
Abstract
We describe a protocol for the precise integration of exogenous DNA into user-defined genomic loci in cultured cells. This strategy first introduces a promoter and a lox site to a specific location via a Cas9-induced double-strand break. Second, a gene of interest (GOI) is inserted into the lox site via Cre-lox recombination. Upon correct insertion, a cis-linked antibiotic resistance gene will be expressed from a promoter introduced into the genome in the first step assuring selection for correct integrants. Last, the selection cassette is excised via a Flp-FRT recombination event, leaving a precisely targeted GOI. This method is broadly applicable to any exogenous DNA to be integrated, choice of integration site, and choice of cell type. The most remarkable aspect of this versatile approach, termed “CasPi” (cascaded precise integration), is that it allows for precise genome targeting with large, frequently complex, and repetitive DNA sequences that do not integrate efficiently or at all with current genome targeting methods.
Introduction
Genome modifications produced by the integration of exogenous DNA fragments have provided important insights into fundamental biological processes, and created avenues for potential cures for genetic diseases. Although the process of homologous recombination (HR) works efficiently in bacteria and yeast, the maximum targeting efficiency by HR achievable in mammalian cells when relying on fortuitous chromosome breaks is ∼1 per 106 transfected cells.1,2 Donor vectors (DV) used for classical gene targeting via HR require homology arms totaling up to 10,000 bp. 3 Efforts to improve targeting frequencies have led to the exploitation of exogenous endonucleases to generate double-strand breaks (DSBs) at desired genomic locations. These endonucleases include the programmable zinc finger nucleases (ZFNs), 4 transcription activator-like effector nucleases (TALENs), 5 and clustered regularly interspaced short palindromic repeats (CRISPR)-associated (Cas) nucleases.6,7 Elements of CRISPR, the prokaryotic adaptive immune system, represent to date the most efficient and widely applied approach for inducing a DSB at a user-determined target site. A homologous, short guide RNA (gRNA) provides the target site specificity for the CRISPR-Cas endonuclease. Cas9-induced DSBs can be repaired by HR or by the error-prone non-homologous end joining (NHEJ) pathway.8,9 Homology-directed repair (HDR) and microhomology-mediated end joining (MMEJ) result in precise repair products. 10 NHEJ predominates in most mammalian cell lines, and can result in nucleotide (nt) insertions and deletions (indels) and substitutions at the cut site. MMEJ harnesses machinery independent from HR, and requires very short homologous sequences for DSB repair, resulting in precise gene knock-ins while using a comparatively more easily constructed DV.10,11 Recently, a seminal publication introduced the Precise Integration into Target Chromosome (PITCh) system, 10 a gene-targeting strategy that uses two short homology arms flanking a gene of interest (GOI) within a DV. While this noteworthy approach has great utility, targeting the genome with a large DNA fragment using PITCh remains a challenge due to low integration efficiency. Another useful targeting strategy, homology-independent targeted integration (HITI), is based upon NHEJ repair. 12 Cas9 recognition sequences used to release a GOI from a DV are identical to the genomic Cas9 DSB integration site, but importantly, the desired integration event is designed to occur when the cleaved cut sites flanking the GOI align with the genomic DSB in the opposite orientation. In this way, NHEJ will not re-create functional Cas9 cut sites at the insertion junctions—an outcome that can occur when cleaved ends align in the same orientation.
The Cre-lox system is a proven and widely used site-specific recombinase technology that has been adapted from P1 bacteriophage for use in genetic engineering.13,14 When two lox (locus of crossover) sites are appropriately juxtaposed, Cre (cyclization recombinase) proteins can carry out DNA deletions, insertions, translocations, and inversions.13,14 The Flp-FRT system, derived from Saccharomyces cerevisiae, is a straightforward and frequently used similar technology. 15 The flippase (Flp) recombinase catalyzes DNA rearrangements via two Flp recognition target (FRT) sites. 16
When exogenous DNA is introduced into cells in the absence of a selectable marker, a laborious screening process is needed to identify recipients of a specific targeting event. The use of selectable markers, for example an antibiotic resistance gene or green fluorescent protein (GFP), greatly eases the process of identifying cells that have integrated the exogenous DNA, but does not guarantee targeted integrants. The use of positive/negative selection schemes improves the process, but random integration can still occur when attempting to perform gene targeting using a single-step targeting approach. If multiple cell lines are to be similarly produced for comparative studies of DNA sequence variants in the same genomic locus, then random integrants will have to be sifted through with each targeting attempt. In the long run, to facilitate the process of producing and selecting for a series of GOI targeted to a common specific genomic locus, we have separated DNA targeting into multiple steps in an approach termed cascaded precise integration, or CasPi. It is a two-part system. The first part entails constructing a recipient cell line containing a transgene docking site at a desired locus, and the second part is the integration of the GOI into the docking site. Our approach is built upon a number of previously described components and protocols, including recombinase-mediated cassette exchange, Cre-lox, Flp-FRT, Flp-In, PITCh, and HITI.
Briefly, CasPi first introduces a promoter and a lox71 docking site to a specific genomic location via a Cas9-induced DSB followed by NHEJ or MMEJ/HDR. The promoter contains an ATG start codon for future use. Second, a GOI is inserted into the lox71 site via Cre-mediated recombination with a lox66 site associated with the GOI. This event results in the formation of two lox sites flanking the inserted GOI: one loxP and one lox71/66 fusion. This arrangement stabilizes the GOI integration against subsequent Cre-lox deletion.17,18 Additionally, upon correct insertion of the GOI, a cis-linked antibiotic resistance gene that lacked a promoter and a start codon will gain function using the promoter/ATG introduced in the first step. 19 Last, following selection, the selection cassette is excised via a Flp-FRT recombination event,20,21 leaving a precisely targeted GOI flanked by one FRT site and one lox site (either loxP or lox71/66, depending on lox orientation in the DV [DV-PRO] and DV-GOI). If recovery of wild-type cells is required, the GOI can be removed via Cas9-mediated cleavage and a wild-type DV. The CasPi strategy combines the merits of CRISPR-Cas, Cre-lox, and Flp-FRT recombination technologies, and can efficiently target large and complex DNA sequences to specific locations in mammalian cell genomes. The protocol, from vector design through a single round of targeting and selection, can be completed in ∼10 weeks for rapidly proliferating cell lines. Each subsequent round of targeting and selection should take an additional 4 weeks.
Methods
Cell culture
HEK 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with high glucose (4.5 g/L) and
Transfection
Unless otherwise indicated in the text, 106 HEK 293T cells were transfected using a 4 μg vector DNA, 5 μL Lipofectamine 2000 (Invitrogen), and 200 μL Opti-MEM (Gibco) mix in one well of a six-well plate. mESCs were transfected using a Mouse Embryonic Stem Cell Nucleofector Kit (Lonza) and Nucleofecor 2b device (Lonza), following the manufacturer's instructions.
Polymerase chain reaction assays
There are four steps in the CasPi workflow where desired outcomes can be confirmed using polymerase chain reaction (PCR; Fig. 2; refer to Tables 1 and 2 for primer sequences and expected amplicon sizes). Quick Extract DNA Extraction Solution (Lucigen) was used for DNA extraction from a small number of cells. A DNeasy Blood & Tissue Kit (Qiagen) was used for routine DNA extractions. JumpStart ReadyMix (MilliporeSigma) was used for most PCR assays. Unless otherwise indicated, the PCR thermal profile was: 95°C for 5 min, followed by 35 cycles of 95°C for 30 s, 56–60°C for 30 s, and 72°C for 30–60 s, followed by 72°C for 10 min in a standard thermal cycler (2720; Applied Biosystems).
Oligonucleotides used in this study
Oligonucleotides are listed in the order they appear in the text. In the AAVS-block oligo, DNA Cas9 recognition sites are indicated in bold/italic, LHA and RHA are shown in bold, and an XmaI recognition sequence is underlined. The lox66-frt-lox66 block contains two lox66 sites (bold/italic), FRT sequence (bold), two BsaI and one XmaI restriction nuclease recognition sites (underlined). In hygrF2 and hygrR2, the BsaI restriction endonuclease recognition site is underlined. The Rosa-block contains Cas9 recognition sequences (bold/italic), LHA and RHA (bold), and an XmaI restriction nuclease recognition site (underlined).
gRNA: guide RNA; DV: donor vector; GOI: gene of interest; PCR: polymerase chain reaction.
Size of PCR amplicons and primers used in validation of genome integration steps
HR: homologous recombination; GFP: green fluorescent protein.
Sequencing
PCR amplicons were analyzed by Sanger sequencing at the Heflin Center Genomics Core of the University of Alabama at Birmingham. Sequencing files were analyzed aligned to reference files by SnapGene (GSL Biotech LLC).
Other DNA manipulations
Plasmids were usually constructed by standard restriction endonuclease digestion and T4 DNA ligase ligation. Calf intestinal alkaline phosphatase and T4 DNA polymerase were used when modification to DNA ends was required. All enzymes were purchased from New England Biolabs, and were used according to the manufacturer's recommendations. Oligonucleotides were acquired from Integrated DNA Technologies (IDT), and when required for cloning purposes, annealing was performed in a thermal cycler by denaturation at 95°C for 5 min followed by slow (1°C per 1 min) cooling for 70 min. Competent Escherichia coli cells Mach1 or DH5α (Invitrogen) were used for transformations. A QIAprep Spin Miniprep Kit and Plasmid Plus Kit (Qiagen) were used for plasmid isolations. All DNA sequencing data for plasmid validation and all vectors constructed in this study are available upon request.
Addgene plasmids used in this study
The pX330-U6-Chimeric_BB-CBh-hSpCas9 was a gift from Feng Zhang (Addgene plasmid 42230; http://n2t.net/addgene:42230; RRID:Addgene_42230). 6 The pBS513 EF1alpha-cre was a gift from Brian Sauer (Addgene plasmid 11918; http://n2t.net/addgene:11918; RRID:Addgene_11918). 22 pCAG-Flpe:GFP was a gift from Connie Cepko (Addgene plasmid 13788; http://n2t.net/addgene:13788; RRID:Addgene_13788). 23 pGGDestSC + ATG was a gift from Joachim Wittbrodt (Addgene plasmid 49323; http://n2t.net/addgene:49323; RRID:Addgene_49323). 24 pAAV-minCMV-mCherry was a gift from Feng Zhang (Addgene plasmid 27970; http://n2t.net/addgene:27970; RRID:Addgene_27970). 25
Imaging
Visualization of reporter gene expression was performed using an Olympus CKX41 microscope (for GFP signal) and a Nikon A1R HD confocal microscope (for mCherry signal).
Results
Design and construction of CasPi vectors
The CasPi approach requires the generation of three vectors: Vector I—“all-in-one” encoding gRNA and Cas9 nuclease; Vector II—DV (DV-PRO) and Vector III—DV with “GOI” (DV-GOI). Although the design and cloning of these constructs appears laborious, most of the work is completed only once. The same vectors can be used to generate multiple cell lines, and simple subcloning experiments are required to change either the genomic target site or GOI to be inserted into the genome. Thus, the CasPi approach is universal and flexible. To date, we have successfully used this strategy to introduce nine different GOIs into three different cell types, including mESCs and human iPSCs. Below, we provide examples and describe in detail a protocol for GOI integration into HEK 293T cells, mESCs, and hiPSCs.
Cas9/gRNA expression vector design and construction (Vector I)
After deciding to introduce a functional gene or a DNA fragment into a specific genomic locus, the nucleotide sequence of a target site is the first issue to consider. Although published reference genome information can be used, DNA sequencing of the target site in the cell line to be engineered is preferred to eliminate potential sequence divergences due to polymorphisms or mutations. The next step is to design a programmable nuclease to create a DSB at the target site, which will force the cell to recruit repair machinery to fix the DNA break. During the repair process, exogenous DNA will be integrated into the break site by HDR, MMEJ, or NHEJ. The CRISPR-Cas9 nuclease is both convenient and reliable for this purpose. There are many free web services available that provide tools to assist in the design of the gRNA required to impart target specificity to the Cas9 nuclease. Several options are listed at Guide Design Resources at http://crispr.mit.edu/. These tools typically output potential gRNAs and corresponding off-target likelihood scores.
As the first example of CasPi workflow, we chose to target the human PPP1R12C safe harbor (AAVS1) locus with the intended integration site sequence TCGATCCGCCCCGTCGTTCC
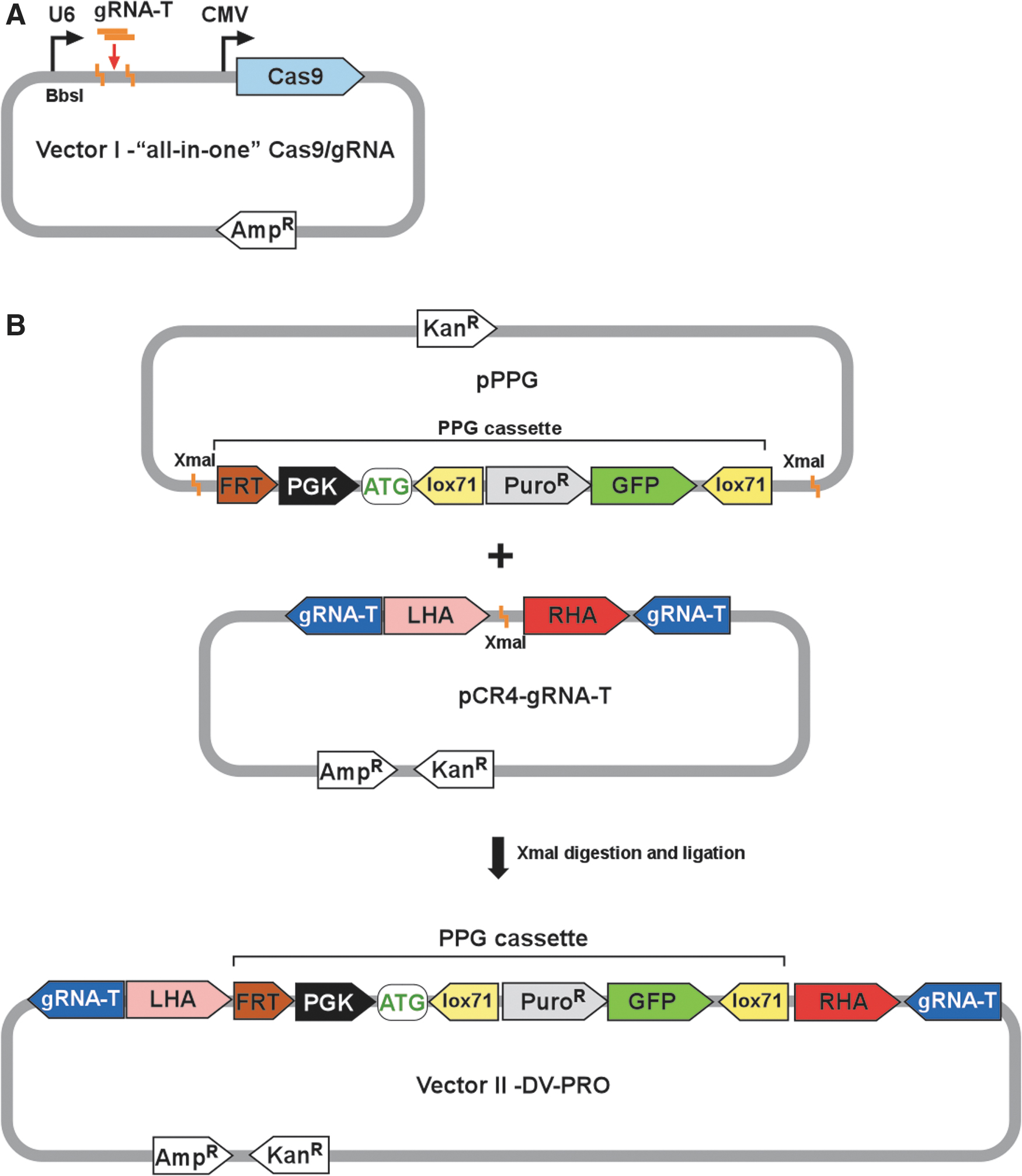
Schematic of Cas9/guide RNA (gRNA) “all-in-one” and donor vector (DV)-PRO vectors used in CasPi genome editing.
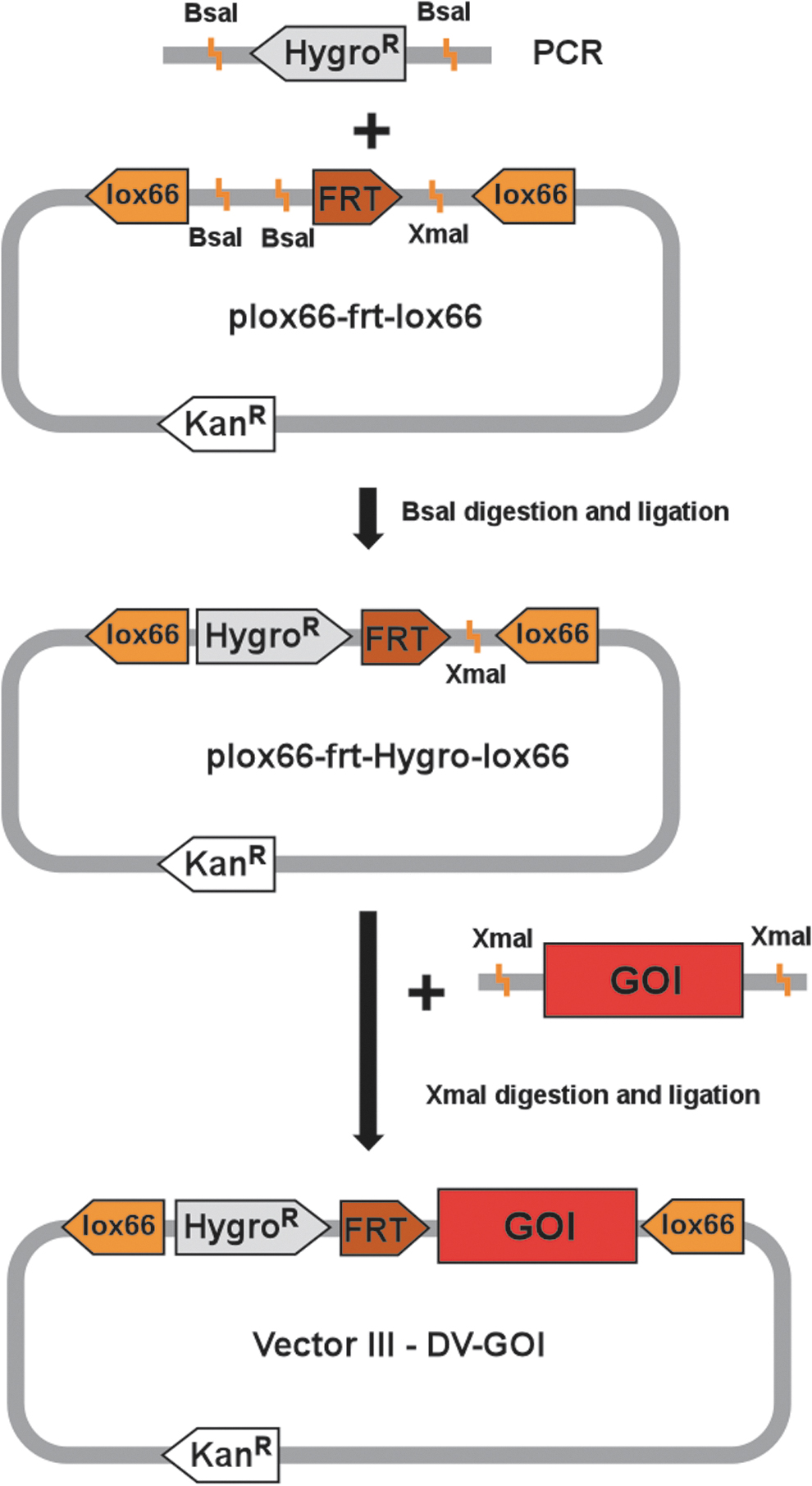
Schematic of DV-gene of interest (GOI) vector construction. The plox66-frt-lox66 vector is created by cloning a DNA block as described in the Results. The hygromycin resistance gene, lacking a promoter and ATG initiation codon, is obtained via polymerase chain reaction (PCR) and subcloned into BsaI sites of plox66-frt-lox66. In the last step, a GOI is cloned into the XmaI site of plox66-frt-hygro-lox66 vector, creating the final DV-GOI plasmid. A GOI can be obtained either by restriction digestion from a desired vector or via PCR amplification with primers containing the XmaI restriction endonuclease recognition sequence.
Design and construction of the promoter and lox71 DV (DV-PRO, Vector II)
In targeting a lox site to a specific genomic location, the Cas9/gRNA vector provides the DSB site specificity and implementation. A DV—DV-PRO—provides the lox71 docking site and a promoter-driven selectable marker. This promoter is critically important for use in a subsequent selection step.
Two components determine proper integration of a GOI into the selected genome location: the gRNA defines the location of DNA cleavage, and sequence of the homology arms facilitates HR. The “all-in-one” Vector I encodes a gRNA (see above), while Vector II (DV-PRO) contains left and right homology arms (LHA and RHA; Fig. 1B) flanking the site of the DSB. Presented here, we chose homology arm lengths of 40 bp. 27 Flanking the homology arms are 23 bp sequences (PAM included) that are identical to the genome-targeted integration site (gRNA-T in Fig. 1A and B). In this manner, in addition to cutting the genomic DNA, the Cas9/gRNA produced from the “all-in-one” vector will also cleave the DV-PRO plasmid. In DV-PRO, both cut site sequences are arranged in an opposite orientation to the genomic cut site. This prevents possible formation of two functional Cas9 cut sites (by NHEJ with complete preservation of all bases) flanking the just-integrated DNA, thereby stabilizing the integration against Cas9-mediated excision. 12 An FRT site is located just upstream of the promoter for subsequent use, and the DV-PRO vector contains a puromycin resistance selection gene (PuroR) linked via an internal ribosome entry site to a downstream GFP gene for visual screening (Fig. 1B).
For constructing DV-PRO, a DNA fragment of ∼132 bp is synthesized, using two complementary oligonucleotides (AAVS-block; Table 1), to include an XmaI restriction site for cloning, left and right arms (LHA, RHA, 40 bp each) homologous to the DSB target region, and two flanking Cas9 cut sites (in the presented example specific for AAVS1 locus). The XmaI restriction site is located between the homology arms for accepting the PPG (PGK-PuroR-GFP) cassette (Supplementary File S1). When designing the homology arm/gRNA target DNA fragment, the two oligonucleotides can also contain restriction sites at the termini to ensure the integrity of the core sequence and for subcloning. Here, A-tailing and TA cloning (Invitrogen) were used to insert the AAVS-block to produce the pCR4-gRNA-T vector (Fig. 1B). However, homology arms/gRNA target sequences can be cloned into several convenient vectors by various cloning methods.
A second part necessary to construct the DV-PRO vector is a PPG cassette composed of a PGK promoter, PuroR, and GFP (Fig. 1B). GFP is not as crucial as PuroR, but it is a convenient screening/selection tool. Lox71 sites flank the PuroR and GFP genes, and they are oriented as direct repeats to allow subsequent Cre-mediated deletion with concomitant formation of a single remaining lox71 site. Directionality of the lox71 sites is also important for final orientation of the GOI. The PGK promoter includes a Kozak consensus sequence that will provide an ATG to serve as an initiation codon after correct integration of the promoterless hygromycin selection gene in the subsequent step. Finally, an FRT sequence is located upstream of the PGK promoter allowing for excision of the selection cassette at the last step of the integration process. The PPG cassette is universal and can be used for all integration projects regardless of the target sequence or cell line. Also, the entire cassette is flanked by XmaI restriction sites (Fig. 1B) to facilitate re-cloning into a final genome targeting vector. After excision, the PPG cassette is inserted into an XmaI site of the pCR4-gRNA-T vector to generate the final DV-PRO integration vector. The cassette can be cloned in two different orientations, allowing for selection of the desired orientation of the GOI relative to the genomic sequences in the targeted locus. Correct construction of DV-PRO must be verified by Sanger sequencing.
Generation of vector encoding for the GOI (DV-GOI, Vector III)
The main feature of the DV-GOI vector is the GOI (DNA fragment) flanked by two identical lox66 sequences. Thus far, we have used the CasPi approach to integrate DNA fragments successfully, varying from 2,600 to 6,900 bp.
In the first step, two oligonucleotides designated lox66-frt-lox66 blocks (Table 1) are synthesized, annealed, and cloned into the pGGDestSC + ATG (Addgene plasmid 49323) 24 to produce plox66-frt-lox66 that contains flanking lox66 sites, two BsaI restriction nuclease recognition sites, an FRT sequence, and an XmaI restriction nuclease recognition site (Fig. 2). Next, an inactive hygromycin B resistance gene that lacks both a promoter and ATG start codon (Fig. 2; HygroR) is amplified by PCR using hygrF2 and hygrR2 primers (Table 1) and cloned into the BsaI restriction sites of the plox66-frt-lox66 vector (Fig. 2). The missing promoter and ATG codon will be provided upon correct integration of the GOI/marker into the genomic docking site, thereby activating expression of the selectable marker and rendering cells resistant to hygromycin. Importantly, the GOI can be cloned in two different orientations relative to the lox66 sequences, and preference is based on the desired experimental design. In this example, the CAG-EGFP cassette represents a GOI that is cloned into an XmaI site to generate the final Vector III DV-GOI construct (Fig. 2).
Importantly, when designing vectors with lox or FRT sites, directionality is a key consideration. Recombination between cis-linked lox site pairs or FRT site pairs in the same orientation results in a sequence deletion, while opposite orientation results in sequence inversion.
Step 1: Integration of DV-PRO into genomic target site
The first step in integration of the GOI into a predefined genome location constitutes generation of the acceptor cell line harboring an appropriate lox docking sequence for subsequent site-specific integration of the GOI. Cells are co-transfected with the DV-PRO and Cas9/gRNA vectors. Expression of gRNA and Cas9 nuclease results in three cuts—two in the DV-PRO vector, and one in the genomic target site (Fig. 3A)—thereby facilitating the integration of the short homology arm-flanked PPG cassette into the genomic target site. The DNA fragment containing the PPG cassette can integrate by MMEJ, HDR, or NHEJ cell repair processes (or a combination of these processes; see below). The integrated product of any of the three processes is suitable for subsequent steps, but HR repair results are preferred due to lack of random sequence alterations. In our experience with CasPi, HR is a predominant repair pathway.
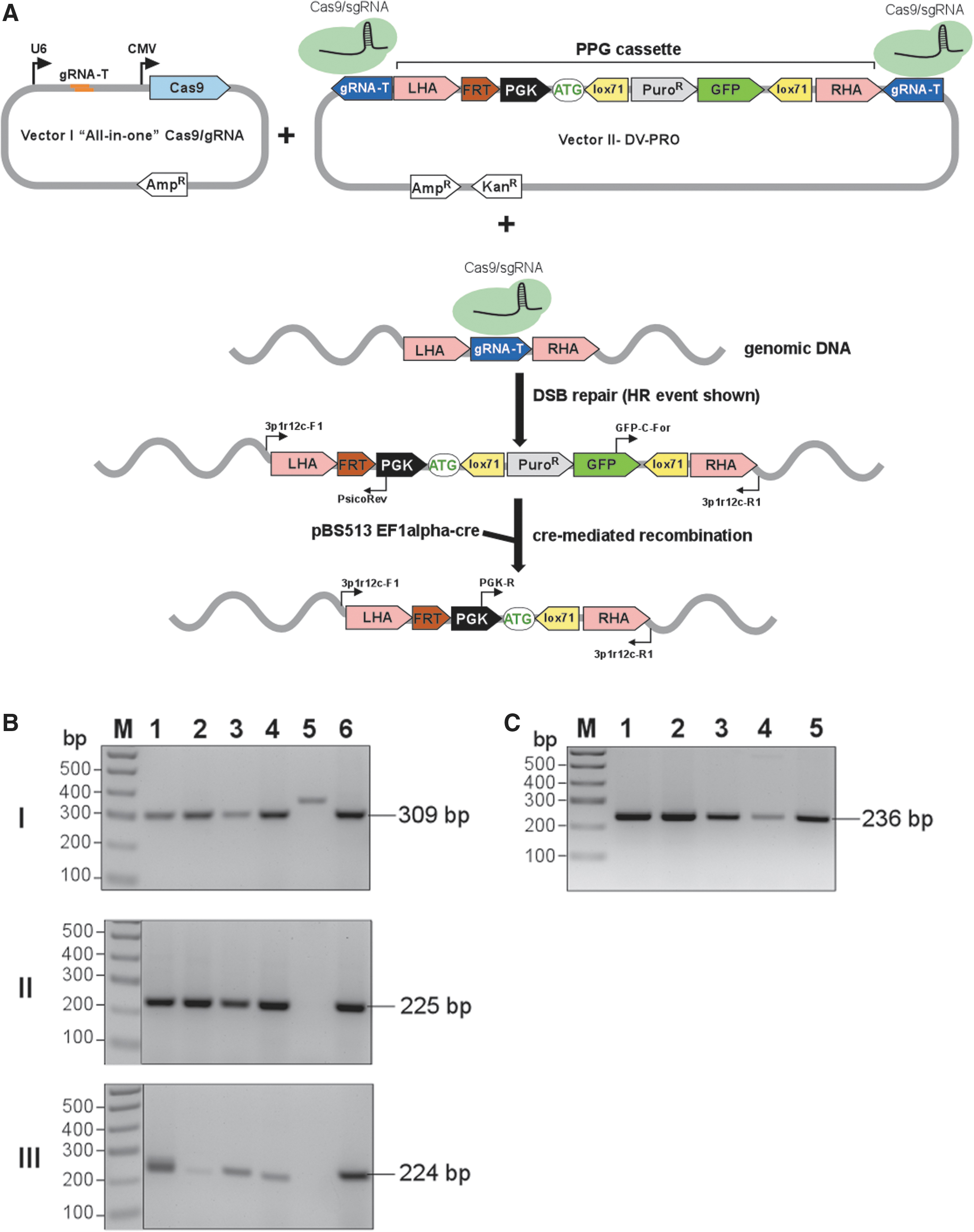
CasPi workflow—integration of DV-PRO into a genomic target site.
After puromycin selection and GFP screening, it is recommended to expand ∼10 clonally derived lines. The number of targeted colonies varies depending on cell type, transfection efficiency, and Cas9 activity, and typically ranges from hundreds (in mES or iPS cells) to thousands (in HEK 293T cells) of clones per 106 transfected cells. Outcomes of the integration process will also vary, and in our experimental setup, we prioritize clones that contain one targeted allele produced by HR and one untargeted wild-type allele. In this work, we did not distinguish between HDR and MMEJ, and we used the terms “HR” or “HDR” to indicate a HR event. We defined the preferred outcome as H-H-1: H (5′ junction via HR) -H (3′ junction via HR), and -1(single modified allele) cell line. The PPG cassette can also be inserted into the genome by NHEJ (N) at either or both ends. Thus, we can potentially find N-N, N-H, H-N, and H-H integrations at one or both alleles (e.g. N-N-2 or N-N-1/N-H-1). In our experience, after antibiotic selection, the majority of junctions (∼70%) are created via homology-mediated repair (MMEJ). More than 90% of the selected clones are edited at the target site, with ∼40–50% representing perfect knock-in clones (mES/iPS cells and HEK 293T cells, respectively).
In the experimental workflow, 48 h after transfection with DV-PRO and Cas9/gRNA vectors, 1–2 μg/mL puromycin is added to select for resistant cells. In the case of HEK 293T cells, efficiency of the integration process is high, and numerous puromycin-resistant colonies appear in the span of 7 days. Prior to the plate becoming confluent, cells should be passaged, diluted, and transferred to 96-well plates for single-cell cloning. 28 Alternatively, individual colonies can be isolated using cloning discs (Sigma–Aldrich) and cultured on 96- or 48-well plates, as we described. 29 After 1 week of culture, cells are ready for PCR analyses. A fraction of the cells are removed for DNA isolation, and the DNA is analyzed for expected integration patterns. As a selected cell line will serve for all subsequent integration steps performed by Cre/Flp recombinases, careful validation at this step is critical.
Routinely, three types of analyses are conducted: analysis of the 5′ and 3′ junctions, and testing the integrity of the second unmodified allele (see Tables 1 and 2 for PCR primer pairs and expected amplicon sizes). For upstream 5′ junction analysis, the genome 3p1r12c-F1 and vector PGK promoter PsicoRev primer pair will produce a 309 bp PCR amplicon from an H-type (HR) integration (Fig. 3B, panel I, clones 1–4 and 6) and a 366 bp amplicon from an N-type (NHEJ) with complete preservation of cleaved ends (40 bp LHA +17 bp residual Cas9 recognition sequence; Fig. 3B, panel I, clone 5). However, any indels will alter this size of the amplicon. The HR is always precise and parallels the reference sequence; NHEJ typically results in indels. Therefore, Sanger sequencing of the PCR product is recommended.
For downstream 3′ junction analysis, the vector GFP-C-For and genome 3p1r12c-R1 primer pair will produce a 225 bp PCR amplicon from an H-type integration (Fig. 3B, panel II, clones 1–4 and 6) and a 271 bp amplicon from an N-type with nucleotide preservation (40 bp right HA +6 bp residual Cas9 recognition sequence; differences in residual Cas9 sequence length results from orientation of the gRNA recognition sequences). Indels will alter this size of the PCR product.
For detection of an unmodified genomic allele, the 3p1r12c-F1 and 3p1r12c-R1 primer pair should produce a 224 bp PCR amplicon (Fig. 3B, panel III). Cas9 cleavage with subsequent indels formation will alter this size (Fig. 3B, panel III, clones 1 and 3). Integration of the DV-PRO sequence on the second allele, large deletions that extend beyond primer binding sites, or translocations can result in no amplicon formation (Fig. 3B, panel III, clone 5). Such clones are eliminated from further analyses. A second copy integration can also be detected using a qPCR approach. Overall, ∼90% of clones are edited on a single allele, and homozygous insertions are rare. The sequence of the second allele should be determined by DNA sequencing (Supplementary File S2). Lastly, due to the use of the Cas9 nuclease at this step, off-target analysis of selected clones should be performed.
Following these PCR analyses, we selected H-H-1 (single copy via HR) edited cells with no indels on the second unmodified allele for subsequent integration steps.
Step 2: Removal of selection cassette to obtain a universal recipient cell line
Removal of the puromycin and GFP genes integrated in Step 1 is required to enable selection for subsequent insertion of the GOI. Deletion is accomplished via Cre recombinase acting on the two lox71 sites that flank the Puro-GFP cassette and, importantly, leaving behind a single lox71 docking site (Fig. 3A).
The H-H-1 cells with an integrated PPG cassette from Step 1 are transfected with a Cre recombinase expression vector. Here, we used pBS513 EF1alpha-cre (Addgene plasmid 11918), 22 but any plasmid expressing functional Cre can be utilized. Progressive appearance of non-fluorescent cells can be observed using routine microscopy examination of the plates. When ∼50% of cells on the plate do not express GFP, splitting and single colony selection is performed, as described above, followed by DNA extraction and PCR analysis. For upstream 5′ junction reconfirmation, the genome 3p1r12c-F1 and vector PGK promoter PsicoRev primer pair should produce a 309 bp PCR amplicon. For downstream 3′ junction reconfirmation and Puro-GFP deletion, the vector PGK-R and genome 3p1r12c-R1 primer pair should produce a 236 bp amplicon (Fig. 3C). PCR products should be also verified by DNA sequencing. In addition, selected cell clones should lack GFP fluorescence and become sensitive to puromycin treatment. The Cre-mediated excision is a very efficient process, reaching ∼70%, irrespective of selection.
An established cell line contains a genome-targeted lox71 docking site and PGK promoter with Kozak consensus sequence/ATG start codon, and can be used as a recipient for GOI targeting. Importantly, this cell line becomes a universal acceptor of any GOI to be integrated in this locus.
Step 3: Integration of a GOI cassette into the genome-targeted lox docking site
The main feature of the DV-GOI vector is the GOI flanked by two identical lox66 sites. When co-transfected with a Cre expressing vector, the DV-GOI prokaryotic sequences are deleted by Cre recombinase acting on the two lox66 sites, and a DV-GOI minicircle is created that contains a single lox66 site (Fig. 4A). The DV-GOI minicircle will subsequently integrate into the genome via Cre-mediated recombination between the vector lox66 site and the genome-targeted lox71 docking site (in the established cell line; see above and Fig. 3A). Integration of the GOI is stabilized due to formation of wild-type loxP and chimeric lox71/66 sites (Fig. 4A). These two lox sites are inefficient for further Cre-mediated recombination, and effectively prevent unwanted excision of the GOI from the docking site (Fig. 4 and 13 ). The DV-GOI vector contains an inactive hygromycin resistance gene that lacks both a promoter and an initial ATG start codon. These missing elements will be provided in cis upon correct integration of the GOI/marker into the docking site of the acceptor cell line, thereby activating expression of the antibiotic resistance gene.
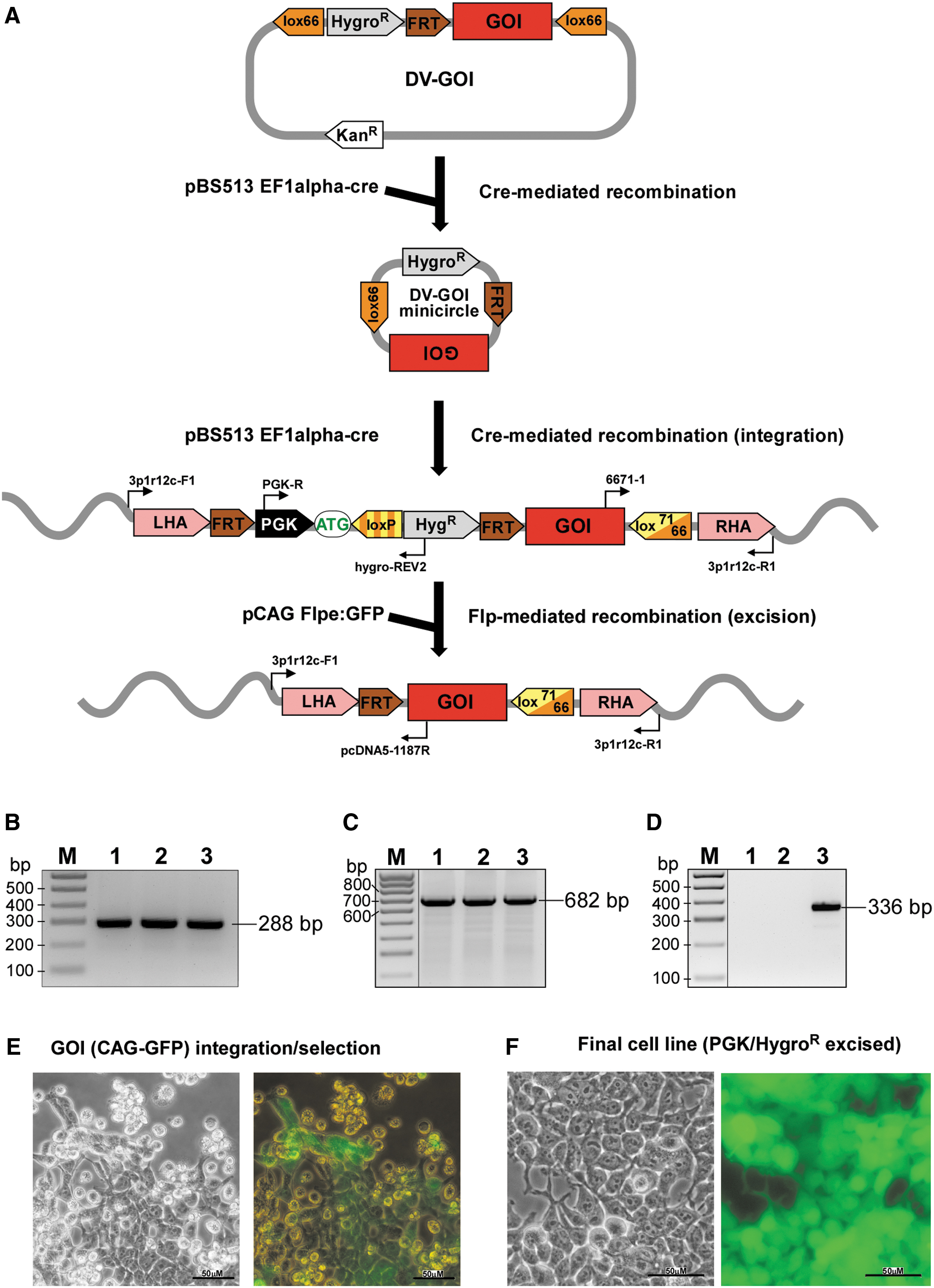
CasPi workflow—integration of a GOI cassette into the genome-targeted lox docking site.
For integration of the GOI, the DV-GOI and Cre-expression vectors (pBS513 EF1alpha-cre) are co-transfected into the docking cell line. After 2–3 days of culturing, hygromycin selection is applied (100–200 μg/mL), and antibiotic-resistant colonies appear after ∼10 days. Typically, ∼10 colonies are analyzed to validate the production of targeted promoter/hygromycin GOI cell lines. At confluence, a fraction of cells from each clone are collected for PCR analyses to confirm correct targeting (Fig. 4B–D). The 5′ junction PCR primers (PGK-R and hygroREV2) should produce a 288 bp amplicon (Tables 1 and 2; Fig. 4B), while 3′ junction PCR primers (6671-1 and 3p1r12c-R1) amplify a 682 bp fragment (Tables 1 and 2 and Fig. 4C). Amplicon sequences should be verified by Sanger sequencing. In this example, GOI encodes for eGFP, allowing for fluorescence as an additional selection criterion (Fig. 4E and F). This is a critical step in the CasPi strategy. Its efficiency depends on multiple factors, including cell type, transfection efficiency, cell survival, and so on. Typically, we obtain 10–20 clones per 106 transfected cells. However, this relatively low efficiency is offset by very high precision (∼100%) of the GOI integration.
Step 4: Removal of the selection cassette to obtain the GOI cell line
The cell line obtained in Step 3 and harboring the GOI also contains a hygromycin selection cassette. In many circumstances, the integrated GOI with selectable marker configuration is the final step in developing a cell line of interest. However, for some applications, exogenous DNA sequences extraneous to the GOI require removal to minimize their potential influences on GOI or other cellular functions. The selectable marker in the GOI cell line is designed to be conveniently removed, as FRT sites flank the promoter/selectable marker (Fig. 4A). Recombination between FRT sites will delete the marker, and result in a final cell line containing a site-specific, genome-integrated GOI flanked upstream by a single FRT site and downstream by a single lox site (loxP or lox71/66; Fig. 4A).
To remove the hygromycin selection gene, cells are transfected with a plasmid expressing Flp recombinase, for example pCAG Flpe:GFP plasmid (Addgene plasmid 13788). 23 Two days post transfection, cells are dissociated for clonal expansion as described above. Established clones are verified by PCR using the 3p1r12c-F1 and pcDNA5-1187R primer pair (Tables 1 and 2). Correct removal of the PGK promoter and hygromycin resistance gene will result in a 336 bp amplicon (Fig. 4D). It is recommended to confirm the integrity of the target locus with GOI by DNA sequencing. The Flp recombinase-mediated removal of the selection cassette is efficient, with ∼60% of clones exhibiting proper excision of the hygromycin resistance gene.
Integration of GOI into mouse ESCs and human iPSCs
The HEK 293T cell line is known for its relative ease of transfection, culturing, and clonal expansion. To test our approach with more challenging cells, we chose to target the Rosa26 locus in mESCs. A Cas9/gRNA “all-in-one” vector was constructed as described for HEK 293T cells, and included a gRNA transcription template for the Rosa26-specific integration site (ACTCCAGTCTTTCTAGAAGA). Two oligonucleotides (mouseRF and mouseRR; Table 1) encoding the gRNA were annealed and cloned into the “all-in-one” vector. The DV-PRO vector incorporates a Rosa-block (Table 1) that contains LHA and RHA (40 bp each) flanked by Cas9 gRNA targets and an internal XmaI site into which the PPG cassette is subcloned.
The Cas9/gRNA and DV-PRO vectors were co-transfected into mESCs, and after puromycin selection, 12 colonies were analyzed for 5′ and 3′ junction amplicons (Fig. 5A). Amplicon sequences were verified by Sanger sequencing. Of the 12 colonies, five (42%) displayed H-H-1 type integrations and were preserved as PPG cell lines.
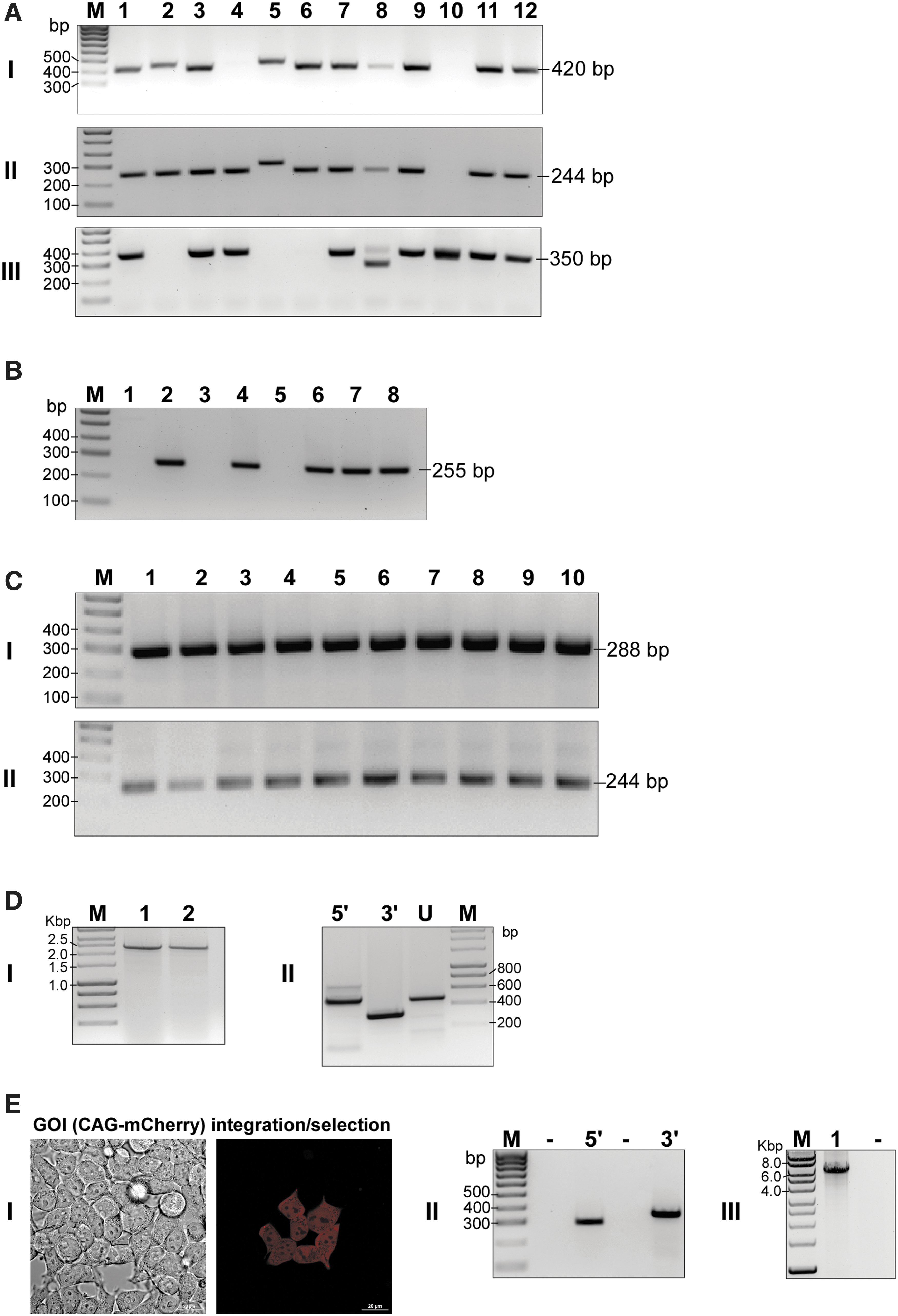
PCR analysis of CasPi-mediated GOI integration in mouse embryonic stem cells, human induced pluripotent stem cells (iPSCs), and HEK 293T cells.
PPG cells were transfected with a Cre-expressing vector to remove the marker cassette and create the genome-targeted lox71 docking site. Clones were analyzed for the predicted size PCR amplicons to ensure removal of the cassette and integrity of the remaining DNA using exactly the same strategy as described for HEK 293T cells, with the exception of genome-specific primers used (MusRosaFOR and MusRosaREV; Tables 1 and 2). PCR products were verified by Sanger sequencing. Five of eight (63%) clones analyzed were correct and were preserved as recipient cell lines for GOI targeting (Fig. 5B). Finally, a DV-GOI vector containing a PGK-GFP GOI was co-transfected with a Cre-expressing vector into recipient cells. Successful targeting of the GOI cassette to the lox71 docking site was verified by PCR and DNA sequencing (Table 2). Ten out of ten (100%) clones analyzed demonstrated correct integration of the GOI, demonstrating the robustness of this approach (Fig. 5C).
Finally, we utilized the CasPi technique to introduce a long repetitive sequence containing ∼700 GAA repeats into the AAVS1 locus in human iPSCs (Fig. 5D) and a large ∼7 kbp DNA fragment expressing the mCherry reporter into the AAVS1 locus in HEK 393T cells (Fig. 5E and Supplementary File S3). Integration of ∼700 GAA triplets was confirmed by PCR using primers flanking the repeat tract in two independent iPSC clones (Fig. 5D, panel I). Validation of 5′ and 3′ junctions and the status of the unmodified allele was performed, as described above (Fig. 5D, panel II). This proof-of-concept experiment demonstrates the utility of the CasPi approach for integrating “difficult” DNA sequences into a fastidious cell type. Similarly, successful integration of the mCherry reporter, derived from pAAV-minCMV-mCherry (Addgene plasmid 27970) 25 was confirmed by imaging of mCherry expressing cells during the integrant selection process (Fig. 5E, panel I) as well as by 5′ and 3′ junction analyses (Fig 5E, panel II; expected sizes of 288 and 335 bp, respectively). The primer pair PGK-R/hygroREV2 was used to validate the 5′ junction, and the primers Ln1-F and AAVs1-cel-I R amplified the 3′ junction (Tables 1 and 2). Amplification of the 5937 bp fragment of the ∼7 kbp mCherry encoding insert (Fig. 5E, panel III) was performed using PGK-R and GFP-C-For primers (Tables 1 and 2). These results show that the CasPi approach can be utilized to integrate large DNA fragments efficiently into the genome.
Discussion
In this report, we present a new genome-targeting strategy (CasPi) to integrate large exogenous DNA sequences reproducibly into a user-determined genome locus. By combining merits of different genome modification methods, including Cas9/gRNA editing and Cre/Flp recombineering, into a multistep, universal workflow, we can achieve desired genome modifications precisely and efficiently.
Classic gene targeting was carried out by introducing linearized, exogenous DNA into a cell and allowing cellular processes to mediate the integration of that DNA into the genome via HR with endogenous DNA. The conventional strategy typically involved constructing a targeting vector containing up to 10 kbp of total homology (e.g., 5 kbp each arm) to the targeted genome location. 3 Even so, more often than not, the targeting DNA would undergo random rather than targeted insertion. Considering that a major obstacle to success was a very low chance of a fortuitous chromosomal break near the intended insertion site that would stimulate recombination machineries to incorporate the targeting DNA, finding the means to create site-specific DSBs in the genome was a major breakthrough in site-specific genomic manipulations. There are now several systems to accomplish this feat reliably, including the ZFN, TALEN, and CRISPR-Cas programmable nucleases.4,5,7 Of the three, CRISPR-Cas, which recognizes its cut site in the genome via a small gRNA, is the easiest to manipulate at the bench, and demonstrates the highest efficiency. 8 As a result, it has become a mainstream tool for genome editing.
Following the formation of a DSB at an intended genomic position, cellular repair machineries will integrate an accompanying exogenous DNA fragment (donor sequence) into the break site by NHEJ, HDR, or MMEJ pathways. These DNA repair pathways operate via different mechanisms, sometimes in different phases of the cell cycle11,30–32 and have different DNA sequence requirements (regarding mainly sequence homology). However, and most importantly from the perspective of genome engineering, they result in different outcomes that can be generalized as error free (HR-based mechanisms) and error prone (NHEJ pathway).
The initial aim of our study was to engineer very long tandem repeat sequences into human and mouse pluripotent cells. We selected the PITCh approach based on MMEJ and the utilization of very short homology arms, from 5 to 40 bp in length, along with a TALEN or CRISPR-Cas9.1,10,27 Notwithstanding the benefits of PITCh, the system is challenged when the goal is to integrate reproducibly long DNA sequences containing highly repetitive elements or that are prone to adopt stable, non-canonical DNA structures. Thus, we were prompted to design a workflow that would efficiently accomplish the goal of targeting long tandem repeats or other long DNA fragments to predetermined sites in the genome. We split the integration of the exogenous DNA into two main steps. The first step is to introduce a promoter and a lox docking site into the genome site specifically with the aid of efficient Cas9-mediated cleavage; the second step is to introduce a GOI and a promoterless selection cassette, via Cre-lox recombination, into the docking site. Using this new approach, we successfully targeted various genomic locations in different cell types, including difficult-to-manipulate hiPSCs, and we were able to introduce different DNA sequences, starting from GFP markers described herein to complex repetitive elements. CasPi has the flexibility to accommodate a variety of promoters, selectable markers, genomic targets, and GOI combinations.
Generally, current precise integration strategies favor the creation of a programmable nuclease-mediated DSB with concomitant insertion of a GOI that is flanked by long homology arms33,34 or microhomology arms. 10 Although relatively rapid, single-step correct insertion of large DNA molecules can be inefficient. In addition, the entire integration protocol has to be repeated every time a new GOI is inserted into a genomic location, and potential indels or off-target effects can differ between independently generated cell lines harboring different GOIs. These concerns are minimized in the CasPi approach by creating a universal acceptor cell line. Our multistep approach takes advantage of the straightforward Cas9-mediated site-specific integration of the DV-PRO sequences to establish a lox docking site, and subsequently makes use of the highly efficient Cre-lox system to integrate a GOI into the docking site. Additionally, by using the lox site variants lox71 (docking site) and lox66 (GOI vector), Cre-mediated recombination strongly favors a one way reaction greatly stabilizing GOI integration. 17 In this regard, CasPi is similar to the commercial Flp-in system (Invitrogen), which introduces a GOI via the Flp-FRT system. 35 Major differences between the Flp-in and CasPi approaches include: (1) the Flp-in system targets a single random location in a selected few available cell lines, while CasPi allows for precise targeting of any genomic locus in any cell line of choice; (2) the Flp-in system utilizes long FRT sequence versions (48 bp) to improve the integration efficiency, while CasPi utilizes a pair of half-mutated lox71 and lox66 sites to favor integration; (3) the Flp-in system results in the integration of all vector sequences, including the vector backbone, into the FRT site, whereas CasPi results in vector backbone-free integration into the lox docking site; and (4) CasPi allows for efficient excision of selection markers from the final cell line, leaving only the GOI integrated into a genomic target site. Finally, our approach creates platform-stable cell lines harboring site-specific genome-targeted lox docking sites that can accept different GOI integrations, which allows for the comparison of GOI function/activity in the absence of position-of-integration effects.
CasPi requires multiple steps to accomplish GOI integration. However, when targeting the genome with large DNA fragments or difficult repetitive and potentially highly structured DNA elements, this drawback is superseded by the efficiency of the approach. In addition, most of the vectors are universal and, once designed, can be utilized in multiple projects for generating numerous cell lines.
Conclusions
Herein, we describe a protocol (CasPi) for the precise integration of exogenous DNA into user-defined genomic loci in cultured cells using a combined Cas9/Cre/Flp strategy. This method is broadly applicable with regard to exogenous DNA to be integrated, choice of integration site, and choice of cell type. This versatile approach allows for precise genome targeting with large, frequently complex, and repetitive DNA sequences that do not integrate efficiently or at all with current genome targeting methods.
Footnotes
Acknowledgments
We thank the Heflin Center Genomics Core of the University of Alabama at Birmingham for DNA sequencing, Dr. Rui Zhao's Lab at UAB for providing materials, Dr. Shondra Pruett-Miller for valuable discussions and advice, and all the members of the Napierala Lab for helpful scientific discussions and assistance with experiments. Research reported in this publication was supported by the UAB High Resolution Imaging Facility.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by National Institutes of Health (R01NS081366) awarded to M.N.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.