
Abstract
Gene manipulations of human induced pluripotent stem cells (iPSCs) by CRISPR-Cas9 genome engineering are widely used for disease modeling and regenerative medicine applications. There are two competing pathways, non-homologous end joining (NHEJ) and homology directed repair (HDR) that correct the double-strand break generated by CRISPR-Cas9. Here, we improved gene editing efficiency of gene knock-in (KI) in iPSCs with minimum components by manipulating the Cas9 expression vector. Either we inserted short hairpin RNA expression cassettes to downregulate DNAPK and XRCC4, two main players of the NHEJ pathway, or we increased cell survival by inserting an anti-apoptotic expression cassette of miRNA-21 into the Cas9 vector. For an easy readout, the pluripotency gene SOX2 was targeted with a T2A-tdTomato reporter construct. In vitro downregulating DNAPK and XRCC4 increased the targeting efficiency of SOX2 KI by around twofold. Furthermore, co-expression of miRNA-21 and Cas9 improved the efficiency of SOX2 KI by around threefold. Altogether, our strategies provide a simple and valuable approach for efficient CRISPR-Cas9 gene editing in iPSCs.
Introduction
Recent advances in engineering DNA nucleases, including ZFN, TALEN, and Cas9, have revolutionized gene-targeting procedures.1–3 These tools introduce a double-strand break (DSB) at the DNA target site that can be repaired by random insertion and deletion (indel)-generating non-homologous end joining (NHEJ) or by homology-directed repair (HDR) pathways using the sister chromatid or an exogenous DNA donor template.4,5 Among these technologies, the CRISPR-Cas9 gene editing is widely used because it is effective, time and cost efficient, and easy to handle. This technology is used for a wide range of gene manipulations, including base pair editing, knockout (KO), and knock-in (KI) applications, and to generate large genomic DNA deletions and/or rearrangements.6–8
Both NHEJ and HDR pathways are active in most cells. However, at the time of a DSB, NHEJ is the predominant pathway to repair the generated gap. Moreover, NHEJ is fast and takes less than one hour, while HDR is much slower and takes several hours to complete.9,10 The major molecular players in the NHEJ pathway are ku70-ku80 heterodimer, DNA-dependent protein kinase (DNAPK), and X-ray repair cross-complementing protein 4 (XRCC4). The KU70-KU80 complex attaches to DSBs, along with DNAPK, bringing both DNA ends into close proximity. Then, XRCC4 and DNA ligase 4 (LIG4) complete the repair process.11–13 As NHEJ generates indels at the DSB, leading to frameshifts, this causes the disruption of genes in loss-of-function studies. 14 However, the basis of HDR for performing an accurate repair is dependent on the presence of a donor template. 15 Hence, HDR is routinely applied for producing targeted gene KI or other precise gene editing, for example introducing/correcting point mutations. Of note, HDR has lower efficiency compared to the NHEJ pathway. To improve precise gene editing, such as introducing coding single nucleotide polymorphisms and KI of reporter genes or corrections of disease mutations in induced pluripotent stem cells (iPSCs), several approaches have been developed to suppress NHEJ and/or promote HDR by small molecule compounds. 16 Accordingly, increase in apoptosis due to cytotoxic stress during lipofectamine transfection is another hurdle for efficient CRISPR-Cas9-mediated gene targeting. In addition, high expression of Cas9 nuclease is toxic for the cell and induces cell death, decreasing the efficiency of gene editing.17,18 However, high-level expression of Cas9 theoretically increases the rate of homologous recombination.
Here, we used a similar concept of downregulating NHEJ and increasing cell survival to improve gene targeting efficiency in human iPSCs. To have an easy and quantifiable readout, we targeted the pluripotency gene of SOX2 in two different iPSC lines using the previous published targeting vector (Addgene ID89991) 19 to generate a SOX2-Thosea asigna virus 2A like peptide-tandem dimer Tomato (SOX2-T2A-tdTomato) reporter iPSC lines. To counteract the NHEJ pathway, we cloned expressions cassettes of short hairpin RNA (shRNA) into the Cas9 expression vector. These shRNAs were directed against the main component of NHEJ repair pathway, XRCC4 and DNAPK. To increase cell survival and counteract stress-induced apoptosis, we inserted an expression cassette of the miRNA-21 into the Cas9 expression vector. Thereby, we established a novel method without increasing the number of components to improve CRISPR-Cas9-based gene KI efficiency by downregulating members of the NHEJ machinery and by increasing cell survival in human iPSCs.
Methods
Single guide RNA design and CRISPR-Cas9 plasmid construction
Single guide RNA (sgRNA) for CRISPR-Cas9 mediating SOX2 gene KI were designed using the CRISPOR web tool (crispor.tefor.net). The sgRNA recognition site was at the 3′ end of the SOX2 open reading frame. To the sgRNA oligos, CACCGGG was added to the 5′ end of the sense oligo and AAAC and CCC to the 5′ and 3′ end of the antisense oligo for better expression and compatibility with the BbsI site of the vector (the sgRNA sequence is listed in Table 1). PU6-(BbsI) sgRNA_CAG-GFP-bpA plasmid (Addgene ID86985) was digested with BbsI used for cloning the sgRNA oligos. To confirm the right sgRNA cloning, the expression plasmid was subjected to Sanger sequencing.
shRNA and miRNA-21 expressing plasmids construction
Invitrogen's Block-It RNAi Designer was used to design four shRNAs for downregulating the main players of the NHEJ repair pathway, XRCC4 and DNAPK. Two shRNAs targeting different positions were designed for each gene, where AgeI and EcoRI sites were added to the 5′ and 3′ sites of the oligos, respectively. Furthermore, a unique palindromic loop of CTCGAG was added between the complementary regions of the oligos. Sense and antisense oligos were annealed and ligated into the AgeI and EcoRI digested pLKO.1 plasmid (Addgene ID8453). Oligo sequences for shRNA and scramble control vectors are listed in Table 1. Furthermore, to construct miRNA-21-expressing plasmid, a 72 bp sequence of pre-miRNA-21 plus 50 nt upstream and downstream of the sequence was amplified by polymerase chain reaction (PCR) using human genomic DNA (NC_000017.11) as a template. AgeI and EcoRI target sites were also included to the 5′ sites of the forward and reverse primers, respectively. The PCR product was inserted into the pLKO.1. Following the cloning, the plasmids were sequenced by Sanger sequencing.
Gibson assembly reaction for generating sgRNA–shRNA and sgRNA–miRNA cassettes
Into the PU6-(BbsI) sgRNA_CAG-Cas9-GFP-bpA plasmid following combinations of cassettes, the sgRNA–shRNAI–shRNAII or sgRNA–miRNA-21 were inserted by Gibson assembly procedure as described. 20 For amplification, this protocol was modified by addition of two more primers, EP1808-BR and EP1809-CR, for amplification of the miRNA (Table 1). Following cloning, the resulting vectors were subjected to Sanger sequencing.
Human iPSC culture and plasmids transfection
The HMGUi001-A and HMGUi002-A iPSC lines were cultured on six-well plates coated with Geltrex (Life Technologies) in the human medium of StemMACS iPS-Brew XF (Miltenyi Biotec). The iPSCs were passaged using 5 mM EDTA in phosphate-buffered saline (PBS) at 37°C for 3 min. At the cell splitting time, 10 μM Y-27632 (Santa Cruz Biotechnology) was used in culture medium. The iPSCs were maintained at 37°C, 20% O2, and 5% CO2. For the lipofectamine-based transfection, SOX2 targeting the plasmid of pUC19-SOX2-T2A-2xNLS-tdTomato-F2A-Puro (Addgene ID89991) was mixed with CRISPR-Cas9 plasmid containing the triple cassette of sgRNA–shRNAI–shRNAII or the dual cassette of sgRNA–miRNA21. The day before the transfection, iPSCs were seeded at a concentration of 2 × 105 cells per well on the six-well plate. The SOX2 targeting plasmid (2 μg) and the CRISPR-Cas9 plasmid (1 μg) were transfected into the iPSCs using 5 μL Lipofectamine™ Stem Transfection Reagent (Thermo Fisher Scientific) and 200 μL OptiMEM medium per each well (Thermo Fisher Scientific) according to the manufacturer's instructions. Three days after the transfection, the cells were dissociated and harvested for flow cytometry analysis as well as RNA extractions.
Fluorescence-activated cell sorting enrichment of transfected iPSCs
Seventy-two hours after the transfection, iPSCs were detached as described above and collected for fluorescence-activated cell sorting (FACS). The cells were re-suspended in 0.5 mL iPS-Brew medium containing 10 μM Y-27632 and DAPI (Sigma–Aldrich). Then, the cell suspension was passed through a cell strainer and used for sorting. The percentage of targeting in the transfected cell population was determined by analyzing GFP and Tomato signal on a FACSAria II (BD Biosciences). For establishing a stable fluorescent SOX2 reporter, iPSC line double-positive GFP/tdTomato cells were FACS sorted and 2,000 GFP+ cells were seeded on Geltrex-coated 10 cm dishes. After 7–9 days, single cell-derived iPSC clones were picked, transferred to new plates, expanded, and subjected to PCR screening on isolated genomic DNA.
Expansion and screening the clones for HDR-mediated SOX2 gene targeting
The appropriate PCR primers for sequencing and clone screening were designed using Clone Manager Molecule software (Table 1). Three-primer PCR using EP1890, EP671, and EP1891 primers for the left arm yields 1,270 or 1,409 bp, representing KI or wild-type (WT) alleles, respectively. Furthermore, PCR for the right arm using EP1892, EP1893, and EP036 primers yields 852 or 1,265 bp, representing KI or WT alleles, respectively. PCR reactions were performed using Taq DNA polymerase enzyme using the following thermal conditions: initial denaturation at 95°C for 3 min, amplification for 40 cycles with denaturation at 95°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 90 s. Direct DNA sequencing was performed to confirm the authenticity of the PCR products.
Cell viability assay
Cell viability was examined at two time points, 8 and 24 h after transfection by 0.4% trypan blue dye (Gibco). Cell supernatant containing dead cells was collected. Adherent cells were dissociated as describe above. Floating dead cells and dissociated cells were centrifuged and re-suspended in 1 mL medium. Re-suspended cells (20 μL) and 0.4% trypan blue (20 μL) were mixed and left for 3 min. Then, the numbers of dead and viable cells were counted on a hemocytometer.
Human iPSCs differentiation toward pancreatic progenitor cells
The SOX2-T2A-tdTomato iPSCs reporter line generated from HMGUi001-A was cultured on Geltrex-coated 10 cm plates in the StemMACS iPS-Brew XF. As the iPSCs were exposed to transient expression of miRNA-21, they were passaged at least five times before starting the differentiation. Then, confluent iPSCs (70–80%) were dissociated into single cells using 1 × Accutase cell detachment solution (Sigma–Aldrich) and cultured at ∼75,000 cells/cm2 in the StemMACS iPS-Brew XF supplemented with 10 μM Y-27632 on ultra-low attachment six-well plates. The next day, the medium was changed, and the differentiation course was initiated. The iPSCs were differentiated toward PDX1 + NKX6.1 + pancreatic progenitor (PP2) cells according to the Rezania protocol. 21 Briefly, the 3D aggregates were treated with Activin A (100 ng/mL), CHIR-99021 (5 and 0.3 μM), vitamin C (0.25 mM), FGF7 (50 and 2 ng/mL), IWP-2 (1.25 μM), SANT-1 (0.25 μM), RA (1 and 0.1 μM), LDN (100 and 200 nM), ITS-X (1:200), and TPB (100 and 200 nM) in S1-2 (containing MCDB131 medium with 0.5% bovine serum albumin [BSA], 1 × GlutaMAX, 1.5 g/L NaHCO3 and 10 mM glucose) and S3-4 (containing MCDB131 medium with 2% BSA, 1 × GlutaMAX, 2.5 g/L NaHCO3, and 10 mM glucose) media. The aggregates were collected and fixed after 10 days of differentiation.
Immunofluorescence imaging
For immunocytochemistry staining, the aggregates after 10 days of differentiation were fixed in 2% paraformaldehyde in PBS for 20 min at room temperature (RT), followed by three washes with PBS. Then, they were embedded in tissue-freezing medium (Leica Biosystems) and sectioned on a cryostat, generating tissue sections 20 μm thick. Sections were permeabilized with 0.1 M glycine and 0.2% Triton in PBS for 30 min, followed by blocking with the blocking solution (3% serum donkey, 0.1% BSA, and Tween20). The first antibodies were added and incubated overnight at 4°C in blocking solution. The following day, the cells were washed three times with PBS. Then, the secondary antibodies were added and incubated for 4 h at RT. Nuclei were stained with DAPI. Finally, images were taken by Zeiss confocal microscope. The following primary antibodies and dilutions were used: goat anti-OCT4 (Santa Cruz, #sc-8628, 1:500), rabbit anti-SOX2 (Cell Signaling Technology, #3579S, 1:500), goat anti-PDX1 (R&D Systems, #AF2419, 1:500), rabbit anti-NKX6.1 (Arcis, #NBP1-82553, 1:500), donkey anti-rabbit Alexa Fluor 555 IgG (Invitrogen, #A31572), donkey anti-goat Alexa Fluor 488 IgG (Invitrogen, #A11055), and donkey anti-rabbit Alexa Fluor 488 IgG (Invitrogen, #A21206). All secondary antibodies were used at a dilution of 1:500.
RNA isolation/cDNA synthesis/quantitative real-time PCR
RNA isolation from the iPSCs was carried out using Trizol Reagent (Invitrogen). cDNA was synthesized using the SuperScript VILO cDNA synthesis kit (Invitrogen). Specific real-time PCR primers (Table 1) were designed for XRCC4, DNAPK, and GAPDH (GenBank accession numbers: NM_003401.5, NM_006904.7, and NM_002046.3, respectively). SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) was used for all quantitative PCR (qPCR) reactions. The expression of target genes was normalized to the expression value of the housekeeping gene GAPDH. All qPCR reactions were carried out on the ViiA 7 system (Applied Biosystems) using the following thermal conditions: initiation at 95°C for 30 s, amplification for 40 cycles with denaturation at 95°C for 10 s, and annealing/extending at 60°C for 1 min.
Statistical analysis
Statistical analyses were carried out using GraphPad Prism (GraphPad Software). All experimental tests were carried out at least two or three times. Data represented are the mean ± standard deviation using a two-tailed Student's t-test or one-way analysis of variance. Furthermore, the qPCR reactions were carried out in duplicate. The criterion for statistical significance was p < 0.01.
Results
CRISPR-Cas9-mediated SOX2 gene-tdTomato KI in human iPSCs
Using a CRISPR-Cas9 genome engineering approach, we hypothesized whether targeting NHEJ molecular machinery by shRNAs, as well as increasing cell survival by miRNA-21 overexpression during the transfection, would improve HDR-mediated gene targeting efficiency. For evaluation, we performed SOX2 gene targeting to generate the SOX2-tdTomato reporter iPSC line. SOX2 is an HMG-box transcription factor, which is essential for the maintenance of pluripotency and the survival of human iPSCs. 22 As SOX2 is highly expressed at the iPSC stage, the gene serves as a perfect locus to monitor gene targeting efficiency of KI reporter constructs by measuring fluorescent activity. Our two established human iPSC lines, iPSC line I: HMGUi001-A; 46, XX 23 and iPSC line II: HMGUi002-A; 46, XY 24 were used for CRISPR-Cas9-mediated gene targeting of the SOX2 locus. We were able to integrate the T2A-tdTomato sequence successfully before the stop codon of the SOX2 gene upon HDR-mediated gene targeting (Fig. 1A). The homologous recombination at the SOX2 locus was confirmed by 5′ and 3′ genomic PCR analysis spanning the homologous recombination border, as illustrated in Figure 1B.

A schematic representation of gene targeting strategy, clone screening, determining the efficiency of gene KI and clone expansion.
To assess random or nonspecific integration of T2A-tdTomato, we performed a different PCR analysis with primers that bind to the T2A-tdTomato sequence and to the backbone of the targeting vector. As we used non-digested targeting vector before transfection, if it is integrated at the right site by homologous recombination at the end of both homology arms, the vector backbone sequence is lost. However, if random integration occurs, most likely the backbone (attached to the homology arms) would be inserted as well. The PCR result showed a 1,888 bp product specific for the targeting vector. Due to the absence of the product (confirming the attachment of the integration sequence with the backbone) in the reporter cells, these data confirm specific integration and no random integration of the T2A-tdTomato targeting vector into the genome (Fig. 1C).
Our strategy to measure the KI efficiency of the SOX2 gene was simple. In summary, the cells receiving CRISPR-Cas9 plasmid express Cas9-GFP. Then, if HDR occurs, the GFP+ cells express SOX2-tdTomato fusion protein. Seventy-two hours following transfection, the GFP+ cells were sorted, and the percentage of tdTomato+ cells was determined (Fig. 1D). The transfection efficiency range was 30–50% in two iPSC lines (Fig. 1E and F). For quality-control purposes, the tdTomato+ cells were expanded and cultured in 2D as well as 3D to form aggregates (Fig. 1G–I).
shRNA-mediated downregulation of NHEJ increases HDR-mediated SOX2 gene-tdTomato KI
To knockdown DNAPK and XRCC4 genes, we used RNAi technology instead of small compounds and modified out CRISPR-Cas9 expression plasmids inserting shRNA expression cassettes (Fig. 2A). For this purpose, we designed four shRNAs to target the mentioned mRNAs (two for each gene). The efficiency of the shRNAs was measured, and qPCR data demonstrated downregulation of DNAPK and XRCC4 transcripts by the shRNAs. DNAPK and XRCC4 were downregulated by 0.15- and 0.11-fold, respectively (Fig. 2B; the data for non-efficient shRNAs is not shown). The most efficient shRNAs were cloned into the CRISPR-Cas9 expression plasmid, resulting in the expression construct Cas9-GFP/sgRNA–shRNAI–shRNAII, whereas each shRNA/sgRNA cassette was driven by its own U6 promoter (we cloned all elements in one expression construct). Next, we analyzed the effect of downregulating the NHEJ repair pathway in the iPSCs on HDR-mediated KI efficiency. The efficiency of KI in HMGUi001-A using scramble shRNA and shRNAI–shRNAII was 8.48% and 15.52%, respectively (n = 3; p < 0.001). The values for the HMGUi002-A iPSC line were 12.5% and 22.48% (n = 3; p < 0.01). On the other hand, targeting of DNAPK and XRCC4 by shRNAs co-expressing with Cas9 and sgRNA increased SOX2 gene KI by nearly twofold when compared to the scramble shRNA control in our two iPSC lines (1.83-fold for HMGUi001-A and 1.79-fold for HMGUi002-A; Figs. 2C and D and 3A and B).
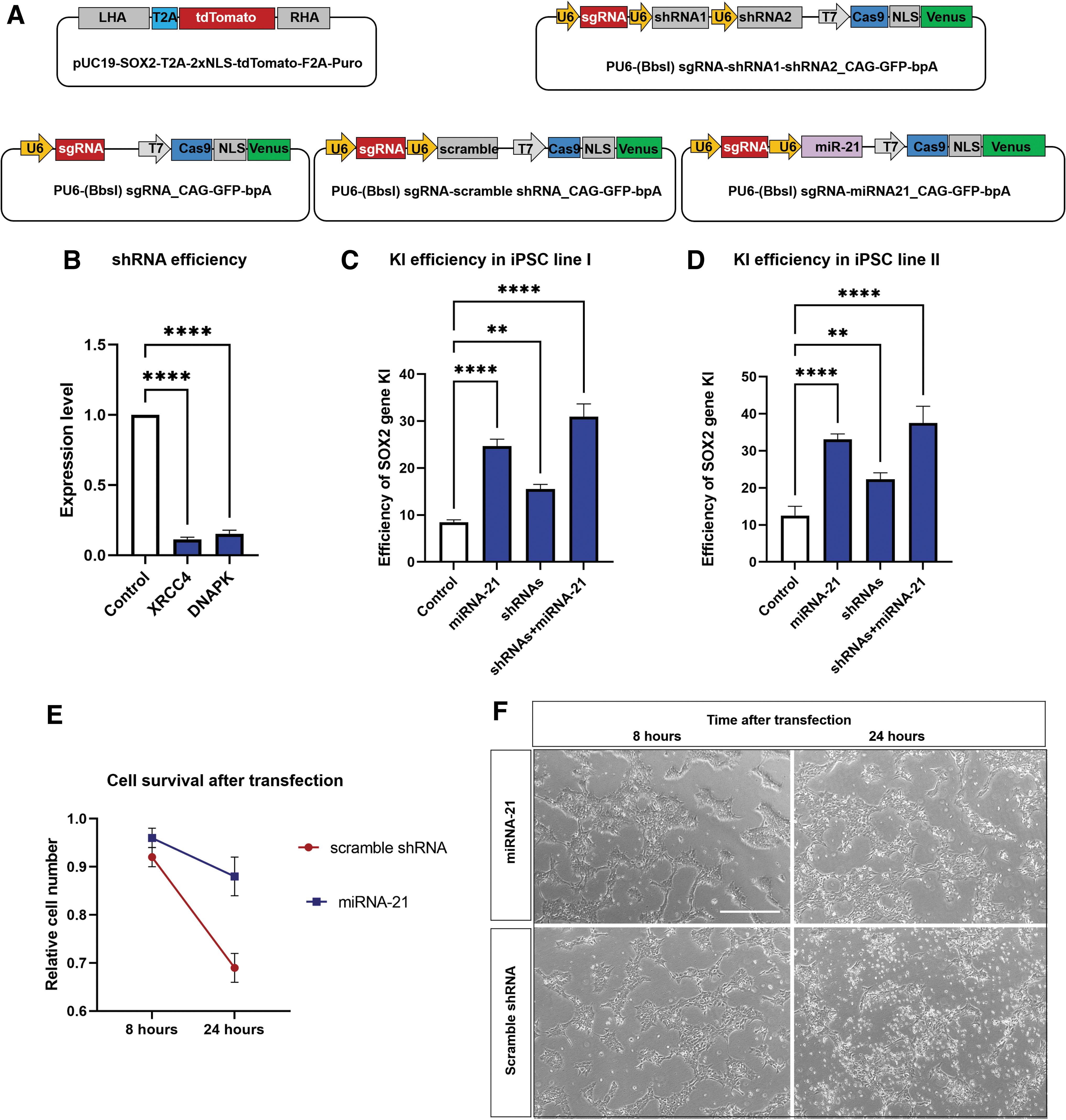
CRISPR-Cas9 plasmids and their HDR editing efficiencies in iPSCs.
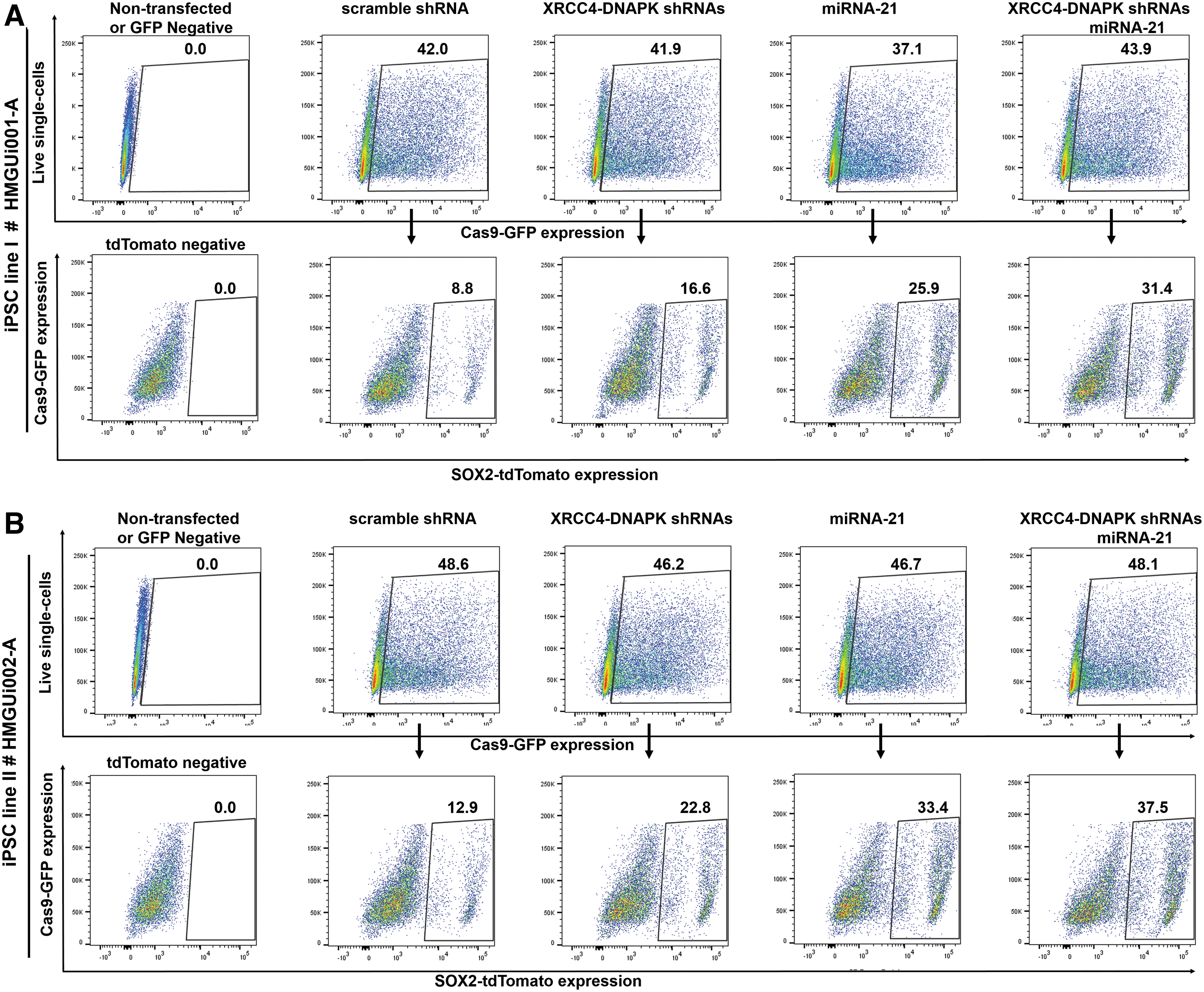
Transfection efficiency and gene KI efficiencies for all plasmids combinations for two iPSC lines by fluorescence-activated cell sorting (FACS).
CRISPR-Cas9/miRNA21 expression system increases the efficiency of SOX2 gene-tdTomato KI
Stress induced by the lipofectamine transfection or by adverse effects of overexpressing Cas9 protein can lead to massive cell death and may hamper efficient CRISPR-Cas9-mediated gene targeting. As the small RNA of miRNA-21 indirectly downregulates apoptotic genes, mainly Caspase3, we predicted its ectopic expression could increase cell survival and consequently enhance the gene targeting efficiency of CRISPR-Cas9. To investigate this, we cloned miRNA21 genomic DNA into the Cas9 expression vector resulting in the Cas9-GFP-sgRNA-miRNA21 vector (Fig. 2A). To evaluate gene targeting efficiency, we used the same transfection and screening conditions as described previously, determining the percentage of SOX2-tdTomato+ cells within the Cas9-GFP transfected cell population. The efficiency of KI using miRNA-21 was 24.66% and 32.4% for the HMGUi001-A and HMGUi002-A iPSC lines, respectively (2.9-fold for HMGUi001-A and 2.59-fold for HMGUi002-A; n = 3; p < 0.0001). On the other hand, the data showed nearly a threefold increase in gene targeting efficiency in the presence of miRNA-21 when compared to the scramble control (Figs. 2C and D and 3A and B). Next, to analyze the effect of miRNA-21 on cell survival of the HMGUi001-A iPSC line, we determined the number of viable cells at two time points: 8 and 24 h after transfection. As expected, the result demonstrated that overexpression of miRNA-21 increased the survival of transfected iPSCs at 24 h after transfection (92% survived cells in the presence of miRNA-21 vs. 69% in its absence; Fig. 2E and F; n = 2; p < 0.01). Consequently, the increased survival of transfected cells improved the efficiency of gene editing mediated by CRISPR-Cas9.
Accumulative effect of miRNA21 and shRNAs on the gene editing efficiency of CRISPR-Cas9 in iPSCs
To see an additive effect of downregulation of NHEJ and increasing cell survival on gene editing efficiency of CRISPR-Cas9, we used miR-21 and DNAPK-XRCC4 shRNAs expression vectors together. To test this, we used a combination of Cas9-GFP-sgRNA-miRNA21, Cas9-GFP-sgRNA-shRNAs, and SOX2-T2A-tdTomato plasmids for transfection of the two iPSC lines. The combination of miRNA and shRNA vectors increased the homologous recombination in transfected cells to 31.4% in comparison to miRNA-21 vector alone (25.9%) or shRNA vectors alone (16.6%). In the other iPSC line, combining vectors increased the targeting efficiency in transfected cells to 37.5% from 22.8% and 33.4% using shRNA or miRNA vector alone (n = 3; p < 0.0001; Figs. 2C and D and 3A and B). We concluded that homologous recombination slightly increased upon combining both strategies.
Differentiation of SOX2-tdTomato reporter iPSC line to pancreatic progenitors
Transient expression of miRNA-21 may affect pluripotency of differentiation potential of edited iPSCs. To test this directly, we explored the expression patterns of the main pluripotency markers, OCT4 and SOX2, by immunostaining. Furthermore, we differentiated the edited HMGUi001-A iPSCs toward pancreatic progenitors to see if they showed any difference when compared to control iPSCs. The differentiation was carried out according to a modified Rezania protocol in 3D (Fig 4A and B). We collected the aggregates at day 10 of differentiation (pancreatic progenitors). Then, aggregates were stained for PDX1 and NKX6.1. miRNA-21 edited iPSCs have high expression levels of SOX2 and OCT4 in most cells, undistinguishable from control iPSCs (Fig. 4C). Furthermore, these cells differentiated efficiently to the pancreatic progenitor stage shown by a high percentage of PDX1 single- as well as PDX1 and NKX6.1 double-positive cells at day 10 of differentiation (Fig. 4D).

Differentiation of SOX2-tdTomato reporter iPSC line to pancreatic progenitors.
Discussion
In the current study, we stablished two simple approaches to increase the efficiency of CRISPR-Cas9-mediated gene targeting for a precise gene integration. The two repair mechanisms that are induced upon a DNA DSB are NHEJ and HDR. As NHEJ is the more effective pathway, we intended to increase HDR by downregulating the main players of NHEJ molecular machinery, XRCC4 and DNAPKs, by shRNA. To test this system, two iPSC lines were transfected with a SOX2-T2A-tdTomato KI reporter construct and a triple expression system of sgRNA–shRNAI–shRNAII to downregulate the two genes XRCC4 and DNAPKs. This approach improved gene targeting efficiency by around twofold. Our approach minimized the number of plasmids used and insured simultaneous expression of shRNAs and sgRNAs. Another strategy to improve the efficiency of CRISPR-Cas9 genome engineering aimed to reduce stress-induced cell death caused by the transfection procedure or by overexpression of Cas9 protein. In the second approach, to increase cell survival and counteract stress-induced apoptosis, we inserted an expression cassette of the miRNA-21 into the Cas9 expression vector. In addition, with this approach, we successfully improved targeting efficiencies at the SOX2 locus, resulting in around a threefold increase in homologous recombination in transfected iPSCs. Finally, we examined an additive effect of miRNA-21and shRNAs together on CRISPR-Cas9 efficiency for precise gene integration in iPSCs. The result yielded more than each single effect, around a 3.5-fold increase in the system's gene targeting efficiency.
By transient downregulating the NHEJ pathway, we tried to increase the efficiency of HDR-dependent applications such as KI or precise gene editing. Modulating the main players of NHEJ by small compounds boosts HDR and alters the gene targeting efficiency. Several studies reported SCR7, a putative inhibitor of ligase IV, increases the gene targeting efficiency of CRISPR-Cas9. The targeting efficiencies for small insertions varied from a 3- to 19-fold increase in different cell types.25,26 However, in one study, SCR7 failed to show improved HDR-dependent gene targeting efficiency. 27 These data demonstrate that the effect of the SCR7 compound on HDR efficiency depends on cell types and the target locus. Furthermore, using several other small compounds termed as RS-1 (activator of Rad51), NU7441 and Ku-0060648 (both are inhibitors of DNAPK) increase the efficiency of HDR and precise gene editing.28,29 Instead of small chemical compounds inhibiting key molecules of the NHEJ pathway, we established a targeting strategy that reduced the components used for gene targeting by inserting shRNA expression cassettes into the Cas9 expression vector that specifically target XRCC4 and DNAPK. Assembling these two shRNAs beside sgRNA in one CRISPR-Cas9 plasmid facilitated their co-expression and thereby improved the efficiency of SOX2 gene KI by nearly twofold.
The stress induced by the transfection procedures as well as the toxicity of Cas9 overexpression oppose efficient gene targeting. Human stem cells suffer rapid cell death after DNA damage-mediated induction of the TP53 signaling pathway. 14 Moreover, human stem cells are sensitive to DSBs induced by the CRISPR-Cas9 system, resulting in activation of TP53 signaling, leading to a decrease of cell survival and an induction of apoptosis pathways.30,31 Accordingly, massive cell death was observed following vector electroporation, which led to a decrease in the efficiency of CRISPR-Cas9 gene targeting. Overexpression of BCL-XL, a major anti-apoptotic gene, led to an increase in iPSC survival and improved gene editing efficiency. Thus, it enhanced cell survival 1–2 days after electroporation and resulted in an increase in the efficiency of both HDR-mediated KI and NHEJ-mediated KO. 32
To find an alternative for BCL-XL to increase cell survival at the time of gene targeting, we tested miRNA-21, one of the best-known miRNAs that it is involved in cell survival and inhibiting apoptosis. APAF1 interacts with free cytochromes, forming an APAF1/pro-caspase9 heterodimer. miRNA-21 suppresses APAF1 expression and thus inhibits further activation of caspases. The activated caspase9 induces caspase3, which in turn triggers apoptosis. Thus, miRNA-21 indirectly inhibits caspase3 activation and downregulates apoptosis. Further studies show that overexpression of miRNA-21 leads to anti-apoptotic actions and increases cell survival.33,34 To overexpress BCL-XL, another overexpression plasmid is necessary in combination with targeting vector and Cas9 expression vector. However, because of the small size of the genomic region encoding miRNA-21, we successfully assembled a dual expression system of sgRNA–miRNA21 on the Cas9 expression vector and guaranteed simultaneous expression in the target cells. Using this approach, co-expression of miRNA-21 and the CRISPR-Cas9 system increased cell survival, and thereby improved gene targeting efficiency of SOX2 KI. To our knowledge, this is the first time it has been shown that a miRNA indirectly improves the gene targeting efficiency of CRISPR-Cas9 system.
Conclusion
Our approaches demonstrated that transient co-expression of shRNAs targeting the main components of NHEJ, XRCC4, and DNA-PK, as well as transient co-expression of miRNA-21 with CRISPR-Cas9 genome engineering system, increase gene targeting efficiency. Our new CRISPR-Cas9 targeting approach dispenses of additional plasmids or small compounds and still counteracts the NHEJ pathway or stress-induced cell death. Improved targeting efficiency reduces the downstream workload needed to screen and identify the cells having an exact gene edit mediated by HDR-based recombination.
Footnotes
Acknowledgment
We thank Prof. Ralf Kuhn for providing the CRISPR expressing plasmid.
Author Disclosure Statement
The authors declare no conflicts of interest related to this paper.
Funding Information
This work was supported by the Helmholtz-Gemeinschaft (Helmholtz Portfolio Theme Metabolic Dysfunction and Common Disease) and Deutsches Zentrum für Diabetesforschung (DZD).