
Abstract
Abstract
In bacterial and archaeal purine biosynthetic pathways, sixth step involves utilization of enzyme PurE, catalyzing the translation of aminoimidazole ribonucleotide to 4-carboxy-5-aminoimidazole ribonucleotide (CAIR) with carbon dioxide. The formation of CAIR takes place through an unstable intermediate N5-CAIR, played by two enzymes—N5-CAIR synthetase (PurK) and N5-CAIR mutase (PurE) that further catalyzes the reaction of N5-CAIR to CAIR. In this study, N5-CAIR mutase (PH0320) from Pyrococcus horikoshii OT3 (PurE) was considered. The three-dimensional structure of Pyrococcus horikoshii OT3 was modeled based on the structure of PurE from Escherichia coli. The modeled structure was subjected to molecular dynamics simulation up to 100 ns, and least energy structure from the simulation was subjected to virtual screening and induced fit docking to identify the best potent leads. A total of five best antagonists were identified based on their affinity and mode of binding leading with conserved residues Ser18, Ser20, Asp21, Ser45, Ala46, His47, Arg48, Ala72, Gly73, Ala75, and His77 promotes the activity of Ph-N5-CAIR mutase. In addition to molecular dynamics, absorption, digestion, metabolism, and excretion properties, binding free energy and density functional theory calculations of compounds were carried out. Based on analyses, compound from National Cancer Institute (NCI) database, NCI_826 was adjudged as the best potent lead molecule and could be suggested as the suitable inhibitor of N5-CAIR mutase.
1. Introduction
Purine biosynthesis is considered as one of the vital metabolisms of any living cell as the survival of the cell will be at peril without it. In general, adenylate and guanylate synthesis from purine biosynthesis pathway requires 10 ubiquitous enzymatic steps through conversion of phosphoribosyl pyrophosphate to inosine monophosphate (IMP; Kappock et al., 2000). Later on, several studies had led to the discovery of divergence in this pathway across different life systems. For instance, in bacteria 11 steps are utilized to synthesize inosine monophosphate (IMP) and involves two additional enzymes, both of which are absent in human (Wolan et al., 2004). These differences between bacterial and human de novo purine biosynthesis make this pathway an ideal target for the antibiotic drug research (Firestine et al., 2009). Purine(s) and pyrimidine(s) are synthesized either by salvage or de novo for the ultimate DNA and RNA production in cell. Salvage pathway utilizes the despoiled nucleotide available in cell, processes and converts into purines and pyrimidines, whereas the de novo pathway Murray (1971) utilizes higher metabolism rate to synthesize new molecules of nucleic acids. Owing to the enormous demand of purine and pyrimidines put forth by cancerous cells, it favors de novo pathway to meet their need for growth (Zhang et al., 2008; Tranchimand et al., 2011). Thus, de novo pathway has been a valid target for drug development against cancer (Guru Raj Rao et al., 2016).
Several noteworthy researches revealed variation among the enzymes across different life domains. Enzymes PurB, PurD, PurF, PurL, and PurM were unswerving, whereas PurT or PurN, PurE/PurK (class I) PurP or PurH, PurE (class II), and PurO or PurJ varied between organisms (Zhang et al., 2008). Our enzyme of interest, N5-CAIR mutase is the only carbon–carbon bond forming reaction in the pathway. PurE is an enzyme that catalyzes the unique chemical transformation of aminoimidazole ribonucleotide (AIR) to 4-carboxy-5-aminoimidazole ribonucleotide (CAIR) with the aid of carbon dioxide (CO2). Till date, PurE enzyme is present across all domains of life but in different nomenclature such as AIR carboxylase in human, PurK and PurE in bacteria, yeast, fungi and archaea were required to convert AIR to CAIR (Guru Raj Rao et al., 2016). The formation of CAIR takes place through an intermediate N5-CAIR, which is short lived and played by two enzymes, namely N5-CAIR synthetase (PurK) that converts AIR to N5-CAIR during scarce level of physiological bicarbonate, whereas N5-CAIR mutase (PurE) class I enzyme catalyzes the reversible interconversion of acid-labile N5-CAIR to 4-carboxy-aminoimidazole ribonucleotide (CAIR) with transfer of CO2 (Watanabe et al., 1989; Mueller et al., 1994; Fig. 1).
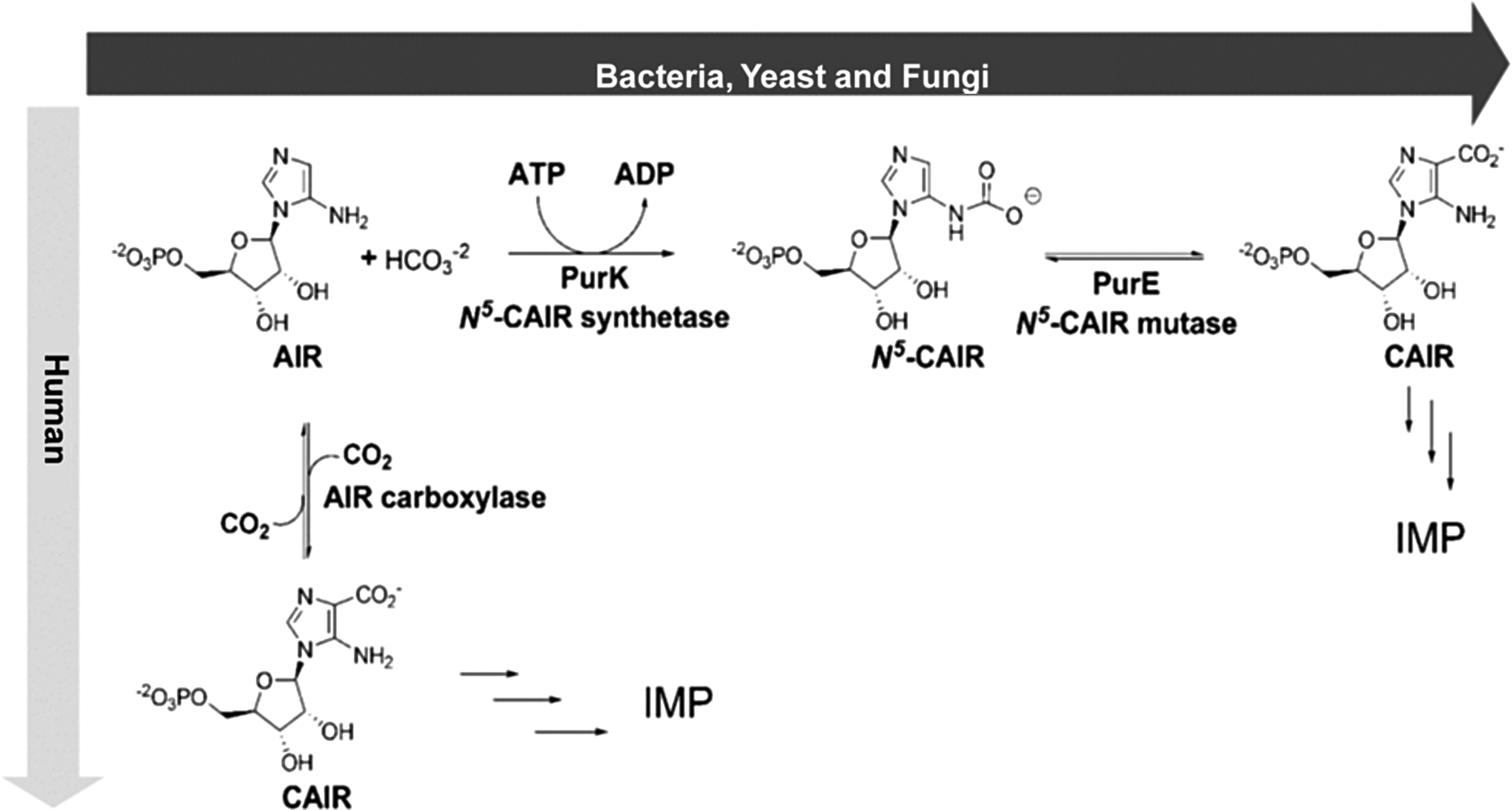
The reaction catalyzed by N5-CAIR mutase. ADP, adenosine diphosphate; AIR, aminoimidazole ribonucleotide; ATP, adenosine triphosphate; CAIR, 4-carboxy-5-aminoimidazole ribonucleotide; IMP, inosine monophosphate.
The proposed mechanism for this chemically limited reaction comprises a decarboxylation–recarboxylation order initiated by protonation of substrate at N5-CAIR carbamate (Meyer et al., 1992). Despite these differences, both enzymes are similar in sequence and perform similar enzyme chemistry (Mathews et al., 1999).
The divergence indicated earlier provides a significant rationale for investigating de novo purine biosynthesis as an antibacterial drug target due to numerous medical interventions that reveal the importance of this pathway and researchers have conducted the experiments that claim the dependence of PurE/K for the virulence (Constantine et al., 2006).
In this study, N5-CAIR mutase (PH0320) of residue length 177 amino acids from Pyrococcus horikoshii OT3 (PurE) was considered due to the absence of structure of the enzyme from this organism. The three-dimensional (3D) structure of Pyrococcus horikoshii OT3 was modeled with reference to the existing experimental structure of N5-CAIR mutase from Escherichia coli using BLASTp against Protein Data Bank (PDB; Meyer et al., 1992). The modeled structure was subjected to 100 ns molecular dynamics simulation (MDS) and the least energy structure was further screened against databases (National Cancer Institute [NCI], Zinc, Binding, Maybridge, Asinex, Specs, HitFinder, Enamine, LifeChem, and TosLab) to obtain suitable lead compounds. Moreover, the top leads based on the clustering method were subjected to induced fit docking (IFD), density functional theory (DFT), and complex molecular dynamics simulation (MDS). Till date, there are no effective inhibitors discovered for the N5-CAIR mutase and, therefore, there is an emergent need to find inhibitors against microorganisms conferring to life-threatening diseases.
2. Materials and Methods
2.1. Template selection Ph-N5-CAIR mutase
The sequence of N5-CAIR mutase from Pyrococcus horikoshii OT3 (PH0320) was obtained from the web server UniProtKB database using the accession number O58058 (www.uniprot.org). The homologous sequences were searched against Brookhaven PDB with default parameters using NCBI-BLASTp. The most homologous structure was considered as the suitable and potential template for homology modeling. The sequences of Pyrococcus horikoshii OT3 and the template were sequentially aligned using ClustalW (McFarland and Stocker, 1987). Furthermore, the sequences were analyzed by predicting the secondary structure based on the alignment using ESPript server (Thompson et al., 1994). The active site residues of Ph-N5-CAIR mutase protein were labeled with different colored legends.
2.2. Homology modeling of Ph-N5-CAIR mutase
The final sequence alignment file of target with the template sequence and the atomic coordinate file of the template structure was used to build the model using the automated homology modeling software Modeler9v11 (Robert and Gouet, 2014) by optimization of the molecular probability density function and discrete optimized protein energy (DOPE) score. A bundle of 50 models from random generation of the starting structure was calculated and among the generated models, the best model with the least root mean square deviation (RMSD) value was selected by superimposing the model with its template 2ATE from E. coli. The stereochemical properties of the modeled protein molecule were further verified by the SAVS server (Eswar et al., 2008) and the Ramachandran plot was created.
2.3. Model validation
The quality of the generated modeled structure of Ph-N5-CAIR mutase was assessed by checking the stereochemical parameters using PROCHECK (Eswar et al., 2008), Verfiy3D (Laskowski et al., 1993), and ERRAT (Colovos and Yeates, 1993) at SAVES server (Eswar et al., 2008) by providing a detailed check on the stereochemistry of a protein structure. This gives an assessment of the overall quality of the structure by comparing with other well-refined structures and also highlighted regions that may need further investigation. The significance of consistency between template and modeled N5-CAIR mutase was evaluated using ProSA server (Wiederstein and Sippl, 2007).
2.4. MDS studies of Ph-N5-CAIR mutase protein complex
MDS of the Ph-N5-CAIR mutase and complex were performed for 100 and 30 ns by GROMACS molecular dynamics package (GROningen Machine for Chemical Simulations) v4.5.6 (Van et al., 2005), which is used consistently for protein, lipid, and nucleic acid dynamics studies. In the preliminary stage, structure of the protein was refined by using simple point charge (SPC) water model and GROMOS96 43a1 force field. Topology of the ligand was generated using PRODRG server (Schüttelkopf and Van, 2004). The small molecule is taken as PDB coordinates, from which it develops different topologies for use in compatible with GROMACS. The unit cell was defined in shape of cube and was packed with SPC water molecules. The solute and the box distance were set to 1.0Å. The solvated system was neutralized with appropriate counterions were added. The gathered system was tolerable to relax through energy minimization with algorithm of steepest descent energy minimization up to maximum force reaches under 1000 kcal/(mol·nm). The complex and protein with the system was equilibrated by two steps. The first step is by simulating for 100 ps NVT ensemble (constant volume, number of particles, and temperature) to stabilize system at 310K and next step, the NPT (constant pressure, number of particles, and temperature) equilibrated till 100 ps that stabilizes the system's pressure using coupling reference pressure of 1 bar. All bond lengths were controlled by the linear constraint solver technique (Hess et al., 1997). The periodic boundary conditions and electrostatic were applied in all directions by Particle mesh Ewald.
The coulomb interactions and van der Waals interactions were restrained till 9Å and 10Å, respectively. MDS was performed for 100 and 30 ns time frame with a time stamp of every 2 fs. MDS was analyzed by Xmgrace, which is a two-dimensional plotting tool.
2.5. Protein preparation
As the crystal structure of Ph-N5-CAIR mutase protein in PDB is not available, the protein was modeled using N5-CAIR mutase from E. coli (PDB: 2ATE) Meyer et al. (1992) with the help of Modeller v9.11 software. Ph-N5-CAIR mutase protein was prepared using protein preparation wizard implemented in the Schrödinger suite (Sastry et al., 2013). H-atoms were added to the protein, including the protons necessary to outline accurate ionization and tautomeric states of residues (amino acid) such as Asp, Ser, Glu, Arg, and His. For each structure, minimization was performed with the impact refinement module, using the optimized potential for liquid simulation-2005 force field to subside steric clashes that may exist in the structures. The minimization was ended when RMSD reached a maximum cutoff of 0.30Å.
2.6. Prediction of active site and receptor grid generation
To determine the binding affinities of Ph-N5-CAIR mutase, the residues in the binding site of the developed model was predicted through the sequence-based structure alignment and binding sites of experimentally determined crystal structures across three domains of life, namely E. coli (Meyer et al., 1992), human (Li et al., 2007), and Pyrococcus horikoshii OT3. The grid box enclosed the active site residues Ser18, Ser20, Asp21, Ser45, Ala46, His47, Arg48, Gly73, Ala75, and His77 that was compared with binding sites of other N5-CAIR mutase proteins. The ligand is permitted to dock flexibly with Ph-N5-CAIR mutase within the area specified earlier. The receptor grid was generated for Ph-N5-CAIR mutase protein using receptor grid generation panel (Halgren et al., 2004; Friesner et al., 2006).
2.7. Virtual screening
Virtual screening (VS) is a high-performance computing procedure to analyze large databases of chemical compounds for screening possible lead molecules. Docking is an important computational method that predicts the best pose of one molecule to the second, bound to form a stable complex. Knowledge of this preferred conformational binding may be used to predict the strength of association or binding affinity between two molecules using a scoring function. The ligand(s) were prepared using LigPrep module in VS workflow Maestro (2015) by minimization using OLPS force field and sieving off the ligands using pharmacological properties (Singh et al., 2011).
A total of 10 different databases from NCI, Specs, Binding, LifeChemicals, Enamine, Asinex, TosLab, Maybridge, HitFinder, and Zinc were used to screen the lead molecules(s) totaling >1,500,000 compounds using Glide high-throughput virtual screening (HTVS). Glide docking uses a series of hierarchical filters to find the best possible ligand binding locations in a previously built receptor grid space. The filters include a systematic search approach, which samples the positional, conformational, and orientational space of the ligand before evaluating the energy interactions between the ligand and the protein. Further the poses generated from Glide SP were again refined in Glide XP (Friesner et al., 2006) resulted in Glide scoring function. Followed by docking, all the compounds were ranked based on the Glide score and energy. The top-ranking ligands from each database are further subjected to MDS for the assessment of protein behavior in presence of the bound complex state for a time frame of 30 ns simulation.
2.8. Docking studies
Initially, the HTVS was done using Glide v6.6 and 50% of top-scored ligands were subjected to Glide SP mode. Finally, all the ligands were subjected to XP (extra precision) docking using Glide module. All the Glide protocols were run using default parameters. An extensive search was conducted to generate all possible conformations. After this all conformations were considered for docking studies. Glide score was used to select the best conformation for each ligand. The top-ranking ligands from each database are manually clustered. Based on docking score, five best ligands were chosen for IFD, DFT studies, and further analysis.
2.9. Induced fit docking
Flexibility of both ligand and receptor in the docking study was crucial for the IFD protocol (Sherman et al., 2006). In IFD, ligands were docked into the rigid protein space using the soften-potential docking in the Glide program with a van der Waals radii scaling of 0.8 for the proteins. This resulted in generation of top 20 poses for each ligand, which was then used to sample the protein plasticity using prime program of Schrödinger suite. Residues having at least one atom within 5Å of any of the 20 ligands poses were subjected to a conformational search and energy minimization process, although residues outside the zone were fixed. In this way, the flexibility of protein was considered. The resulted 20 new receptors conformation were taken forward for re-docking. In the re-docking stage, Glide docking parameters were set to the default hard potential function. The Glide XP (extra precision) was used for all the docking calculations. The binding affinity of each complex was reported in the Glide score.
2.10. DFT calculation
DFT was applied to the top screened compounds obtained from VS and IFD studies. DFT calculates the molecular electronic features such as electron density (ED), frontier molecular orbitals (highest occupied molecular orbital [HOMO] and lowest unoccupied molecular orbital [LUMO]) and molecular electrostatic map to predict the biological activity and molecular property of the compounds (Binkley et al., 1980). To obtain the detailed molecular features, a complete geometric feature optimization was performed using hybrid DFT at B3LYP (Berke's three-parameter exchange potential and the Lee–Yang–Parr correlation functional) with basis set 6-31G** level defining flexibility to the electrons. The Poisson–Boltzmann solver was used to calculate the energy in gaseous phase. The molecular electrostatic potential (MESP) V(r) at point r, r of a molecular system with nuclear charges {ZA} located at {RA} and the ED (r) were derived.
Here, the total number of nuclei denoted as N in the molecule and the two terms refer to the bare nuclear potential and electronic contributions, respectively. The molecular electrostatic properties such as LUMO, HOMO, MESP, dipole moment, and energy were calculated by using Jaguar v8.7 module. The electrostatic potential energy of the compounds was calculated and the positive–negative regions in the compounds were denoted by color variance. The positively charged electrostatic potentials are denoted by dark blue and negatively charged electrostatic potentials by red. Orange, green, and yellow indicate the intermediate range of reactivity.
3. Results and Discussion
3.1. Structural analysis and validation of Ph-N5-CAIR mutase
The sequence and structural homology studies revealed Ph-N5-CAIR mutase (177 amino acids) have high degree of similarity with E. coli-N5-CAIR mutase. The BLASTp search of Ph-N5-CAIR mutase (PH0320) amino acid sequence against PDB database identified E. coli-N5-CAIR mutase (PDB ID: 2ATE) as the most appropriate template with good query coverage of 66% and sequence similarity of 53%, respectively. The monomer structure of Ph-N5-CAIR mutase generated by MODELLER was selected based on the least DOPE score is shown in Figure 2. The RMSD between model and template was found to be 0.52Å (Fig. 3). The structure of Ph-N5-CAIR mutase had similar features with E. coli-N5-CAIR mutase (Fig. 4) such as the central domain encompassed by four (β1–β4) in the order of β2, β1, β3, and β4 surrounded by three α helices (α2, α3, and α4) on one side and two helices α6 and α7 at the C-terminal representing a three-layer (αβα) sandwich. The central domain adopts a Rossmann fold and the 55% of the residues in the model have α-helices, 16% of residues form β-sheet, and 28% consist of other structures similar to loops. To validate the homology modeled Ph-N5-CAIR mutase structure, a PROCHECK Ramachandran plot was generated; it revealed that 94% of residues lie in core region, 5% lie in allowed region, and none found in disallowed regions (Supplementary Fig. S1).
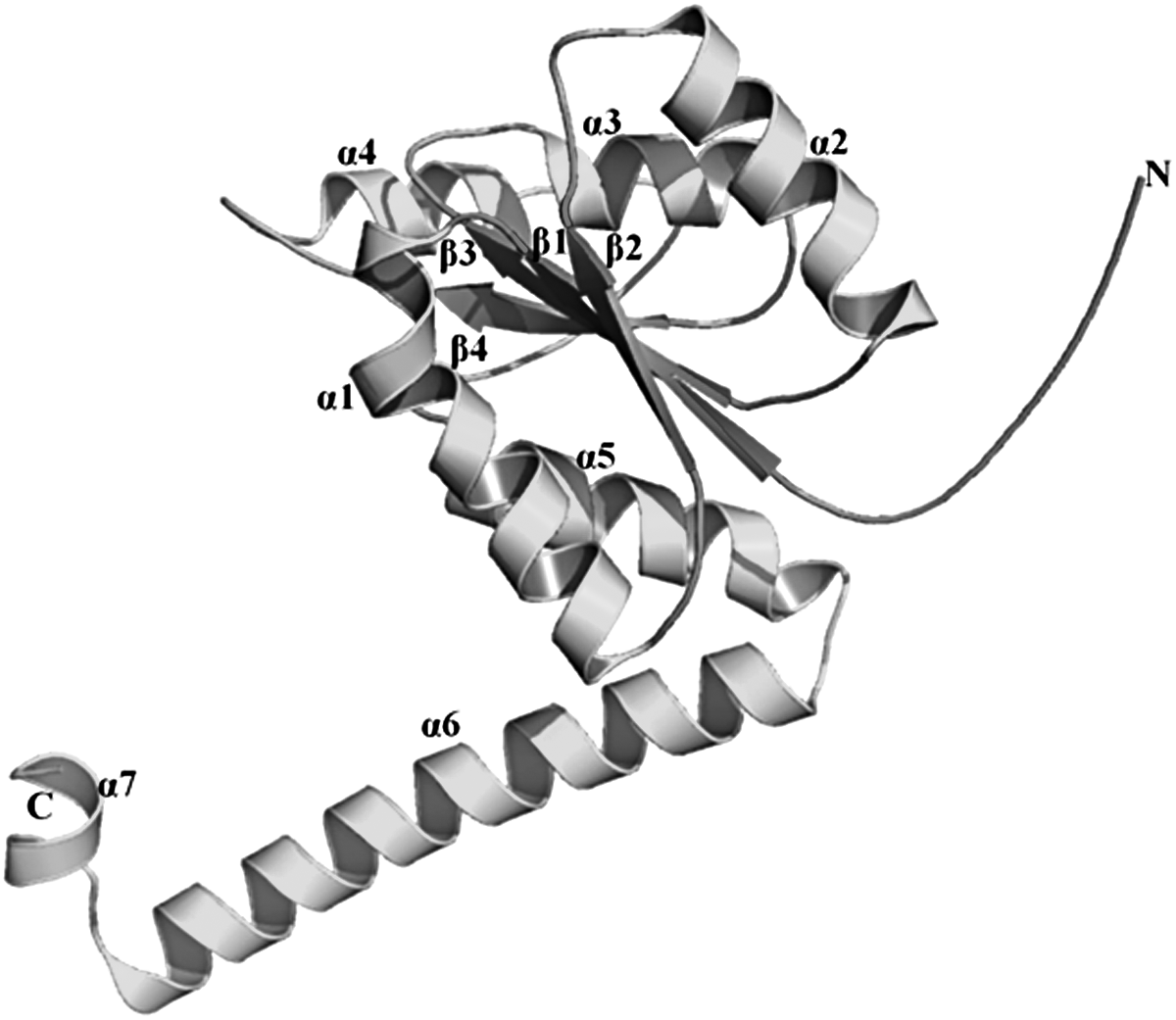
The modeled structure of Ph-N5-CAIR mutase.
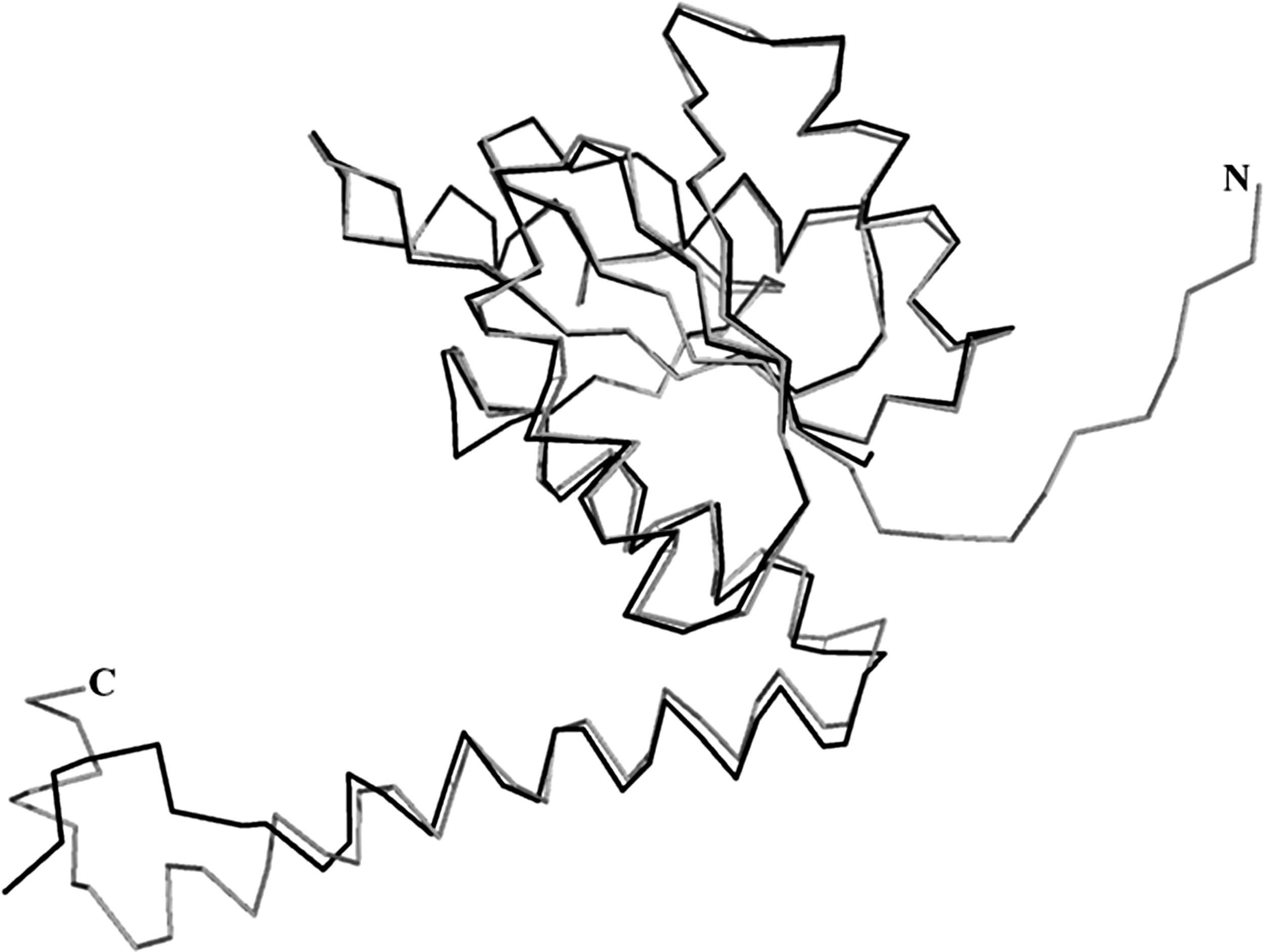
Structure comparison overlay of Ph-N5-CAIR mutase and Ec-N5-CAIR mutase by superimposition using Cα traces.
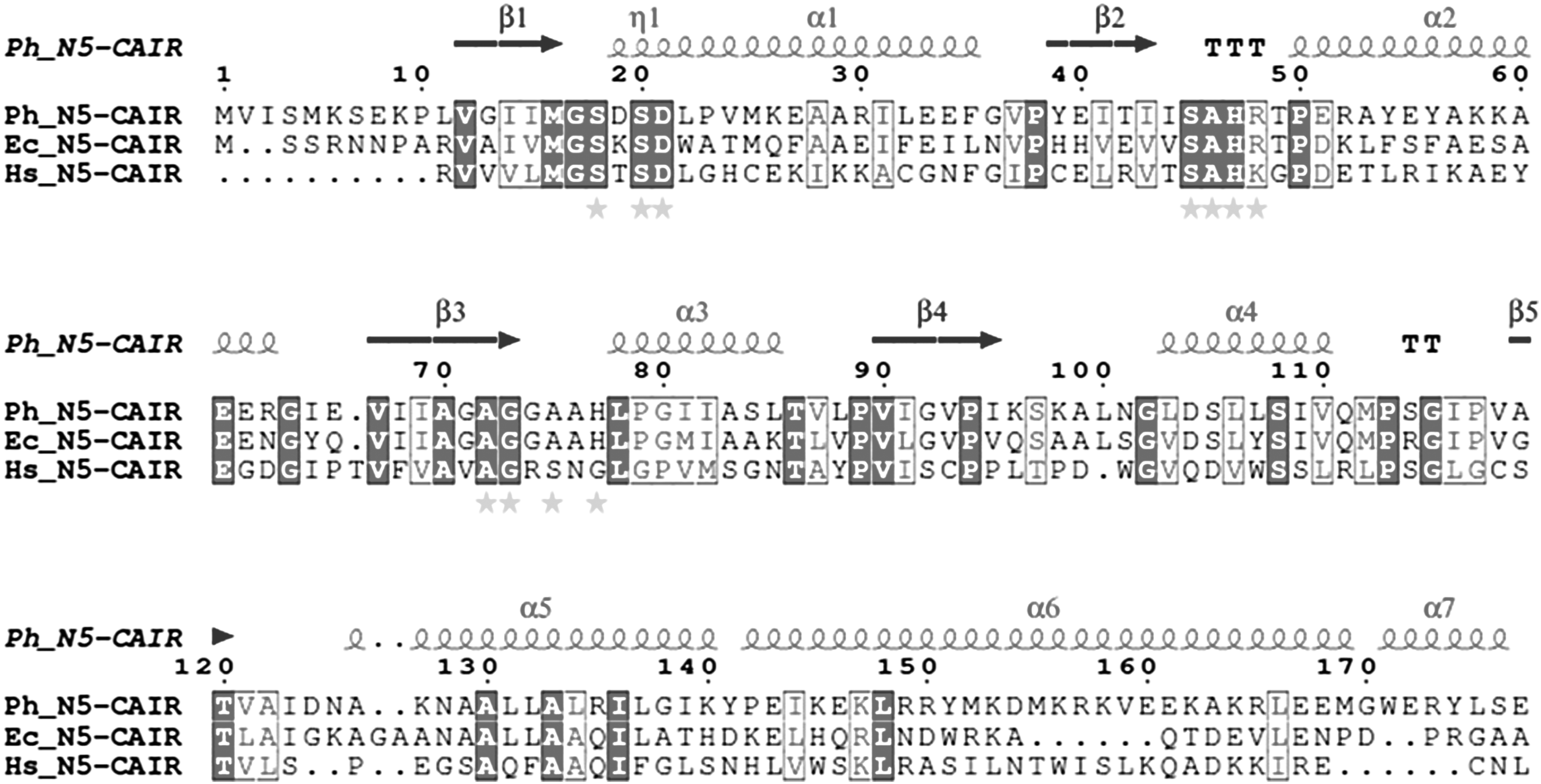
Sequence alignment of Ph-N5-CAIR mutase with Ec-N5-CAIR mutase and Hs-N5-CAIR mutase using ClustalW and the figure was produced using ESPript (Gouet et al., 1999). The secondary structural elements of Ph-N5-CAIR mutase and residue numbering are shown above the sequence alignment. Sequence identities are highlighted in dark and similarities are shown as dark letters. Both identical and similar sequences are shown in boxes. The active sites of Ph-N5-CAIR mutase, Ec-N5-CAIR mutase and Hs-N5-CAIR mutase are indicated by stars, respectively.
The Goodness factor (G-factor, obtained from PROCHECK) is a log-odds score based on the observed distribution of stereochemical parameters such as nature of covalent and whole bond distances. The G-factors obtained for modeled Ph-N5-CAIR mutase are within the permissible range as −0.16 for dihedrals, −0.10 for covalent bonds, and −0.06 overall. The modeled protein Ph-N5-CAIR mutase had negative interaction energies (−7.84) that is similar to the template, which is evident from PRoSA analysis.
3.2. Structural assessment of Ph-N5-CAIR mutase through MDS approach
To understand the stability of the modeled Ph-N5-CAIR mutase structure, MDS was performed. The modeled structure was energy minimized using GROMOS96 43a1 force field and the preliminary energy minimization was done to eliminate the high-energy intramolecular interactions, in similar way steric clashes are reduced by optimizing the overall geometry and atomic charges of the molecule. The dynamic properties of the protein motions are determined based on the topology of native contacts in biological functions. Ph-N5-CAIR mutase was solvated in a cubic box using the SPC water model system to mimic the physiological behavior of the biological system. The whole simulation was carried out for 100 ns and the system was stabilized by a number of default parameters present in the MDS studies with minor modifications. The key conditions for reviewing the stabilized systems were evaluated by plotting temperature, pressure, potential, and total energy. Investigations of these constraints have displayed that the system was stable during the MDS time frame. One of the most important criteria to analyze the stability of the protein structure was to measure the RMSDs. The deviations from the original starting structure have been measured over the course of simulation.
Overall, RMSD fluctuation was analyzed and the least energy structure of the modeled Ph-N5-CAIR mutase was retrieved from the time frame of 33rd ns. The RMSD has fluctuation of about 2Å to 3Å during the time frame of 10–30 ns, which is negligible. Initially, the protein converges as much as around the area of 3Å to 5Å (Supplementary Fig. S2A) and reaches stability from 6Å to 7Å at 50 ns timescale, which represents that the model was more stable in that region of timescale. The root mean square fluctuation (RMSF) of individual residues of Ph-N5-CAIR mutase was fluctuated up to an average of 6Å throughout the 100 ns simulations time period except residues at terminal region that possess the tendency to be flexible due to the presence of loop region compared with the rest (Supplementary Fig. S2B). As known, fluctuations up to 4Å are negligible and the loosely bound terminal residues especially at the N-terminal regions are responsible for the observed gradual increase at the end.
3.3. Structural and binding mode analysis of inhibition against Ph-N5-CAIR mutase identified from top leads
One of the most important challenges of VS workflow is the ability to find molecules with the required activity but without trivial similarity (in terms of chemical structure) to known active compounds. Thus, to determine from the 2032 potential N5-CAIR mutase inhibitors predicted by VS workflow from 10 different databases could be considered as new lead molecules that may be of use in drug design and development, we have merged 2032 top leads that can be further used for validation purposes, and the resulting set was classified into clusters. The best poses were identified using the following criteria in the given order of preference: (1) lowest binding energy in the largest sized cluster, (2) number of hydrogen bonds with the active site residues, and (3) conservation of interactions with those from predicted active site residues. Preference is given to the largest sized least binding energy cluster and then examined to verify if one or more hydrogen bonds are conserved. The top five lead compounds from each database with their Glide energy, Glide score, interacting residues, and emodel score were tabulated upon manual clustering in Schrödinger (Table 1). The top compounds in the clustering list are follows: The compound 689 from NCI database has a glide score of −8.286 kcal/mol followed by Zinc database—compounds 420 and 101 and compounds 841 and 826 from NCI database with docking score ranging from −8.044 to −7.863 kcal/mol were further selected for IFD studies among which the compound 826 from NCI database has the highest glide score of −11.618 kcal/mol and subjected to 30 ns MDS.
Top Five Compounds from Each Database with Glide Score, Energy, and Emodel Score
NCI, National Cancer Institute.
The docking results of the best five compounds post-IFD were obtained for the compounds from NCI and Zinc database with NCI ID—826 4-(3,4-dihydroxyphenyl)-3-(3,4,5-trihydroxyphenyl)-2(5H)-furanone, NCI_689 [1-(3,4-dihydroxybenzyl)-6,7-isoquinolinediol], NCI_841 [1-[(3,4-dihydroxyphenyl)methyl]-1,2,3,4-tetrahydro-6,7-isoquniolinediol], Zinc_420 2-[4-(benzylsulfanoyl)-2-chlorophenoxy] acetate, and Zinc_101 (2E)-3-{[(1R)-1-carboxylato-2(1H-indol-3-yl)ethyl]carbamoyl} prop-2-enoate are shown in Table 2. The compounds NCI_826, NCI_689, NCI_841, Zinc_420, and Zinc_101 were docked at the active site pocket of Ph-N5-CAIR and the binding pocket comprises of residues Gly17, Ser18, Asp19, Ser20, Asp21, Ser45, His47, Arg48, Gly71, Gly73, Gly74, His77, Ile95, Lys96, Ser97, Lys98, Ala99, Asp104, and Ser105.
Induced Fit Docking Results of Selected Five Active Compounds
The docking results revealed that the aforementioned share the same mode of intermolecular interactions with the N5-CAIR mutase and also have high glide score ranging from −11.618 to −8.528 kcal/mol, whereas the glide energies from −52.684 to −38.725 kcal/mol, glide emodel score ranges from −81.799 kJ/mol to upper limit of −51.633 kJ/mol, and IFD score ranging between −361.255 and −356.228 kcal/mol. In the compound NCI_826, H-bond formation involves 3,4,5-trihydroxyphenyl—hydroxyl, carboxy, and amine component of residues Ser20, Lys96, and Lys98 at a distance of 2.95Å, 2.89Å, 3.05Å, and 3.27Å, respectively. Apart from that, dihydroxyphenyl is involved in the formation of H-bond with amine and hydroxy group of residues Ser18, Asp21, and His77 with distance of 2.92Å, 2.86Å, and 2.67Å, whereas the interactions in the compound NCI_689 involves amine and carboxy group of residues Ser20, Asp21, and His77 at a distance of 2.80Å, 2.48Å, and 2.94Å. In addition, dihydroxyl groups of isoquinoline diol showed hydrogen bond interactions with Ile95 and Lys96 at a distance of 2.63Å and 2.59Å, respectively.
Whereas in the compound NCI_841, the dihydroxybenzyl showed hydrogen bond interactions with amine group of Ser20, His47 and carboxy of Asp21 at a distance of 3.03Å, 2.65Å, 2.51Å, and 3.10Å. The interactions involve with hydroxyl groups of isoquinoline diol with carboxy group of Ile95 at a distance of 2.60Å and 2.83Å. As for the compound Zinc_420, the sulfoxy group interacts with amine of Ser20 and Lys98 with a distance of 2.84Å and 3.06Å tailed by residual interactions of acetate with that of amino and amine group of Arg48 with distance of 2.65Å and 2.69Å, and lastly the interaction of amine with carboxy group of Asp21 with a distance of 3.16Å, respectively. Finally, the compound from Zinc_101 formed interactions with hydroxy and amine of Ser18 and Arg48 at a distance of 2.68Å and 2.67Å followed by the interaction with amine of indole toward carboxy group of Lys96 with distance of 3.05Å and finally the interaction of amine of Lys98 with carboxy at a distance of 2.78Å (Fig. 5). The key basis is to employ the best effective compounds from Pyrococcus horikoshii OT3 to human N5-CAIR mutase that could inhibit the onset of cancer activity, and the results reveal that the compounds from this archaeal can be thought to be applied in human to prevent the disorders caused by the expression of Ph-N5-CAIR mutase, and are provided in Supplementary Figure S6A–C.
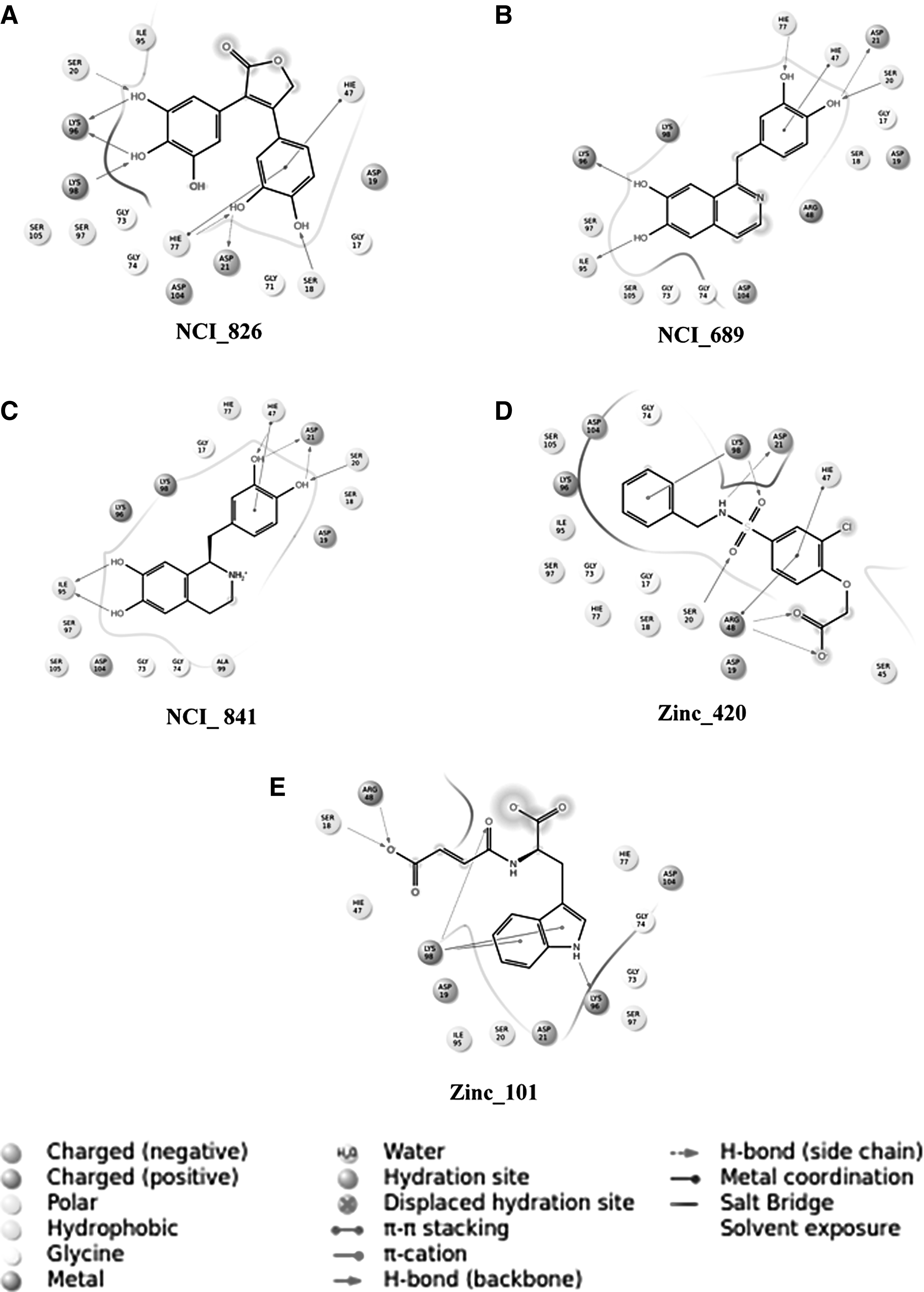
Two-dimensional schematic ligand representations of protein–ligand interactions between Ph-N5-CAIR mutase and top five hits
3.4. ADME/T predictions for Ph-N5-CAIR inhibitors
In this study, we have assessed the top five compounds from IFD to check the drug likeliness and pharmaceutical relevant properties. The QikProp module implemented in the Schrödinger software suite estimated the drug likeliness of the compound using Lipinski's rule of five and other necessary pharmacokinetic parameters such as absorption, distribution, metabolism, and excretion (ADME). The calculated ADME properties for the top five compounds are given in Tables 3 and 4. The top five compounds of molecular weight range from 355.792 to 283.283 Da, which is in accordance with Lipinski's rule of five and also has <10 hydrogen bond acceptors, <5 hydrogen bond donors, and satisfying the criteria, and are also in permissible range of assessment for to be drug-like molecules.
Principal Descriptors Calculated by QikProp of the Compounds
Molecular weight, in daltons (range for 95% of drugs: 130–725 Da).
Molecular weight of the volume.
Van der Waals surface areas of polar nitrogen and oxygen atoms.
No. of hydrogen bonds donated by the molecule (range for 95% of drugs: 0–6).
No. of hydrogen bonds accepted by the molecule (range for 95% of drugs: 2–20).
Physicochemical Descriptors Calculated by QikProp of the Compounds
Predicted octanol/water partition co-efficient log P (acceptable range: −2.0 to 6.5).
Predicted aqueous solubility; S in mol/L (acceptable range: −6.5 to 0.5).
Apparent Caco-2 permeability (nm/s; <25 poor, >500 great).
Log HERG, HERG K+ channel blockage (concern less than −5).
Apparent Madin Darby canine kidney (MDCK) permeability (nm/s; 25 poor, >500 great).
Percentage of human oral absorption (<25% poor and >80% is high).
HERG, human ether-a-go-go-related gene; MDCK.
The key constraints for assessing the absorption and distribution of drugs in human body is partition coefficient (QPlogPo/w) and water solubility (QPlogS), that ranged between approximately −0.248 to ∼2.242 and approximately −3.322 to approximately −1.839, cell permeability (QPPCaco), an important factor in regulating the drug metabolism and its transport across membranes, ranged from ∼47 to ∼98, whereas the bioavailability and toxicity were from approximately −7 to approximately −4, and the IC50 values for blockage of human ether-a-go-go-related gene K+ channels ranged from −5.484 to −1.786. The human oral absorption percentage of all compounds ranged from ∼40% to 70%. All these pharmacokinetic parameters as calculated in Qikprop 4.5 are in the adequate range well defined for human and thereby demonstrating their potential as drug-like molecules.
3.5. DFT calculations
3.5.1. Analysis of orbital energies
Structures of top five compounds obtained from the IFD process were optimized at B3LYP/6-31G** level to have a better insight in the electronic features that determine the inhibitory effect on Ph-N5-CAIR mutase inhibitors. The stereoelectronic properties (MESP) and statistically relevant descriptors were calculated. The frontier orbitals HOMO and LUMO of chemical species are associated with the donor/acceptor properties of the molecules and are furthermore indicative of the possible nucleophilic/electrophilic attack sites on the molecules that in turn indicate possible reactions the molecule might undergo in the receptor active site. Small values of both HOMO and LUMO energies, extending between −0.23 to −0.19 and −0.06 to 0.01 eV, respectively, specify the delicate nature of bound electrons (Table 5). These values were in the similar range at higher basis set of 6-31G**. Changes in the charge distribution through rapid electron transfer between HOMO and LUMO are possible for such molecules.
Frontier Orbital Energies of the Five Identified Compounds
HLG, HOMO-LUMOGAP; HOMO, highest occupied molecular orbital; LUMO, lowest unoccupied molecular orbital; QM, quantum mechanical.
Most often, the heteroaromatic rings, which contain heteroatoms such as nitrogen and oxygen, are the regions in all these compounds that can act as electron donors or acceptors to the active site of Ph-N5-CAIR mutase. For instance, in the compound NCI_826 the electron donor rings can be identified in accordance with the greatest electron density (ED) from the HOMO. In this case, the HOMO is scattered mostly over the 3,4,5-trihydroxyphenyl and in parts with the 3,4-dihydroxyphenyl and furanone, whereas LUMO is spread over the furanone region containing heteroatom such as oxygen. Docking results also showed that the regions having scattered HOMO densities are involved in interactions with crucial residues Ser18, Asp21, His77, Lys96, and Lys98. In the case of NCI_689, HOMO is composed of dihydroxybenzyl, which is involved in the interaction with residues Ser20 and His77 donates electrons and the LUMO spreads over dihydroxyl groups of isoquinoline diol showed hydrogen bond interactions with electron acceptor residues Ile95 and Lys96. Whereas the HOMO plot is scattered on dihydroxybenzyl group of compound NCI_841 as both the hydroxyl groups are involved in donating electrons to Asp21 and some of the electron densities concerning HOMO are also found in both the hydroxyl groups of isoquinoline diol with Ile95. Particularly, in this compound the hydroxyl groups are involved in donating electrons thereby establishing crucial interactions with residues.
The LUMO densities are mostly found in the regions of dihydroxyl groups of isoquinoline diol because of the electron accepting tendency of Ile95. And also, in the dihydroxyl groups of dihydroxybenzyl ring as residues Ser20 and His47 donate electrons. In Zinc_420, the HOMO densities are scattered over acetate group and is also involved in accepting the electrons from Arg48, whereas as for the LUMO is concerned the scattered densities are found in the regions of chlorophenoxy and sulfoxy because of the tendency of accepting electrons from Ser20 and Lys98 from the latter group. Lastly, HOMO is spread over indole ring of Zinc_101 as the amine group is involved in donating electrons to Lys96 and the LUMO intensities are scattered over prop-2-enoate as it accepts electrons from Ser18 and Arg48. In addition, there are HOMO orbitals located in the rings systems of the compounds which are involved in π–π, cation–π interactions with residues His47, Arg48, and His77 (Supplementary Fig. S4A–J). The effect of the orbital energies on the inhibition activities can be associated with the charge transfer, π–π, or cation–π stacking between inhibitors and aromatic amino acid residues in the binding site of Ph-N5-CAIR mutase. The result of molecular docking studies on Ph-N5-CAIR mutase inhibitors also proved the presence of such kind of interactions.
3.5.2. MESP profiles
Electrostatic potential is extensively used in characterizing molecules, particularly biomolecules, and takes distinct outcome in the biomolecular recognition and in prediction of functional sites (Kenny, 2009). Considering the discoveries comprehensively, it has been supposed that the electrostatic potential of the inhibitor also played a significant role in the binding and interaction with Ph-N5-CAIR mutase together with orbital energy and consequently influenced the inhibition effect (Daga and Doerksen, 2008; Dehez et al., 2008; Nam et al., 2008). As is well known, the electrostatic potential is defined as the interacted energy of a positively remote charge point with the nuclei and the electrons of a molecule. The 3D plots of ED and the MESP for compounds NCI_826, NCI_689, NCI_841, Zinc_420, and Zinc_101 are shown in Supplementary Figure S3A–E.
The coloring area of the surface represents the overall molecular charge distribution with the electrostatic potential. As for the compounds in this study, the electronegative potential (MESPmin) was coded with red in a range from −52.50 to −208.04 kcal/mol indicating strongest attraction, whereas the interpolated blue map represents the electropositive potential (MESPmax) of strongest repulsion varying from 165.42 to 18.89 kcal/mol. The predominance of green region in the MESP surfaces corresponds to a potential halfway between the two extremes that are indicated in red and blue, respectively.
MESP plotted onto constant ED surface for the compound NCI_826, the blue/red electropositive/negative maps are located at the 3,4-dihydroxyphenyl where the blue electropositive potential associates with the electron deficit property thus accepting electrons from the O-H of Ser20 and amine of Lys98 where an attractive force acts upon to pull the electrons toward the lacking region. In addition, the presence of blue electropotential maps describes the loss of electrons toward Lys96 due to the occurrence of repulsive oxygen atoms.
Similarly, in furanone moiety the acceptance of electrons from amide of Ser18 and His77 characterizes the attractive force between them thereby thus establishing the formation of hydrogen bond. And also, the presence of repulsive force at the hydroxyl group facilitates in the electron transfer to Asp21. For compound NCI_841, the most electronegative potentials are mapped onto the oxygen atoms of isoquinoline diol, which happens to be the electron-rich region thereby donating electrons to Asp21 and accepts electrons through strong attraction of amine group of Ser20, His47, and oxygen atom of dihydroxyl groups, whereas some part of the electronegative potential is situated at hydroxyl groups of dihydroxybenzyl that participates with giving away the electrons to the oxygen of Ile95. Whereas in compound NCI_689, appearance of negative potentials on the oxygen atoms from dihydroxyl groups of isoquinoline diol are consistent with the docking results, which recognized this region as hydrogen bond acceptor for the residues Ile95 and Lys96.
Moreover, one more prominent localized negatively charged region protruding over the dihydroxybenzene was oriented adjacent to Ser20 and His77, to be recognized as a hydrogen bond acceptor. Zinc_420 showed the most electronegative potential region (red) over the oxygen atom of the acetate group. The strong electrostatic interaction of negative potential with key residue Arg48, namely the formation of the hydrogen bond, will enhance the inhibition effect substantially. However, in the case of the compound Zinc_101 most of the negative potentials are found on the oxygen atoms especially the presence of lone pair of electrons at the prop-2-enoate is due to the strong attraction and repulsive forces between oxygen amine and Ser18 and Arg48. In addition, localization of the electronegative and electropositive maps on negatively charged oxygen of carboxylate and amide of indole accepts/donates electron from Lys98 and to Lys96 to achieve the full valence status of the interacting atoms. Owing to the accumulation of positive potential, these moieties exhibited π–π and π–σ interactions with the aromatic residues of active site. These MESP features are also in concert with VS of databases for the identification of new potent Ph-N5-CAIR mutase inhibitors. Thus, electrostatic potential of the inhibitors can play a significant role in the binding and interaction with Ph-N5-CAIR mutase together with orbital energies, and consequently influence the inhibition effect.
3.5.3. MDS of the Ph-N5-CAIR mutase-NCI_826 complex obtained from IFD
The best scoring compound from IFD, NCI_826, was further considered for MDS to analyze the structural stability in terms of RMSD, and the potential interactions for the inhibition of the molecule were identified during 30 ns time period. The RMSD, RMSF, and hydrogen bond plots of the Ph-N5-CAIR mutase-NCI_826 complex are collectively shown in Figure S5. Moreover, it is necessary to understand the interaction of docked complex during 30 ns time periods for inhibiting mechanism. In case of Ph-N5-CAIR mutase-NCI_826 complex, the backbone RMSD values were found in the average range of 0.1–0.15 nm at the initial 5000 ps time period, revealing that the complex was highly stable followed by the deviation of values that are increasing up to 0.3–0.35 nm and attain stable conformation throughout the 30 ns time period.
However, RMSD behavior showed a marked increase during 27–30 ns time period, indicating more structural variations. Subsequent analysis of RMSF by residues indicated fluctuations of the residues at the N- and C-terminal due to their flexible nature of loops (Supplementary Fig. S5B). To understand the inhibitory mode of interactions, each trajectory was observed. The potential H-bond forming residues Gly17, Ala46, His47, Pro50, Ala76, and His77 were identified throughout 30 ns at a distance of 0.5Å–1.25Å (Supplementary Fig. S5C), respectively. These residues are catalytically important residues and the ligand molecule inhibiting throughout 30 ns time period. An overall trend for bound and unbound systems indicated that systems are well equilibrated and validated the stability of complex during simulations.
4. Conclusion
In this study, Ph-N5-CAIR mutase was targeted using rational approach accorded with in silico predictive ability for preventing the proliferation and growth of cancer cells. The study harnessed the potential of structure-based modeling to explore the molecules with strong binding affinity and biologically relevant interactions. The sequence alignment and superimposition of Cα traces reveals the similar function of Ph-N5-CAIR mutase protein through conserved residues Ser18, Ser20, Asp21, Ser45, Ala46, His47, Arg48, Ala72, Gly73, Ala75, and His77 that occupy the same active site pocket postulating the similar structural fold between Pyrococcus horikoshii OT3, E. coli and Human. The 3D structure of Ph-N5-CAIR mutase was modeled through homology modeling approach. MDS was performed to assess the stability and reliability of the modeled protein. Analysis of homology modeling and MDS results indicated that the theoretical predictions were consistent with the reported structures. VS was performed from a library of compounds retrieved from several databases to obtain the potent lead molecules against Ph-N5-CAIR mutase protein. Based on the clustering and least glide score, top 50 hits were narrowed down to 5 leads, further IFD was carried out to ascertain the most promising compound NCI_826 from the NCI database.
The compound NCI_826 had the highest glide score of −11.618 kcal/mol and interacted with the key residues Ser18, Ser20, Asp21, His77, Lys96, and Lys98. DFT calculations also revealed similar results, and the compound NCI_826 had the ability to inhibit, which was reflected through the HOMO and LUMO orbitals at the interacting hydroxyl group of the residues Ser18, Ser20, Lys96, and Lys98, and least HOMO-LUMO gap value −0.160 eV indicating the stability of the compound facilitated through the strong binding interactions with the Ph-N5-CAIR mutase. The backbone scaffolds of Ph-N5-CAIR mutase—NCI_826 interactions were analyzed by RMSD, RMSF, and distance between interacting residues that explained the binding mode and stability of the complex through MDS. Moreover, in future, experimental validation will be carried out to study the curative and assessment of the compound's biological activity against the Ph-N5-CAIR mutase protein.
Footnotes
Acknowledgments
Authors are grateful to the computational facilities provided by DST, New Delhi, India (Grant No. SR/SO/BB-0079/2012 dated September 7, 2013). J.J. greatly acknowledges DST (F. No. EMR/2016/000498), ICMR (No. BIC/12(07)/2015), and BRNS (No. 35/14/02/2018-BRNS/35009) for providing financial assistance through research projects. The authors thank DST-FIST (SR/FST/LSI-667/2016), DST-PURSE Scheme (No. SR/PURSE Phase 2/38 (G)), MMRD-RUSA2-D, New Delhi [F.24-51/2014-UPolicy (TNMulti-Gen), Dept. of Edu., Govt. of India, 9/10/18], and Department of Bioinformatics, Alagappa University, Karaikudi, for the financial support and infrastructure facilities.
Author Disclosure Statement
The authors declare that no conflicts of interest exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.