
Abstract

A formal comparison of the two largest published pan-cancer CRISPR-Cas9 knockout screens finds good concordance between data sets.
In the postgenomic era, it is maddening that most currently available cancer therapies are still broad-based chemotherapy treatments designed to target rapidly dividing cells whether normal or malignant cells. The mutations that cause cells to become oncogenic also confer specific vulnerabilities that normal cells lack and can hopefully be exploited for targeted cancer therapies.
In order to expand precision oncology treatments, we must gain a better understanding of these cancer dependencies and the underlying genetic alterations. To this end, several groups have conducted large-scale screens to identify cancer dependencies and hopefully novel therapeutic targets.1–7
As part of the cancer dependency map effort,8,9 the Broad and Sanger Institutes have independently conducted pooled CRISPR-Cas9 genome-wide dropout screens in hundreds of cancer cell lines.1,2,4 Writing in Nature Communications, 10 Joshua Dempster et al. conducted a formal comparison of these two pan-cancer CRISPR-Cas9 screens in order to assess the reproducibility of these experiments and the concordance of resulting data sets.
These screens were conducted by transducing Cas9-expressing cancer cell lines with a library of single guide RNAs (sgRNAs) targeting most genes in the genome for knockout. The sgRNAs were delivered via lentiviral transduction and at a low multiplicity of infection such that only one sgRNA was integrated per cell. Because the sgRNAs are integrated into the genome, they can also be used as barcodes to identify and quantify if sgRNAs are enriched or depleted during the screen.
Once expressed, each sgRNA will, in principle, complex with Cas9, find its target, and induce a knockout of the target gene (one gene knocked out per cell). Cells are subsequently collected and genomic DNA harvested and sequenced to identify which sgRNAs have been depleted. If a cancer cell line is dependent on a given gene for survival, cells containing a sgRNA targeting that gene will have a growth disadvantage and/or die, and correspondingly that sgRNA will be depleted or drop out.
By conducting these dropout screens in multiple cancer cell lines, researchers are able to compile a list of essential genes that are common across different cell lines as well as gene dependencies for a specific cell line. Cell lines can be divided into different cancer types and common dependencies identified for different subtypes.
Apples to Apples or Oranges?
Although the Sanger and Broad screens were conceptually the same, there were significant differences in the experimental details (Fig. 1). For example, one could argue that the two most fundamental reagents were different: the Cas9-expressing cells and the sgRNA libraries. For comparison, Dempster et al. only included common cancer cell lines that were screened at both locations, but each group independently generated pools of Cas9-expressing cells for each cell line via lentiviral transduction. Moreover, the sgRNA libraries used in each screen were different and designed using different rule sets. Other variables include the duration of the screen (14 days for Sanger, 21 days for Broad), the selection protocol for enriching for cells with successful sgRNA integration events, and the culture medium used for many of the tested cell lines.
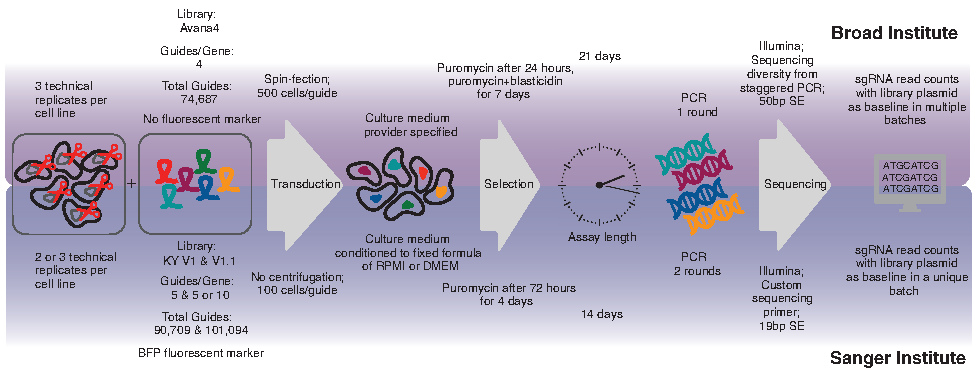
Schematic depicting experimental details for CRISPR-Cas9 dropout screens performed at the Broad and Sanger Institutes (adapted from Dempster et al. 10 ).
Despite these experimental differences, Dempster et al. were able to show that the two data sets have surprisingly good correlations using a variety of statistical methods and comparisons. Because data-processing pipelines also varied between groups, the authors included comparisons at different levels of data processing, which all showed consistently good correlations between data sets overall. One noteworthy statistic showed that using unprocessed data, both screens jointly identified more than 70% of the common dependencies that were identified in each individual screen. Additionally, and therapeutically relevant, the data sets had good agreement in identifying which cell lines are dependent on selective dependencies (genes with score variances across cell lines).
Because there was some discordance between the data sets, the authors ventured to elucidate the root causes of the discrepancies. Using the unprocessed data from the original screens, Dempster et al. were able to show that the sgRNA library composition with associated sgRNA on-target efficacies and different experimental time points had a measurable impact on the resulting data sets. To confirm the batch effects between screens experimentally, replication experiments were independently performed at both centers in which the Broad used the Sanger sgRNA library and vice versa. Additionally, cells were collected at both the 14- and 21-day mark. Of the tested variables, the sgRNA library composition had the biggest batch effect. The authors also point out that such batch effects can be corrected with standard methods, such as ComBat, 11 without sullying data quality.
Stronger Together
In order to create a complete cancer dependency map, it has been suggested that more than 5,000 screens are likely needed to model confidently and predict most cancer dependencies fully. 12 Ideally, cancer dependency screens conducted at different geographical sites are robust and reproducible. The comparison by Dempster et al. demonstrates that it is possible for two well-designed and independently derived screening data sets to have strong agreement and thus pave the way for possible integration and/or joint analysis of data sets obtained from large independent screening efforts. In order to fill in the topography of the evolving cancer dependency map, more cancer cell lines with broader genetic diversity need to be analyzed. The potential for combining larger data sets will allow us to complete the dependency map sooner and hopefully unveil a treasure trove of new therapeutic cancer targets.