
Abstract
Extrinsic, experimental information can be incorporated into thermodynamics-based RNA folding algorithms in the form of pseudo-energies. Evolutionary conservation of RNA secondary structure elements is detectable in alignments of phylogenetically related sequences and provides evidence for the presence of certain base pairs that can also be converted into pseudo-energy contributions. We show that the centroid base pairs computed from a consensus folding model such as RNAalifold result in a substantial improvement of the prediction accuracy for single sequences. Evidence for specific base pairs turns out to be more informative than a position-wise profile for the conservation of the pairing status. A comparison with chemical probing data, furthermore, strongly suggests that phylogenetic base pairing data are more informative than position-specific data on (un)pairedness as obtained from chemical probing experiments. In this context we demonstrate, in addition, that the conversion of signal from probing data into pseudo-energies is possible using thermodynamic structure predictions as a reference instead of known RNA structures.
INTRODUCTION
Many functional RNAs exhibit well-conserved secondary structures (Gardner et al., 2011). Sequence variations on large evolutionary timescales are constrained such that they preserve base pairs. These selective constraints can be identified as consensus structures in multiple sequence alignments (MSA) (Bernhart et al., 2008; Hofacker et al., 2002; Tagashira and Asai, 2022; Will et al., 2012). Covariance and mutual information measures are suitable to detect consensus base pairs directly in sufficiently large MSAs. In MIfold a column-wise score of this type is used in place of an energy model (Freyhult et al., 2005). Direct evidence for the presence of individual helices is computed in ShapeSorter using a probabilistic model (Tsybulskyi and Meyer, 2022).
Different members of an RNA family nevertheless show structural variations that occasionally also violate consensus base pairs. To account for larger structural variations, the Rfam database collects multiple families in “clans” (Gardner et al., 2011) that share common ancestry and some common structural features. The consensus structure of a few related sequences is usually much more accurate than the secondary structure prediction for a single sequence (Gardner and Giegerich, 2004; Hajiaghayi et al., 2012). However, it does not account for the structural variability within a family.
The variability within an RNA family can be accommodated by using the consensus structure inferred from an alignment of family members as a constraint for the prediction of an individual structure. The consensus structure of an MSA can be computed, for example, with RNAalifold, which extends the thermodynamic model from single sequences to alignments by averaging energy contributions over alignment columns (Bernhart et al., 2008; Hofacker et al., 2002). As an alternative, Pfold (Knudsen and Hein, 2003; Sükösd et al., 2012) uses an MSA and a phylogenetic tree to compute a global consensus structure using the Stochastic Context Free Grammar (SCFG) instead of the Turner energy model of RNA structures (Turner and Mathews, 2010). The construction of the MSA and the inference of the consensus structure are combined in structure-based alignment tools such as locarna (Will et al., 2012; Will et al., 2007). Alignment and consensus structure of an RNA family can be combined and summarized in a covariance model (CM) (Eddy and Durbin, 1994). The CM can then be aligned to a sequence of interest using cmalign (Nawrocki and Eddy, 2013), resulting again in a projection of the consensus structure. The R2DT pipeline (Sweeney et al., 2021) implements this workflow to predict and visualize RNA secondary structures in RNAcentral. The projection of the consensus structure to an individual sequence of interest can be realized using hard constraints (Lorenz et al., 2016) that enforce all base pairs of the consensus when computing the secondary structure. This procedure is implemented in the script refold.pl, which is part of the utilities distributed through the ViennaRNA GitHub site.1 This approach is problematic, however, if individual structures do not fit well with the consensus structure or if the consensus structure covers only part of the sequence.
Early implementations of both mfold (Zuker et al., 1991) and the ViennaRNA Package (Hofacker et al., 1994) used pseudo-energies to force or exclude base pairs based on external evidence and as a means of handling exceptions to the general energy model. An early example included bonus energies for extra-stable tetraloops (Mathews et al., 1999). In RNAalifold, bonus energies are used to reward co-variation of base pairs and to discourage the inclusion of nonstandard pairs into consensus structures (Hofacker et al., 2002). In TurboFold (Harmanci et al., 2011), pseudo-energies are used to incorporate conservation information in a way that avoids a fixed MSA. Instead, for each potential base pair (i, j) in sequence x, a “proclivity” is computed by aggregating the probabilities that sequence positions potentially homologous to i and j in the related sequences are paired. This “proclivity” is then converted into a pseudo-energy that is added to every secondary structure of x in which (i, j) is paired. The computation is iterated until the “proclivities,” and thus the pseudo-energies and the resulting structure predictions, converge. This amounts to position-specific modifications of the standard energy model (Turner and Mathews, 2010) for each of the input sequences.
Pseudo-energies have become the method of choice to include chemical probing data into secondary structure prediction algorithms. Position-specific stabilizing energies for paired bases, for instance, are derived from SHAPE reactivities (Deigan et al., 2009; Hajdin et al., 2013; Zarringhalam et al., 2012). Also other types of chemical probing methods, such as DMS probing (Cordero et al., 2012), and lead probing (Kolberg et al., 2023) as well as enzymatic probing methods, such as PARS (Kertesz et al., 2010; Wan et al., 2014), yield signals that are readily converted into pseudo-energies for either paired or unpaired sequence positions. In a more elaborate approach, position-specific pseudo-energies are computed indirectly by minimizing a total error estimate for both experimental signal and thermodynamic parameters (Washietl et al., 2012). SCFG-based folding algorithms, such as PPfold (Sükösd et al., 2012) and ProbFold (Sahoo et al., 2016), are similarly modified to incorporate external evidence. In Eddy (2014), a fully probabilistic approach has been suggested, which was implemented in (Deng et al., 2016).
The performance of RNA structure predictions with empirical pseudo-energy contributions is a consequence of the accuracy of the standard energy model (Turner and Mathews, 2010). As it provides an excellent approximation, the predicted energy of a biologically relevant RNA structure cannot differ drastically from the predicted ground state, albeit the predicted structure might be very different. Small modifications of the energy model are therefore sufficient to nudge the ensemble of structures toward the structures that harbor the features supported by external empirical evidence.
In this contribution we consider pseudo-energy contributions as a means of representing extrinsic information on conserved secondary structures in a systematic manner in Section 2. Despite the generally good performance of TurboFold and RNAalifold, pseudo-energies derived directly from MSAs do not seem to have been used for the folding of single RNA sequences so far. Such an approach is easy to implement in the ViennaRNA package because it features a generic interface for the position- and base pair-specific pseudo-energies (Lorenz et al., 2016). The ensemble of alignment-based consensus structures computed by RNAalifold can be used to drive pseudo-energies for base pairs in a very natural manner. We show in Section 3 that these pseudo-energies yield substantial improvements in the accuracy of secondary structure predictions. We then proceed to comparing the consensus-based information with the evidence obtainable from chemical probing data.
THEORY
Secondary structures
We consider an RNA sequence of interest X. Moreover, we denote by
A secondary structure s is compatible with X, if every base pair (i, j) in s adheres to certain base pairing rules. In the standard RNA model, only the pairs GC, CG, AU, UA, GU, and UG are allowed, whereas all other combinations of nucleotides are forbidden. For alignments
RNAalifold, for instance, requires that
Features in secondary structures
Consider the set of secondary structures Ω for a fixed sequence X. Intuitively, a feature μ in an RNA structure s is a pattern of base pairs and/or unpaired bases. Examples for features are paired positions (i), unpaired positions
Two features
Paired and unpaired nucleotides (i) and
Denote by p(s) the probability of observing a particular secondary structure s for a fixed sequence X. A feature μ is then observed with the probability
A feature μ is dominating if
Let
Proof. Suppose
Lemma 1 generalizes the observation in Ding et al. (2005) that the centroid structure of a Boltzmann ensemble, which by definition consists of all base pairs with
The dominating set of base pairs
The standard energy model for nucleic acids (Turner and Mathews, 2010) comprises contributions for stacked base pairs and loops that depend on their sequence. These energy contributions derive from a large number of precise thermodynamic measurements. The free energy
Chemical probing experiments typically provide position-specific pseudo-energies
The recursions appearing in the dynamic programming algorithms for RNA folding can incorporate certain types of pseudo-energy contributions very easily. This pertains, in particular, to unpaired positions
The conversion of extrinsic information to pseudo-energies usually uses empirical expressions (Deigan et al., 2009; Harmanci et al., 2011), often motivated by some fitting procedure. In case the extrinsic information can be quantified as the probability
For chemical probing data,
Whenever the secondary structures s can be associated with an energy(-like) quantity
The pseudo-energy
Thus, secondary structures are stabilized by pseudo-energy contributions only for dominating features.
Given an MSA, probabilities
It is also possible to convert empirical base pair propensities into corresponding feature probabilities
In Section 3 we will restrict ourselves to the base pairing probabilities provided by RNAalifold as a measure of phylogenetic conservation.
In models such as RNAalifold that computes a global consensus structure, a sequence position i appears as unpaired for two very different reasons, namely, (1) if i is unpaired in the structure of each of the sequences in the MSA and (2) if i is paired with different, and therefore in general incompatible, pairing partners in the different sequences. In other words, an unpaired position in a consensus structure may simply reflect the absence of a conserved base pair. A large value of
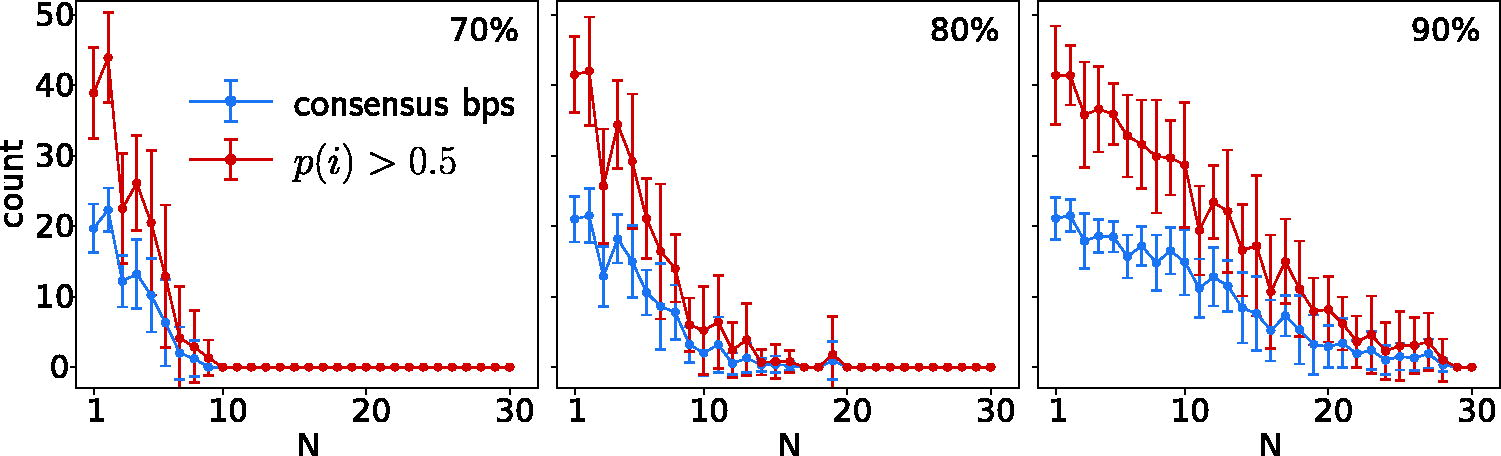
Larger MSAs of similar sequences with randomly placed substitutions (here 70%, 80%, and 90% identity with a common reference of length 80 nt) do not exhibit consensus base pairs. Both the number of base pairs in the minimum energy structure (consensus bps) by RNAalifold and the number of positions that are paired with a probability
A similar argument can be made for probing experiments. It is impossible to distinguish from the data whether a position—for which a probing reagent designed to detected, say, unpairedness does not yield a signal—is truly not unpaired or whether the signal is missing for technical reasons. From this point of view, the commonly used way of analyzing SHAPE-MaP data in terms of pseudo-energies that explicitly stabilize paired positions with low reactivity (Low and Weeks, 2010) might not be optimal.
In most situations it will be plausible to assume that an experiment can determine the presence of a feature μ, whereas the absence of μ cannot be distinguished from missing data. As there is positive support for μ only if
In the case of the RNAalifold model, therefore, we consider only the set
Datasets
In order to test and benchmark conservation-based pseudo-energies, we selected nine seed alignments
Reference structures for the focal sequences X were retrieved from RNAcentral. By definition these structures are free of pseudoknots. Positions of pseudoknots therefore remain “unpaired” in the reference structures. Details can be found in the Supplementary Data S1. The accuracy of a predicted secondary structures was quantified by Matthew’s correlation coefficient (MCC) based on the comparison of predicted base pairs and the base pairs of the reference structures.
In order to compare the information provided by chemical probing data with the impact of phylogenetic information, we used published datasets generated with different chemical probing methods for Escherichia coli. Comparisons of structure predictions with and without inclusion of probing data were restricted to transcripts with sufficient coverage.
DMS-seq comprises DMS probing data generated by Burkhardt et al. (2017) to investigate the link between operon structure, translation efficiency, and RNA structure. Data were obtained through the RASP atlas of transcriptome-wide RNA secondary structure probing data (Li et al., 2020). We selected 19 sequences of lengths
Led-Seq comprises lead probing data generated by Kolberg et al. (2023). Comparisons with reference structures were carried out for 24 sequences of lengths
SHAPE-MaP comprises SHAPE-Map data generated by Mustoe et al. (2018) for a transcriptome wide survey of mRNA structures. The dataset was downloaded from RASP (Li et al., 2020). A comparison with reference structures was possible for 12 sequences of lengths
Details on the investigated transcripts and reference structures can be found in the Supplementary Data S1.
Software
RNAsoftcons implements the following workflow:
Using a MSA
The alignment columns
RNAfold is used to compute the secondary structure of X using
The source code and implementation details for RNAsoftcons can already be accessed at https://www.github.com/ViennaRNA/softconsensus. Starting with the next release (v2.6.5), it will further be included into the ViennaRNA Python bindings available from https://pypi.org/project/ViennaRNA. A full integration into the ViennaRNA Package and more detailed example code in the official ViennaRNA Manual (https://viennarna.readthedocs.io) follow later.
The script refold.pl uses the consensus structure
For comparison we also used PETfold (Seemann et al., 2008), which identifies base pairs that have high probabilities of being conserved and of being energetically favorable and extracts the consensus structure using a maximum expected accuracy scoring. Data were processed using the PETfold web service (Seemann et al., 2011).
In order to compare with TurboFold, we extracted the individual sequences for the (possibly augmented) MSAs
The inclusion of phylogenetic information consistently improves the accuracy of the prediction (measured in terms of base pairs) from about 65% for the pure thermodynamic model to well above 90% (see Table 1). The differences between the different consensus and consensus-aware methods are small. We observe a small advantage for the approach advocated here, presumably because it uses the consensus information as efficiently as the alternative and, in addition, accounts for the small sequence-specific deviations from the consensus structure.
Performance Metrics for RNA Secondary Structure Prediction for Different Approaches That Incorporate Phylogenetic Information
See text for details on the dataset.
MCC, Mathew’s correlation coefficient; PPV, Positive Predictive Value.
Bold represents best performance.
Using RNAsoftcons, there is an improvement in accuracy in the majority of predicted structures. We consistently reach a level of at least 80% and sometimes perfect predictions. The average improvement in MCC is 0.29 [ standard deviation (SD) = 0.28) overall and 0.45 (SD = 0.23) for sequences where unconstrained folding yields an
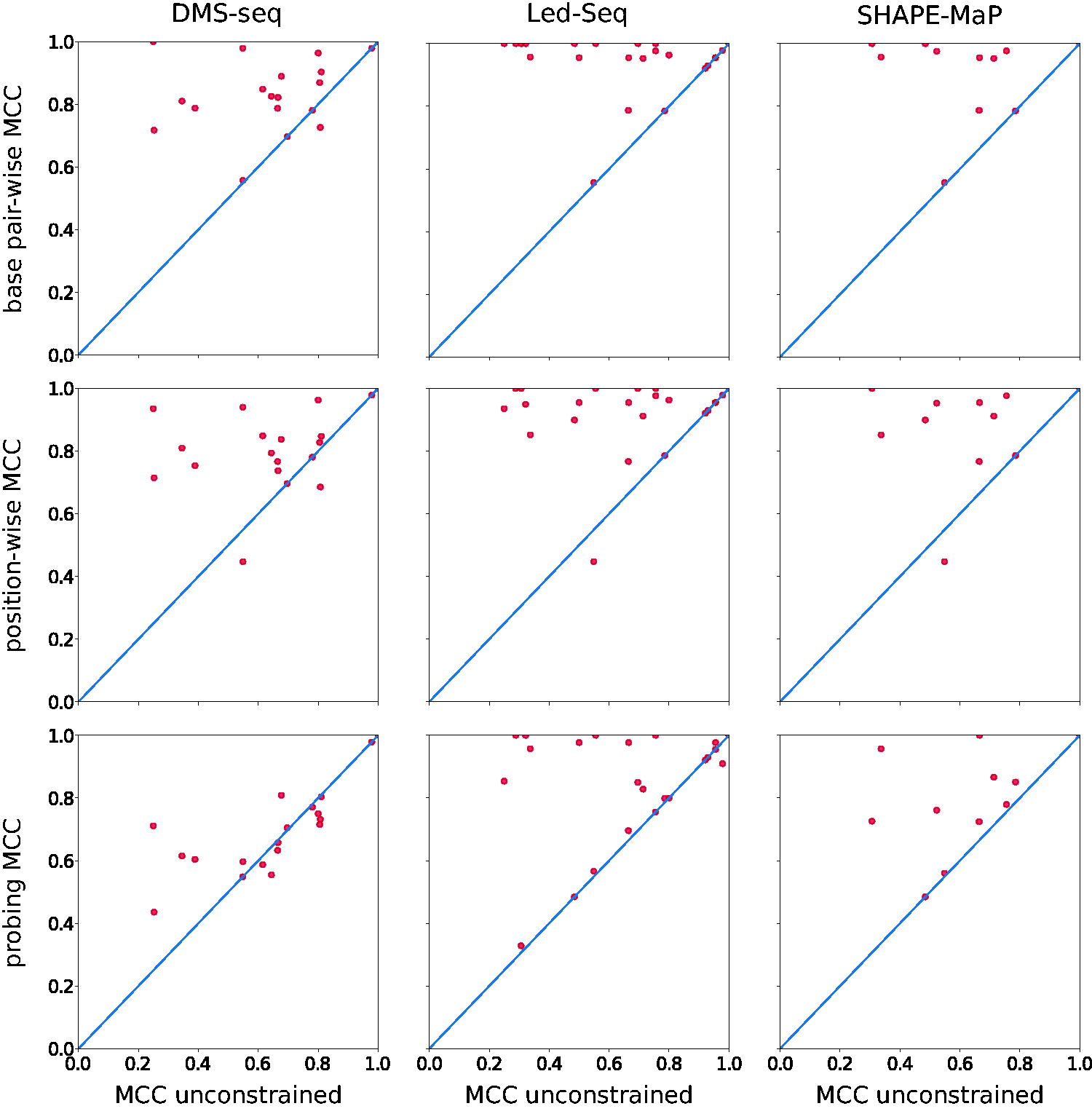
Scatter plots comparing the Mathew’s correlation coefficients (MCC) of secondary structure predictions with and without extrinsic information. Each data point is an RNA with known reference structure with sufficient coverage in one of the three chemical probing datasets (DMS-seq, Led-Seq, SHAPE-MaP). Data points in the upper-left triangle indicate an improvement because of the inclusion of pseudo-energies. Top: Structure predictions using pseudo-energies from base pair-wise phylogenetic conservation. Middle: Structure predictions using pseudo-energies from position-wise conserved pairedness. Note that the data in these two rows are independent of chemical probing and differ only in the selection of RNA transcripts. Bottom: Structure predictions pseudo-energies from chemical probing data.
In contrast to phylogenetic information, which identifies consensus base pairs, chemical probing methods, reviewed, for example, by Strobel et al. (2018), Mitchell et al. (2019), and Solayman et al. (2022), yield position-wise information and thus also position-wise pseudo-energy contributions. Knowledge on paired positions, however, does not necessarily imply knowledge of base pairs. Consider, for example, the following artificial RNA sequence, which can form two structures (even with comparable free energies) that do not share a single base pair but have exactly the same paired and unpaired positions.
By definition, the two structures cannot be distinguished my means of position-wise probing signal. This example begs the question how much structural information can be inferred from position-wise data compared with pair-wise constraints. For the phylogenetic approach outlined previously, this question can directly be answered empirically: It suffice to compute position-wise bonus energies according to Eq. (6) and to compare the accuracy of the structure prediction with the results of prediction with pseudo-energies for RNAalifold centroid base pairs.
On the entire reference set, we observed an improvement for position-wise pseudo-energies that is slightly more moderate than for base-pairing information, with an average increase of the MCC by 0.26 (SD = 0.25) overall and 0.41 (SD = 0.20) for RNAs with an unconstrained MCC < 0.8. The data are shown in this form in the preliminary version of this work (von Löhneysen et al., 2023). The middle row in Figure 2 shows the same effect separately for the reference structures in the three chemical probing data sets. Notably, the number of sequences with perfect predictions decreases in comparison with the use of base pairing information.
It is not unexpected that predictions that are fairly accurate already without extrinsic information (accuracy
Most currently available probing methods provide direct evidence only for cleavage at flexible, and thus preferentially unpaired, positions. The raw data, for example, counts of read ends, are first normalized over a locus or transcript such that the local expression level is taken into account. The normalized position-dependent probing signal Si must then be converted into a probability
The bottom row in Figure 2 shows that chemical probing data of all three datasets result in a systematic improvement of the secondary structures. However, the beneficial effect of the corresponding pseudo-energies is, in general, smaller than the inclusion of the structural conservation, even if only (un)pairedness is considered. Although this may be surprising at first glance, it is worth recalling that chemical probing methods are by no means a fail-safe method to distinguish paired from unpaired signals, that is, p(S) is not even close to a step function.
The excellent performance in particular of the pairing status inferred from phylogenetic information is at least, in part, the consequence of highly accurate, manually curated Rfam seed alignments. Pure sequence alignments recomputed with mafft reached comparable performance only when including about a dozen homologous sequences. We expect that pairwise sequence similarity will play a crucial role in particular for alignments covering only a moderate number of sequences. For details, we refer to von Löhneysen et al. (2023).
The set of RNAs with accurately known structures is rather limited, in particular if transcripts are studied in a system other than a small number of model organisms. As noted in Stadler et al. (2024), it is possible to circumvent the need for a large enough set of reference structures by defining p(S) as the average probability that a position with signal in
The predictive power of this alternative approach hinges on the accuracy of the standard energy model for RNA folding, which is far from perfect even though it is sufficient for some applications (see, e.g., Bugnon et al., 2022; Xu et al., 2012). Thus,
The key idea is now that a(S) and b(S) are less strongly dependent on the signal strength S. One can estimates a(S) and b(S) for each experimental protocol using datasets for which reference structures are readily available and then transfer these estimates to other systems, where reference structures are sparse or even unknown.
Empirically, we observe that p(S) can be estimated well using Eq. (10) from the predicted pairing probabilities and technology-dependent estimates a(S) and b(S) that positions predicted to be unpaired are unpaired and paired, respectively (see Fig. 3). In general, a(S) saturates quickly at values above 0.8, indicating that bases that are both predicted as unpaired and exhibit large chemical probing signal are very likely to be unpaired in reality. This is true even when the signal S becomes very small. On the contrary, the b(S) curve suggests that positions predicted as unpaired but showing large signals S are still unpaired with probabilities of about 60%.
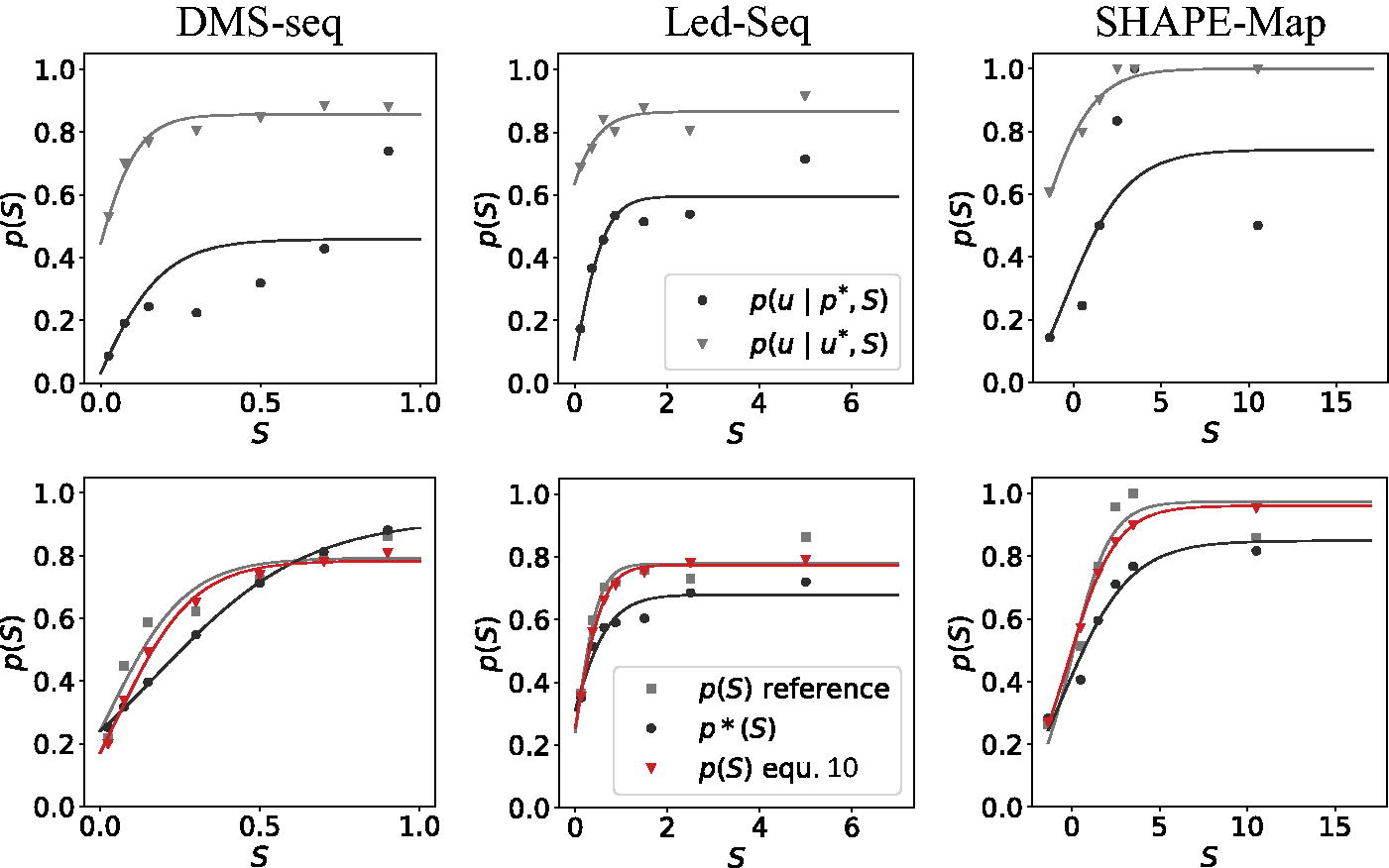
Inference of the probability p(S) that a position with probing signal S is unpaired. Top: Conditional probabilities that bases are unpaired given that they are predicted to be paired and unpaired according to the thermodynamic model and a signal strength S is observed in probing experiments. Below: Estimates of p(S) from reference structures and with the help of Eq. (10). The curves
Both phylogenetic information an chemical probing data can be used to refine and improve secondary structure predictions. Figure 4 summarizes the accuracies that are achieved for the secondary structures using various sources of extrinsic information.
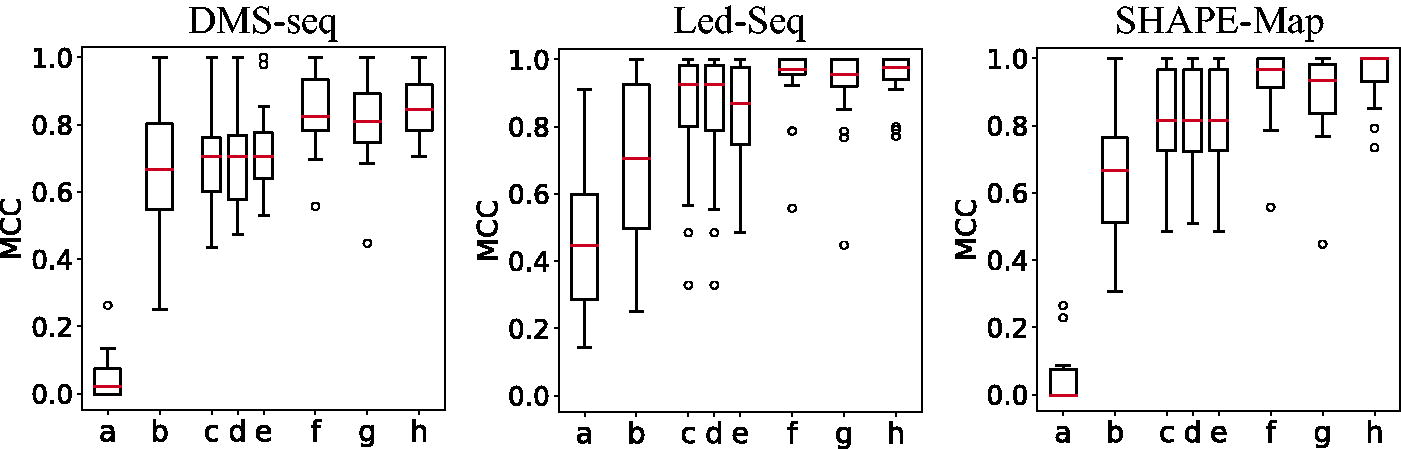
Comparison of different methods for secondary structure prediction:
The Led-Seq data provide two independent measurements for unpairedness. Although the 5′-OH data completely cover the 5′-end of each transcript, there is no signal for about the last dozen nucleotides. Complementarily, the 2′,3′-cP library covers the 3′-end and has a blind spot at the 5′-end. We computed the pseudo-energies as the average for all positions for which data were available from both libraries. The complete coverage may explain the good performance of Led-Seq in comparison with the other probing methods. We emphasize that the data shown here should not be misconstrued as a benchmarking of probing technologies. The datasets differ in age and coverage, and no effort has been made to account for such differences. The point we do wish to make here is that all of the probing methods fall short of the performance of the phylogenetic information by a quite substantial margin. At the same time, there is not much to be gained by combining probing and phylogenetic data.
Conservation of RNA secondary structures over evolutionary timescales causes sequence covariations through which consensus base pairs become detectable. Although reliable detection of covariation requires alignments of large numbers of RNA sequences, it is possible to identify likely consensus base pairs by extending secondary structure prediction algorithm to operate directly on the alignment. Embedding the sequence of interest into the alignment yields a projection of the consensus on the query. These data can then be used to improve the structure prediction specifically for the query sequence. This principle has long been known, and several tools have been available to compute consensus-based structures. Here we have shown that the conversion of the consensus structure into pseudo-energy contributions yields accurate predictions in an efficient and transparent manner. This conceptually simple approach is competitive in comparison with complex workflows, such as TurboFold, or more sophisticated statistical models, such as PETfold.
Revisiting the theoretical foundations, we observe that only positive information, that is, paired position and base pairs in the consensus structure, can meaningfully be used to derive pseudo-energy contribution. Unpaired positions, in contrast, may also arise simply from the lack of a structural consensus. The benefits of phylogenetic pseudo-energies, moreover, depend critically on the quality of the alignments used to derive the consensus base pairs. This is not surprising: alignment errors imply conflicts between predicted base pairs and thus a reduction of signal-to-noise ratio in the consensus prediction.
Information on conserved base pairs is somewhat more informative than position-wise data on the (un)pairedness, although the differences appear to be moderate. It is advisable, nevertheless, to make use of conserved base pairs, even though the use of pairedness status may seem easier from a data analysis point of view.
A closely related topic is the comparison of the relative effects, and possibly the combinations, of structural information inferred from conservation and chemical probing measurements. As a first step, we compare examples of probing data obtained with different chemistries: DMS-Map, Leq-Seq, and SHAPE-Map. In all cases we observe on average less accurate secondary structure from probing data than from conservation. It should be noted that the RNAs for which reference structures are available are expected to have conserved structures covering the entire transcript. In mRNAs and long non-coding RNAs, however, such global consensus structures cannot be expected, and hence, the consensus-based approach is not applicable.
Moreover, some structured RNAs require transitions between different conformations for their biological function. Transcriptional riboswitches, for example, operate by forming alternative terminator and anti-terminator hairpins (Helmling et al., 2018; Scull et al., 2020). These structures are conserved but incompatible with each other. The conservation-based pseudo-energies as used in RNAsoftcons cannot account for such situations. A very general probabilistic framework to incorporate such evidence into structure prediction has been proposed by Eddy (2014). In a simpler setting, Washietl et al. (2012) proposed to determine position-wise pseudo-energies such that a loss function is minimized that combines the discrepancy between observed and predicted probabilities of (un)pairedness and the magnitude of the pseudo-energy terms. Assuming sufficiently accurate data, alternative structures are suitably represented in the resulting Boltzmann ensemble. As noted, for example, by Ritz et al. (2013), conserved alternative conformations sometimes can be found as suboptimal local minima in the Boltzmann ensemble. For chemical probing data, several methods for deconvolving structural ensembles have been developed (reviewed by Spitale and Incarnato, 2023), which may also be applicable to covariance data. It is also possible in some cases to obtain direct evidence for the conservation of helices that are incompatible with the most stable structure (Tsybulskyi and Meyer, 2022). A promising framework for aggregating information on incompatible conformations, possibly supported by different sources of information, is the ensemble tree proposed by Li and Reidys (2020). Alternative structures thus remain an interesting topic for future research.
Footnotes
AUTHORS’ CONTRIBUTIONS
S.V.L.: Conceptualization, software, formal analysis, visualization, and writing. T.S.: Software. Y.V.: Software. H.-T.Y.: Software. R.L.: Conceptualization, validation, and writing. I.H.: Conceptualization, validation, and writing. P.F.S.: Conceptualization, validation, supervision, and writing.
AUTHOR DISCLOSURE STATEMENT
The authors declare that they have no conflicting financial interests.
FUNDING INFORMATION
This work was funded by the Deutsche Forschungsgemeinschaft (grant number STA 850/48-1) and by the Austrian Science Fund (grant number F-80 and I-4520).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.