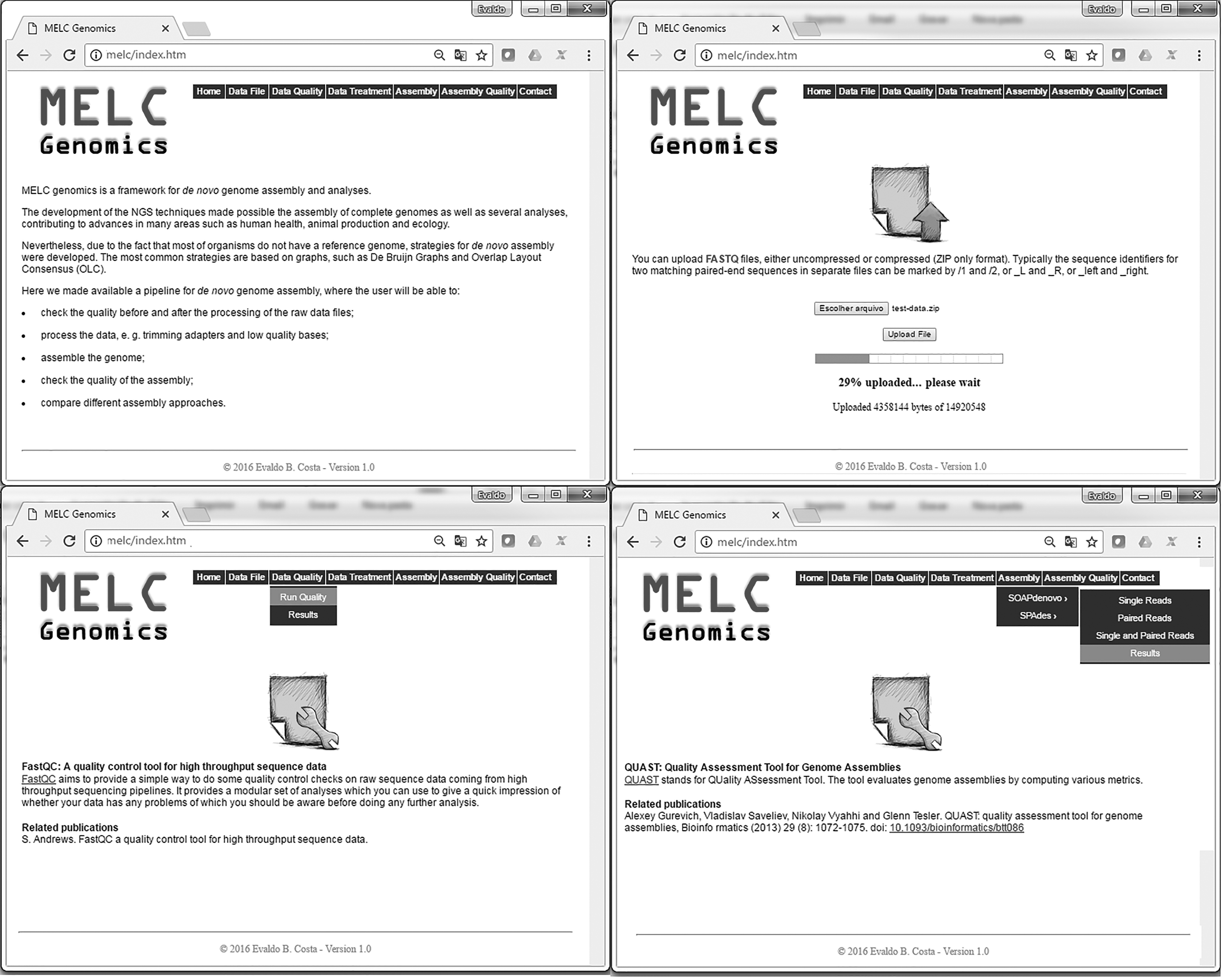
MELC genomics interface. The navigation bar provides links to the major components of this framework for genome assembly.
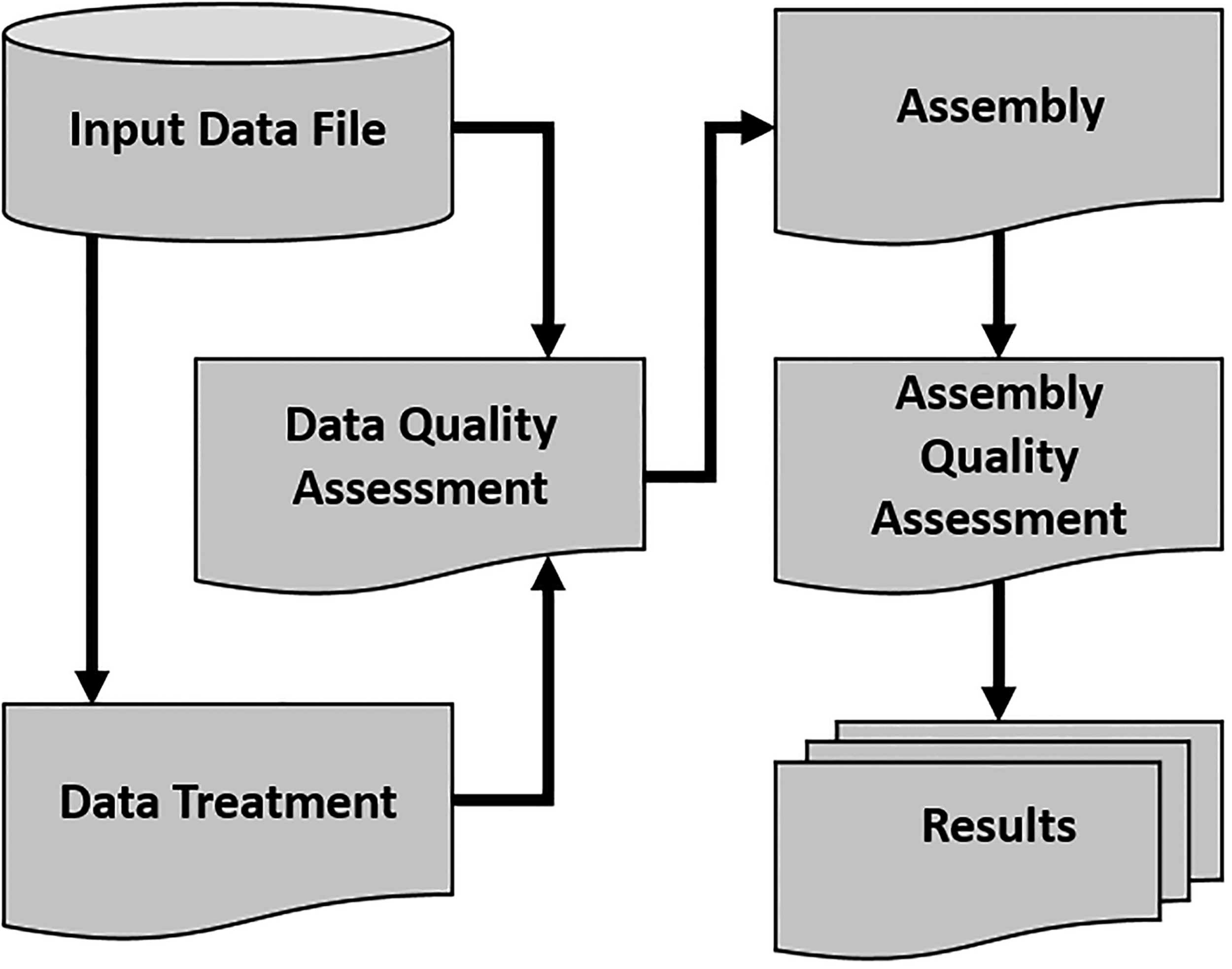
MELC genomics workflow.
The first step for MELC genomics workflow is the input of the fastq files generated during sequencing. After that, users are able to perform sequencing quality ascertainment, treatment for low-quality bases and reads, genome assembly, and also to verify the quality of the assembly. Besides, the output data generated at each executed task follow the default of the available programs.
3. Modules
3.1. Data quality
During DNA sample preparation and sequencing, some errors may occur due to polymerase mistakes during polymerase chain reaction (PCR) process, cluster amplification, sequencing cycles, and image analysis, which can result in incorrect variant calls (Fox and Loeb, 2014). Thus, even though sequencing platforms perform a previous data quality checking, it is recommended the performance of a new quality check for raw data, that is, the reads generated during the sequencing.
This process will aid the reliability of the assembly results, and MELC genomics made available a simple step to perform data quality assessment using the software FastQC v. 0.11.5 (Andrews, 2010).
At the Menu tab, select Data Quality and then Run Quality to start the quality analysis. Once finished, users are able to download and check the FastQC results on their own computer. FastQC generates HTML files containing the evaluation of several parameters, such as GC content, per base and per sequence quality, amount of bases not identified (N), and remaining adapters, among others.
This initial step is important to direct the choice of the parameters for the next task: data treatment.
3.2. Data treatment
During sample preparation, adapters are added on both ends of each DNA fragment, which allow the stability of the fragments on the flow cell, for cluster generation and sequencing. These adapters refer to small pattern DNA sequences specific for each sample preparation kit. Therefore, the extraction of these adapters from each sequenced read is important to avoid assembly errors. This screening is highly recommended even if the sequencer has done it automatically.
In the present framework, data treatment is performed using the software Trimmomatic v.0.36 (Bolger et al., 2014), accessed on the tab Data Treatment. To perform this step, users must first select the files containing the appropriate adapter sequences, in other words, the adapters used during library preparation. The results will be displayed on Data File menu, on the tab List. Trimmomatic is a fast, multithreaded command line tool that can be used to trim and crop low-quality bases, as well as to remove adapters.
A new data quality check is recommended before continuing the workflow.
3.3. K-mer estimating
The term k-mer typically refers to all possible substrings of length k, contained in a string. This case refers to all possible subsequences of length k (seeds) from a read obtained through DNA sequencing to perform the assembly by overlapping and extension of the seeds. Identifying the best k-mer to be used is very important for the improvement of the genome assembly.
K-mer prediction can be performed on the menu Data Treatment. For this analysis, MELC genomics made available KmerGenie v. 1.7016 (Chikhi and Medvedev, 2014), which estimates the best k-mer length for each data set. For a given set of reads, KmerGenie computes the k-mer abundance and displays histograms for many values of k, indicating the one that fits better the data set.
3.4. Assembly
The genome assembly can be performed guided by a reference genome or in a de novo strategy, when a reference is not available. MELC genomics pipeline is structured to perform a de novo assembly from small to large genomes, requiring low computational resources, as follows.
To assemble your sample accessed on the MELC tab Assembly, choose the best assembler for your characteristic sample. You can assemble your sample files using single reads, paired reads, or single and paired reads. The results will be displayed on the MELC tab Assembly; choose assembler to be used on the tab Results.
3.4.1. SOAPdenovo assembler
SOAPdenovo2 v. 2.0.4 is widely used to do short reads assembly and was developed to work with data sequenced on Illumina platform and it is efficient for larger genomes. SOAPdenovo2 algorithm demands less memory usage for the construction of the de Bruijn graphs (Earl et al., 2011).
De Bruijn graph strategy is to first break the sequences into smaller fragments (K-mers) and then by overlapping (k-1) generate graphs, rebuilding the sequences contained in the genome (Pevzner et al., 2001).
During that phase, the asynchronous read operation aio read is used to read the sequences generated by Illumina platform. The function aio read informs the system which file must be read, the offset to begin the reading, how many bytes to read, and where to store the read bytes. With this asynchronous operation together with I/O operations, the assembly of de Bruijn graphs becomes faster (Costa et al., 2015).
3.4.2. SPAdes assembler
SPAdes v. 3.8.0 is intended for both standard isolates and single-cell multiple displacement amplification bacteria assemblies, but was specially developed for single-cell data (Bankevich et al., 2012). The current version of SPAdes works with Illumina or IonTorrent sequenced reads and is able to provide hybrid assemblies, using PacBio, Oxford Nanopore, and Sanger reads. Moreover, additional contigs can be provided and used as long reads. This assembler also uses the de Bruijn graph strategy and, different from SOAPdenovo, SPAdes calculates and tests different k-mers, based on the data set provided.
3.5. Assembly quality
Some parameters are considered for assembly quality evaluation, such as N50, which is defined as the shortest contig length needed to cover 50% of the entire genome. However, the quality of the data is related to the assembler's processing, as well as, and it is noteworthy, to genomes themselves. In other words, the degree of data contiguity varies not only according to the used assembler but also between different genomes (Salzberg et al., 2011).
Thus, different methods must be used to obtain better results, and this decision does not mean a simple work. Therefore, programs like Quast v. 4.1 (Gurevich et al., 2013) evaluate the essential metrics from different assembly results, providing tables and plots making easier the comparison among different assembly strategies.
On MELC genomics, users can access Quast after run assembly. The result files can be run to check the quality of the assembly. In this MELC version, Quast runs only files created after run assembly. The new version can compare files already assembled.
4. Conclusion
The primary purpose to create MELC genomics was to make simpler and faster the genome assembler. MELC genomics environment is presented in a user friendly interface wherein users can choose and perform the module of their interest or even perform the whole assembly pipeline.
Optionally, users are able to download the file containing the results for each performed task. In addition, the genome assembly performance through MELC genomics framework does not require any previous knowledge on programming languages or on Linux operating system language by the users.
Footnotes
Acknowledgment
The author thanks Microway Incorporated for providing the computing resources used to conduct the experiments presented in this article.
Author Disclosure Statement
No competing financial interests exist.