
Abstract

A study from CRISPR Therapeutics in this Toolbox issue evaluates three commonly used genome-wide off-target assays and finds remarkable congruency in sequence-confirmed off-target sites.
The use of targeted genome editing in somatic cells to treat disease should be both efficient and precise. Accordingly, numerous assays to detect on- and off-target editing have been developed. The chances of creating an off-target edit in a targeted clone are quite low when using a well-designed guide RNA (gRNA). 1 However, for therapeutic applications in which millions of cells are exposed to genome editing reagents either in vivo or ex vivo, a much different set of standards is needed to mitigate risk.
Off-target prediction assays can be binned into one of three categories: in silico, biochemical, and cell-based (see Fig. 1):
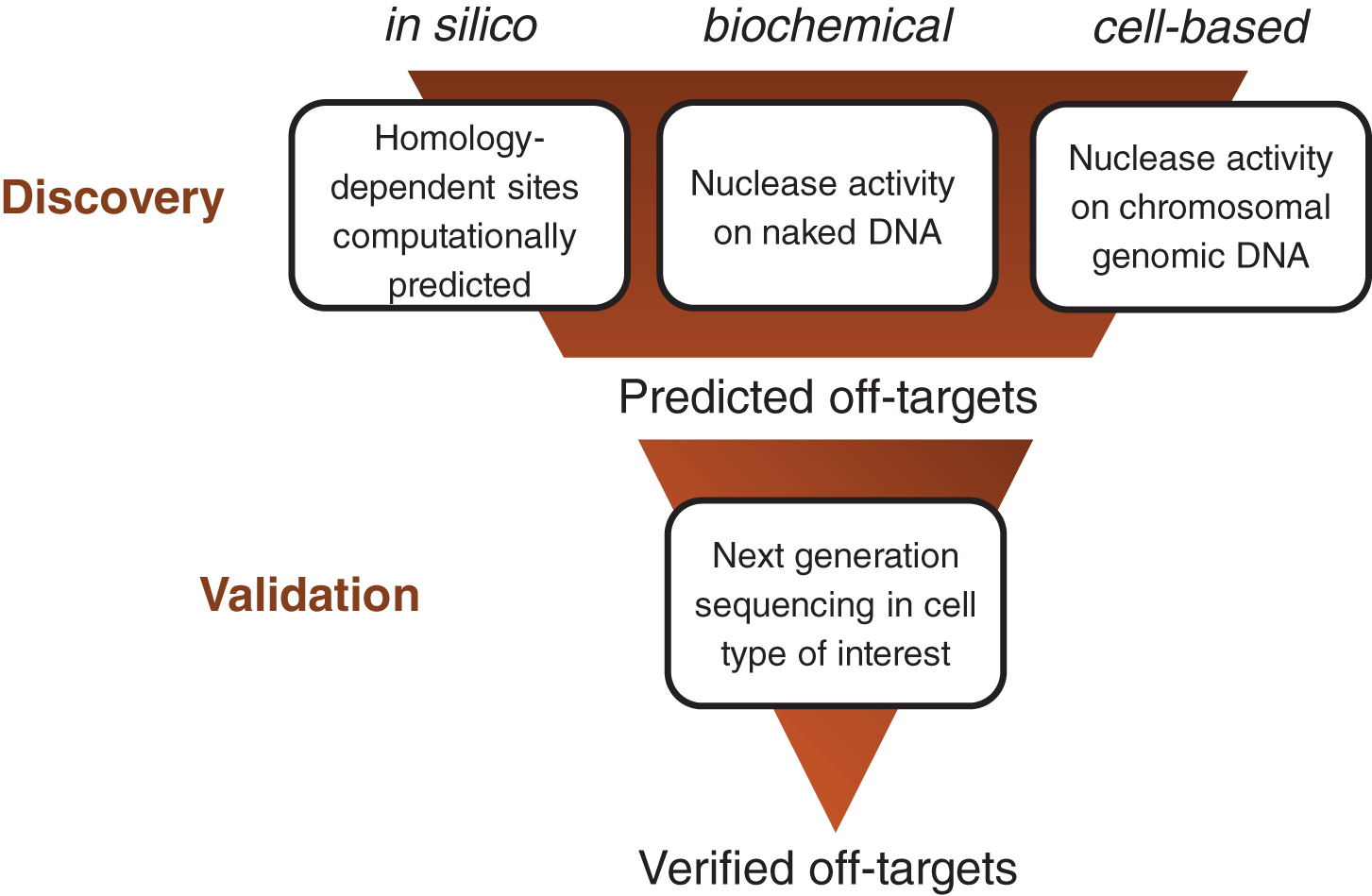
Off-target nomination and verification workflow. Potential off-target edit sites are nominated in the discovery phase by in silico, biochemical and/or cell-based assays. Predicted off-targets are further evaluated in the validation phase by next-generation sequencing to determine bona fide off-target sites.
In silico-based strategies are homology dependent and computationally nominate potential off-target sites based on mismatch and gaps (termed “the editing distance”) between the gRNA spacer and sites in the genome of interest.
Biochemical off-target detection assays use purified genomic or synthetic DNA as a template for nuclease cleavage to identify potential off-target events and are homology independent.
Cell-based assays are also homology independent but are performed in cells using genomic DNA in a chromosomal context as the template for nuclease cleavage. These assays rely on the subsequent DNA repair by the endogenous cellular machinery to mark the location of the break.
All three types of assays or a combination thereof can be used in the off-target discovery phase to nominate a list of potential off-target sites, which are then further evaluated using next-generation sequencing (NGS) in the cell type of interest during the validation phase (Fig. 1). Targets are considered bona fide if they have sufficient sequence coverage and show statistically significant differences in indel rates between control and nuclease-treated samples.
In this issue of The CRISPR Journal, Andrew Kernytsky and coworkers 2 from CRISPR Therapeutics compare three popular homology-independent assays: GUIDE-seq, 3 CIRCLE-seq, 4 and SITE-seq. 5 Guide-seq is a cell-based, genome-wide, off-target detection method, which relies on the incorporation of a short (34 bp) double-stranded oligodeoxynucleotide (dsODN) into the site of DNA break. Using the dsODN as an anchor for primer binding enables the identification of the neighboring genomic DNA sequence and off-target site. 3 CIRCLE-seq and SITE-seq are both biochemical, homology-independent assays. In brief, CIRCLE-seq depends on purified and circularized genomic DNA for nuclease cleavage. The resulting linearized fragments of DNA are amplified and sequenced with NGS to determine potential off-targets. 4 SITE-seq uses purified genomic DNA as the nuclease template, which is then adenylated, enriched using biotinylated adaptors, and amplified prior to NGS. 5
In the new study, 2 Chaudhari et al. performed GUIDE-seq, CIRCLE-seq, and SITE-seq in triplicate for eight non-therapeutic gRNAs of varying specificity on genomic DNA from or in HEK293T cells stably expressing Cas9. The ensuing sequencing data were filtered to remove nominated sites that had an edit distance of more than six. GUIDE-seq predicted the lowest average number of sites per gRNA at 40 sites, while CIRCLE-seq and SITE-seq predicted more than 5,200 and 2,000 sites per gRNA, respectively. This is an expected result, as the chromatin state inside a cell can affect the accessibility to CRISPR-Cas9 cleavage and subsequent dsODN incorporation, resulting in fewer nominations by GUIDE-seq.
By performing each assay in triplicate, the authors were able to compare the reproducibility of each assay. They found CIRCLE-seq and SITE-seq to be more reproducible with more than 70% and 60%, respectively, of sites nominated in all three replicates compared to about 30% with GUIDE-seq. Additionally, CIRCLE-seq and SITE-seq both predicted approximately 86% of the sites nominated by GUIDE-seq, and sites that were nominated by any two assays had lower edit distance than sites nominated by a single assay.
The authors further benchmarked the assays by measuring the editing at computationally predicted, homology-dependent sites with NGS. More than 75,000 sites with up to five mismatches or three mismatches with one gap were included and sequenced in triplicate via hybrid-capture Illumina sequencing on control and gRNA-treated samples, with a median sequencing coverage of ∼2,300 × . Sites with a statistically significant difference between control and treated samples, at least 500 × coverage, and at least 0.2% indel frequency in any replicate were annotated as verified off-target sites.
Using these filters, 51 sites were identified across all eight gRNAs, including nine sites with perfect complementarity to the gRNA spacer sequences. Fifty of the sites were nominated by at least one of the genome-wide assays, with 45 of the sites nominated by all three replicates of all three methods. The one site that was not nominated by any of the three assays was later shown by rhAmpSeq to likely be a false-positive of the sequence confirmation step (the site contained a long homopolymeric stretch near the cleavage site that can cause sequencing errors).
Interestingly, 158 sites were nominated by all three genome-wide methods but were not annotated as verified hits during the sequence confirmation step. The authors followed up 152 of these sites with rhAmpSeq; only six of the sites had indel frequencies >0.2% and <0.36%, suggesting that if these are true off-target sites, the editing indel frequency is <0.2% and near the limits of NGS detection. The authors also observed the highest correlation of read counts and editing rates with GUIDE-seq. CIRCLE-seq had some correlation as well, but SITE-seq had no correlation between indel frequencies and read counts in the tested conditions.
Overall, this study is the most comprehensive evaluation of genome-wide off-target assays reported to date. Remarkably, the authors would have been able to predict all sequence-confirmed edited sites for all test gRNAs with a combination of any one of the genome-wide homology independent assays and an in silico nomination space of up to two mismatches and one gap. Accordingly, they suggest using both homology-dependent and homology-independent methods to nominate potential off-target sites and to follow up on predicted sites using sequence confirmation with a read depth greater than 5,000 × . Moreover, combining hits from multiple replicates of any one assay improves the reliability and likelihood of identifying low-frequency sites. In addition, the authors recommend considering the genomic context of an off-target edit. This is perhaps the most important and most difficult recommendation to follow, as most off-target edits are likely benign—a notion that is quite challenging to demonstrate experimentally and conclusively.
This report from CRISPR Therapeutics appears shortly after a groundbreaking clinical study from the same company (plus academic and industry collaborators) reporting some exciting results for sickle cell disease and beta-thalassemia. 6 While it might not trigger the same giddy headlines as the clinical study, this CRISPR Journal report is a significant study in its own right. As a community, it is essential that we come together to determine empirically the best approaches to mitigate the risk of off-target editing for therapeutic applications. This exhaustive study by Chaudhari et al. provides an informative data set with well-supported recommendations.