
Abstract
Prime editors (PEs) were developed to induce versatile edits at a guide-specified genomic locus. With all RNA-guided genome editors, guide-dependent off-target (OT) mutations can occur at other sites bearing similarity to the intended target. However, whether PEs carry the additional risk of guide-independent mutations elicited by their unique enzymatic moiety (i.e., reverse transcriptase) has not been examined systematically in mammalian cells. Here, we developed a cost-effective sensitive platform to profile guide-independent OT effects in human cells. We did not observe guide-independent OT mutations in the DNA or RNA of prime editor 3 (PE3)-edited cells, or alterations to their telomeres, endogenous retroelements, alternative splicing events, or gene expression. Together, our results showed undetectable prime editing guide RNA–independent OT effects of PE3 in human cells, suggesting the high editing specificity of its reverse-transcriptase moiety.
Introduction
Prime editors (PEs) are engineered gene-editing tools that combine a CRISPR-Cas9 RNA-guided gene targeting system with a reverse transcriptase (RTase) that generates precise genetic alterations at target genomic DNA loci dictated by a template carried on the prime editing guide RNA (pegRNA).1,2 By using a nicking single-guide RNA (sgRNA) together with the Cas9-RTase fusion protein, the prime editor 3 (PE3) system was developed to induce high levels of on-target editing.1,2 PEs have been successfully applied to induce targeted base substitutions, small deletions, and insertions in mammalian and plant cells.1,3
Distinct to previously reported base editors (BEs), which can generate on-target C-to-T changes with the combination of CRISPR-Cas9 and APOBEC cytidine deaminase, PEs are much more versatile and able to deliver many different types of edits. As PEs can theoretically correct most of the mutations associated with genetic disorders, whether PEs induce off-target (OT) effects is of great importance for their potential future clinical applications. 4 Previous studies have shown that PEs can induce low levels of pegRNA-dependent mutations at OT sites that have sequence similarity to on-target sites, similar to the guide-dependent OT mutations seen with virtually all CRISPR effectors.4,5
Additionally, cytosine base editors (CBEs) have been shown to introduce guide RNA (gRNA)-independent OT mutations in genomic DNA and cellular RNA,6–9 and adenosine base editors (ABEs) have been shown to introduce gRNA-independent OT mutations in cellular RNA,8,9 owing to the activity of the deaminase moiety of CBEs and ABEs that can act on nucleotides independent of the Cas9 component's binding to target sites specified by the gRNA.10,11
A recent study reported that PE induced no significant pegRNA-independent OT mutations in the genomic DNA of plants. 12 However, whether the effector moiety (RTase) of PEs induces pegRNA-independent OT effects on genomic DNA or transcriptomic RNA in mammalian cells has not been systematically examined, and remains a key open question for the potential therapeutic applications of PEs.
In this study, we performed genomic and transcriptomic sequencing analyses to profile any pegRNA-independent OT effects of a commonly used prime editing effector, PE3, in human cells. We found that PE3 did not induce detectable OT mutations, alterations of telomeric regions, or changes to endogenous retrotransposons in genomic DNA, or OT mutations, altered splicing patterns, or altered gene expression in the transcriptome. All these data together indicate the high editing specificity of the RTase moiety of PE3.
Methods
Plasmid construction
Oligonucleotides hRNF2_FOR/hRNF2_REV were annealed and ligated into BsaI linearized pGL3-U6-gRNA-PGK-eGFP to generate the vector psgRNF2 for the expression of sgRNF2. Oligonucleotides +41_nicking-hRNF2 _FOR/+41_nicking-hRNF2 _REV were annealed and ligated into BsaI linearized pGL3-U6-gRNA-PGK-puromysin to generate the vector pnsgRNF2 for the expression of nicking sgRNF2(+41). Other sgRNA and nicking sgRNA expression vectors were constructed by a similar strategy. The primer sets (pegRNF2_F/pegRNF2_R) were used to amplify the fragment scaffold-pegRNF2 with the template pU6-pegRNA-GG-Acceptor.
Then, the amplified fragment scaffold-pegRNF2 was cloned into BsaI and EcoRI linearized pU6-pegRNA-GG-Acceptor to generate the vector pU6-pegRNF2. Other pegRNA expression vectors were constructed by a similar strategy. The primer set (pTRE3G_PE2_F/pTRE3G_PE2_R) was used to amplify the fragment SV40NLS-Cas9(H840A)-MMLV-bGH poly(A) with the template pCMV-PE2. Then, the amplified fragment SV40NLS-Cas9(H840A)-MMLV-bGH poly(A) was cloned into MluI and SmaI linearized pTRE3G-Bio-PupE-IRES-BFP to generate the vector pTRE-PE2.
The sequences of the oligos used for plasmid construction are summarized in Supplementary Table S1.
Cell culture and transfection
HEK293FT (from Invitrogen) or HEK293FT A3–/– cells were maintained in Dulbecco's modified Eagle's medium (DMEM; 10566; Gibco/Thermo Fisher Scientific) +10% fetal bovine serum (FBS; 16000-044; Gibco/Thermo Fisher Scientific) and regularly tested to exclude mycoplasma contamination.
For base editing with hA3A-BE3 and gene editing with Cas9, the cells were transfected with 250 μL serum-free Opti-MEM that contained 2.52 μL Lipofectamine LTX (Life, Invitrogen), 0.84 μL Lipofectamine PLUS (Life, Invitrogen), 0.5 μg pCMV-hA3A-BE3 (or pCMV-spCas9) expression vector, and 0.34 μg sgRNA expression vector.
For prime editing with PE3, the cells were transfected with 250 μL serum-free Opti-MEM that contained 3.9 μL Lipofectamine LTX, 1.3 μL Lipofectamine PLUS, 0.9 μg pCMV-PE2 expression vector, 0.3 μg pegRNA expression vector, and 0.1 μg nicking sgRNA expression vector.
For enhanced green fluorescent protein (eGFP) expression, the cells were transfected with 250 μL serum-free Opti-MEM that contained 1.5 μL Lipofectamine LTX (Life, Invitrogen), 0.5 μL Lipofectamine PLUS (Life, Invitrogen), and 0.5 μg pCMV-eGFP expression vector.
For prime editing with PE3 in the Tet-On system, the cells were transfected with 250 μL serum-free Opti-MEM that contained 6.6 μL Lipofectamine LTX, 2.2 μL Lipofectamine PLUS, 0.9 μg pTRE3G-PE2 expression vector, 0.9 μg prtTA expression vector, 0.3 μg pegRNA expression vector, and 0.1 μg nicking sgRNA expression vector.
Isolation and expansion of edited single-cell clones for whole-genome sequencing
293FT A3–/– cells expanded from a single-cell clone with successful A3 knockout were maintained in DMEM (10566; Gibco/Thermo Fisher Scientific) +10% FBS (16000-044; Gibco/Thermo Fisher Scientific) and regularly tested to exclude mycoplasma contamination. The single-cell clone-derived 293FT A3–/– cells were transfected with genome editors (e.g., PE3, hA3A-BE3, and Cas9) or eGFP-expressing plasmids, and 72 h after transfection, single cells were sorted onto 96-well plates with BD FACSAria III.
For testing the effect of PE expression time on pegRNA-independent OT mutations, the single-cell clone-derived 293FT A3–/– cells were co-transfected with TRE-PE2 expression vector, reverse tetracycline-controlled transactivator (rtTA) expression vector, pegRNA expression vector, and nicking gRNA expression vector and then cultured with or without doxycycline (1 μg/mL) induction for 72 h.
Alternatively, doxycycline was added into the media of transfected cells for the first 24 h and then changed to fresh media without doxycycline for another 48 h culture. After a total of 72 h, single cells were sorted onto 96-well plates with BD FACSAria III. After 18-day clone expansion, the genomic DNA derived from transfected single-cell clones was extracted with QuickExtract™ DNA Extraction Solution (QE09050; Epicentre) for Sanger sequencing, and the genomic DNA with bi-allelic editing was further subjected to whole-genome sequencing (WGS). On average, one bi-allelic edited clone can be obtained from around six to eight clones when the editing efficiency of bulk setting is ∼30–50%.
DNA library preparation and amplicon sequencing
Target genomic sequences were polymerase chain reaction (PCR) amplified by high-fidelity DNA polymerase PrimeSTAR HS (Takara) with primer sets flanking examined target sites. The target sequences and PCR primer sequences are summarized in Supplementary Table S2.
Indexed DNA libraries were prepared by using the NEBNext Ultra II FS DNA Library Prep Kit for Illumina. After quantitated with Qubit High-Sensitivity DNA kit (Invitrogen), PCR products with different tags were pooled together for deep sequencing by using the Illumina Hiseq X Ten (2 × 150) at Omics core of Bio-Med Big Data Center, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai, P.R. China. Raw read qualities were evaluated by FastQC (v0.11.8, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/; parameters: default).
For paired-end sequencing, only R1 reads were used. Adaptor sequences and read sequences with a Phred quality score lower than 30 were trimmed. Trimmed reads were then mapped with the BWA-MEM algorithm (BWA v0.7.17) to target sequences. After being piled up with Samtools (v1.9), base substitutions and insertion/deletion (indel) frequencies at on-target sites were calculated according to previously published literature.13,14
Base substitution frequency calculation
Base substitution of every position at the target sites of examined sgRNAs and pegRNAs was piled up with at least 1,000 independent reads. Base substitution frequencies were calculated by the published CFBI pipeline as: count of reads with substitution at the target base/count of reads covering the target base. Counts of reads for each base at examined target sites are described in Supplementary Table S3.
Intended indel frequency calculation
Intended indel frequencies were calculated as: count of reads with only intended indel at the target site/count of total reads covering the target site. These counts are described in Supplementary Table S4.
Unintended indel frequency calculation
Unintended indel frequencies for base substitution were estimated among reads aligned in the region spanning from upstream 8 nt to the target site to downstream 19 nt to the protospacer adjacent motif site (50 bp). Unintended indel frequencies for base substitution were calculated as: count of reads containing at least one unintended inserted and/or deleted nucleotide/count of total reads aligned in the estimated region. Unintended indel frequencies for targeted indels were estimated among reads aligned at the target site. Unintended indel frequencies for targeted insertion/deletion were calculated as: count of reads containing unintended indels/count of total reads covering the target site. The counts for on-target are described in Supplementary Table S4.
WGS and data analysis
Genomic DNA was extracted from transfected 293FT A3–/– single-cell clones by using cell DNA isolation kit FastPure® (DC102-01; Vazyme). Indexed DNA libraries were prepared by using NEBNext Ultra II FS DNA Library Prep Kit for Illumina. A total of 12 Tb WGS data were obtained by using Illumina Hiseq X Ten (2 × 150) at Omics core of Bio-Med Big Data Center, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai, P.R. China. The average coverage of sequencing data generated for each transfected 293FT A3–/– single-cell clone sample was 14 × with the published BEIDOU 15 toolkit to call high-confident base substitution or indel events that could be identified by all three different callers, GATK, 16 Lofreq, 17 and Strelka2. 18
Briefly, to reduce the impact of varying sequence depth among samples, 120M reads were randomly sampled by Seqtk (v1.3, https://github.com/lh3/seqtk; parameters: sample -s100 120000000) from raw data for further analyses.
After quality control by FastQC (parameters: default), WGS DNA-seq reads were trimmed by Trimmomatic (v0.38, parameters: ILLUMINACLIP:TruSeq3-PE-2.fa: 2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36) 19 to remove low-quality read sequences. BWA-MEM algorithm (v0.7.17; parameters: default) was used to map clean reads to the human reference genome (hg38). Samtools (v1.9; parameters: -bh -F 4 -q 30) was used to select reads with mapping quality scores ≥30 and convert SAM files to sorted BAM files. After marking duplicate reads by Picard (v2.21.2; parameters: REMOVE_DUPLICATES = false) in the BAM file, GATK (v4.1.3.0) was employed to correct systematic bias by a two-stage process (BaseRecalibrator and ApplyBQSR; parameters: default).
Single-nucleotide variations of OT mutations were individually computed by the BEIDOU toolkit with three algorithms—GATK, Lofreq (v2.1.3.1; parameters: default), and Strelka2 (v2.9.10; parameters: default)—with workflows for the germline variant calling. Genome-wide indels were also detected by the BEIDOU toolkit with GATK, Strelka2 (parameters: default), and Scalpel (v0.5.4; parameters: –single –window 600). 20
For GATK, genome-wide de novo variants were determined by three GATK commands: HaplotypeCaller (parameters: default), VariantRecalibrator (parameters: “–resource:hapmap,known = false,training = true,truth = true,prior = 15.0 hapmap_3.3.hg38.vcf.gz –resource:omni,known = false,training = true,truth = false,prior = 12.0 1000G_omni2.5.hg38.vcf.gz –resource:1000G,known = false,training = true,truth = false,prior = 10.0 1000G_phase1.snps.high_confidence.hg38.vcf.gz –resource:dbsnp,known = true,training = false,truth = false,prior = 2.0 dbsnp_146.hg38.vcf.gz -an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR -an DP –max-gaussians 4” for SNVs; “-resource:mills,known = true,training = true,truth = true,prior = 12.0 Mills_and_1000G_gold_standard.indels.hg38.vcf.gz -an QD -an MQRankSum -an ReadPosRankSum -an FS -an SOR -an DP –max-gaussians 4 -mode INDEL” for indels), and ApplyVQSR (parameters: “-mode SNP -ts-filter-level 95” for SNVs; “-mode INDEL -ts-filter-level 95” for indels).
VCF files used for VariantRecalibrator were downloaded from https://ftp.ncbi.nih.gov/snp/ and https://console.cloud.google.com/storage/browser/genomics-public-data/resources/broad/hg38/v0. Of note, overlaps of SNVs/indels called by these three algorithms were considered as reliable variants by the BEIDOU toolkit. Further, to obtain de novo SNVs/indels, we filtered out the background variants, including: (1) SNVs/indels in nontransfected cells of this study and dbSNP (v151; http://www.ncbi.nlm.nih.gov/SNP/) database; (2) SNVs/indels with allele frequencies <10% or depth fewer than 10 reads; and (3) SNVs/indels overlapped with the UCSC repeat regions. Analyses were only focused on SNVs/indels from canonical (chr 1–22, X, Y, and M) chromosomes.
Genome-wide base substitutions and indels are described in Supplementary Table S5.
Telomere length calculation and variant calling from WGS data
Telseq 21 (parameters: -k 2) was used to calculate the telomere lengths. BAM files containing mapped WGS DNA-seq reads processing from the BEIDOU toolkit 15 were as the input of Telseq. Telomeric repeat variants, the sequence fragments within telomeric reads that differ from the canonical telomeric repeat pattern (TTAGGG in human), were identified by Computel 22 (v1.2; parameters: default) with trimmed WGS FASTQ files as input. Telomere length and the numbers of telomeric repeat variants are described in Supplementary Table S5.
Copy numbers analysis of endogenous retrotransposons
After trimming and sampling, WGS reads were mapped to Alu and L1 sequence by BWA-MEM algorithm (v0.7.17; parameters: default). Reads mapped to the terminal end of Alu and L1 and singleton mapped reads are remapped to hg38 reference genome to locate the genomic position. Note, only uniquely mapping reads are taken into consideration. Copy numbers of Alu and L1 are described in Supplementary Table S5.
RNA extraction, whole-transcriptome sequencing, and data analysis
The conditions of PE3, hA3A-BE3, Cas9, and eGFP transfection were same as the ones for genomic DNA extraction described above. Forty hours after transfection, transfected cells in the top 10% of the fluorescence intensity were sorted by BD FACSAriaIII, and total RNAs of sorted cells were extracted by using the Rneasy Mini Kit (Qiagen #74104).
RNA-seq libraries were prepared using Illumina TruSeq Stranded Total RNA LT Sample Prep Kit. Size-selected libraries were subjected to deep sequencing with Illumina Hiseq X Ten (2 × 150) at Omics core of Bio-Med Big Data Center, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai, P.R. China. RNA-seq reads were trimmed by Trimmomatic (v0.38; parameters: ILLUMINACLIP:TruSeq3-PE-2.fa: 2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36) to remove low-quality read sequences, and read qualities were evaluated by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). RNA editing sites were called by the published RADAR pipeline. 23
Gene expression was determined by fragments per kilobase of transcript per million mapped reads (FPKM) with featureCounts 24 (v1.6.3,–fraction -O -t exon -g gene_id) using the bam file processing from RADAR pipeline as input. Transcriptome-wide mutations and Pearson correlation coefficients of gene expression are described in Supplementary Table S6.
For alternative splicing, rMATS 25 (v4.1.0; parameters: -t paired –libType fr-firststrand –tophatAnchor 8 –cstat 0.0001 –tstat 6) was used to calculate the p-value and false discovery rate (FDR) of differential splicing with the trimmed FASTQ as input, using 5% as the threshold for between-group difference in exon inclusion levels, 0.05 as the threshold for FDR, and 20 as the threshold for JC read coverage (|Δψ| >5%, FDR ≤0.05, and JC_Read_coverage >20). Alternative splicing events are described in Supplementary Table S7.
Statistical analysis
All statistical analyses were performed with R v3.6.2 (The R Foundation for Statistical Computing, Vienna, Austria). p-Values were calculated from one-tailed Student's t-tests in this study. In this study, p-values <0.05 were considered to be significant.
Availability of data and material
The original WGS data can be accessed in the NCBI Sequence Read Archive (accession: PRJNA731612). The original DNA deep-sequencing and RNA sequencing data from this study can be accessed in the NCBI Gene ExpressionOmnibus (GEO: GSE178117). The BEIDOU toolkit that calls high-confident base substitution or indel events from WGS data is available at https://github.com/YangLab/BEIDOU. 15 The custom Perl and Shell scripts for calculating frequencies of base substitution and indels (CFBI) are available at GitHub (https://github.com/YangLab/CFBI).
The RADAR pipeline that detects and visualize all possible 12 types of RNA editing events from RNA-seq data is available at https://github.com/YangLab/RADAR. 23
Results
Developing a platform for sensitive detection of guide-independent OT mutations in human cells
Because gRNA-independent OT mutations are induced by the effector moiety of genome editors (e.g., the cytidine deaminase component of CBE) at random sites in the genome of individual cells, 10 traditional methods for OT mutation detection that work by sequencing targeted amplicon(s) from a population of edited cells are not able to identify this type of OT mutation. Previous studies have detected BE3-induced gRNA-independent OT mutations by expanding and then WGS-edited single mouse embryonic cells or human induced pluripotent stem cells (iPSCs), both of which have relatively low levels of background mutations.7,26 However, these experiments in murine embryonic cells and human iPSCs require special expertise to perform.
Thus, we sought to set up a practical way to evaluate gRNA-independent OT mutations induced by genome editors in cultured cell lines and to apply our method to evaluate gRNA-independent OT mutations instilled by PE3 genome and transcriptome wide.
Recently, we used WGS to analyze whether BEs induce the gRNA-independent OT mutations on cytosines when BEs were used to correct a Pgm3 mutation in 293FT cells. 15 In order to reduce the background mutations occurring on cytosine, the endogenously expressed APOBEC3 (A3) gene cluster was knocked out in that cell line. 15 In this study, we also knocked out the endogenously expressed A3 in wild-type (WT) 293FT cells (Supplementary Fig. S1A–D) and then examined the effect of A3 knockout on all types of background mutations, including all types of base substitutions and indels. We first transfected an eGFP-expressing plasmid into WT 293FT or 293FT A3–/– cells, and 72 h after transfection, single cells were sorted onto 96-well plates.
After clonal expansion, the genomic DNA of single-cell clones was subjected to WGS (Fig. 1A). WGS data analyzed by a previously published BEIDOU toolkit (https://github.com/YangLab/BEIDOU) 15 showed that the knockout of the A3 cluster significantly reduced the number of genome-wide base substitutions (Supplementary Fig. S1E; from ∼4,900 to ∼1,600; p = 6 × 10–5). Interestingly, genome-wide indels (Supplementary Fig. S1F; from ∼400 to ∼20; p = 2 × 10–8) in 293FT A3–/– cells were also significantly fewer than those in WT 293FT cells, consistent with a previous study showing that the expression of APOBEC3 genes can also induce indel formation. 27
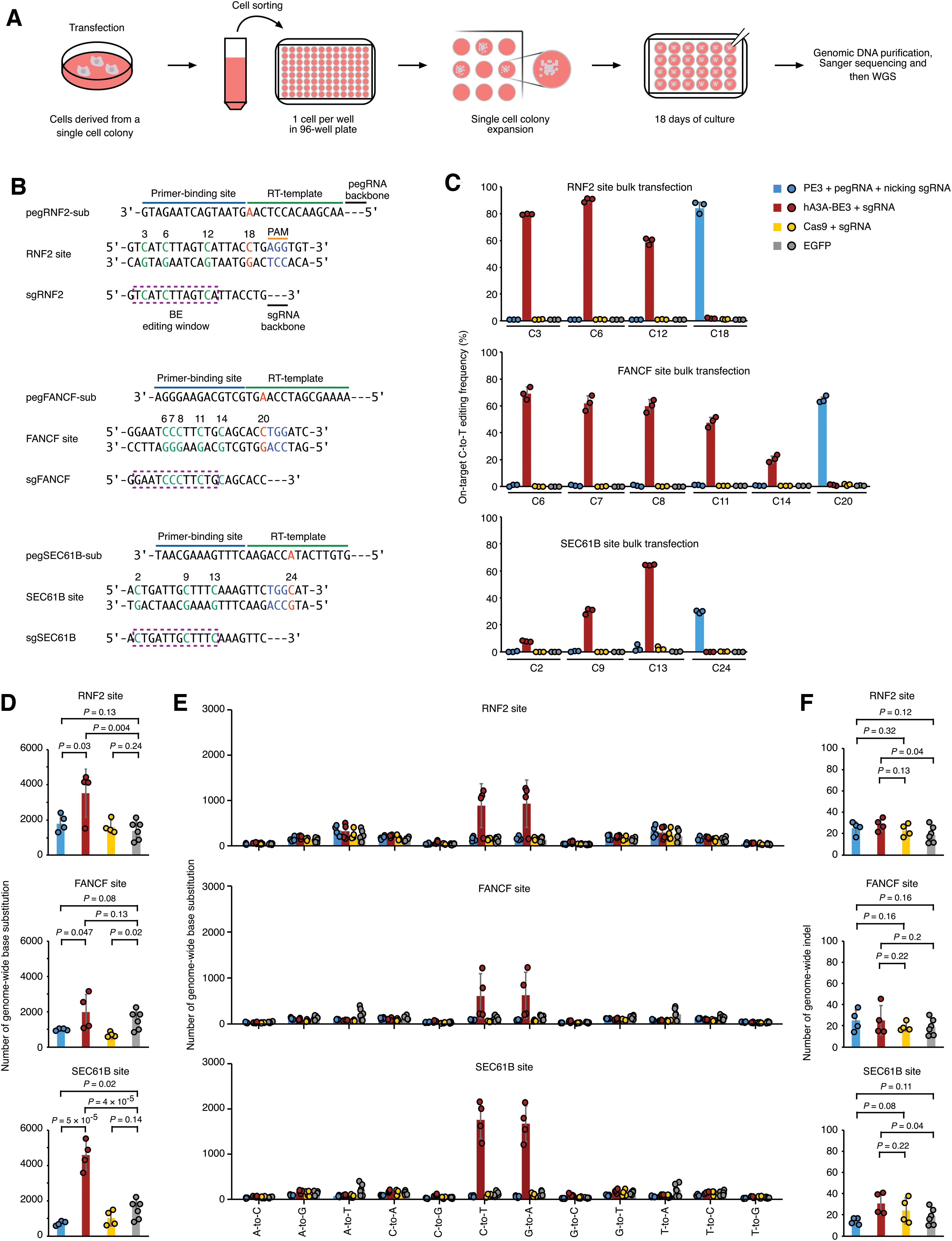
Prime editor 3 (PE3) induced no detectable prime editing guide RNA (pegRNA)-independent off-target (OT) mutations in genomic DNA when generating targeted base substitutions.
These results indicated that the knockout of A3 significantly reduced the background mutations, including base substitutions and indels in 293FT cells. We then used the 293FT A3–/– cell line with a low mutation background in subsequent experiments to evaluate the pegRNA-independent OT effects induced by PE3 genome wide.
No evidence for pegRNA-independent OT mutations in genomic DNA with PE3 generating base substitutions
First, we sought to determine whether PE3 induces pegRNA-independent OT mutations genome wide. For this analysis, we evaluated 293FT A3–/– cells treated with PE3 and pegRNA targeting several different loci and compared them to eGFP-treated cells as the negative control or cells treated with a previously reported BE (hA3A-BE3) 14 as a positive control.
To confirm the editing efficacy of PE3, we compared the on-target editing efficiencies of PE3 to hA3A-BE3 with amplicon sequencing in a bulk transfection setting. 293FT A3–/– cells were transfected with (1) three plasmids expressing PE, pegRNA (Fig. 1B), and the optimized nicking sgRNA 1 (Supplementary Fig. S2A); (2) two plasmids expressing hA3A-BE3 and sgRNA to serve as a positive control, since this combination is known to induce sgRNA-independent OT mutations; (3) two plasmids expressing Cas9 and sgRNA to serve as a negative control, since this combination is known to induce no sgRNA-independent OT mutation; or (4) one plasmid expressing eGFP as another negative control.
Seventy-two hours after transfection, a portion of transfected cells was lysed to extract genomic DNA to examine on-target editing frequencies. As specified by the provided pegRNAs, PE3 generated C-to-T substitutions at RNF2, FANCF, and SEC61B target sites, with editing efficiencies (∼30–80%) similar to those of hA3A-BE3 (Fig. 1C). Consistent with previous studies showing that both PE3 and hA3A-BE3 triggered indels,1,14 we found that both editors induced indels at these on-target sites (with ∼1–10% indel frequencies), but fewer than those by Cas9 (with ∼50% indel frequencies; Supplementary Fig. S2B).
Another portion of transfected cells was sorted to single cells and expanded on 96-well plates. After culturing, the single-cell clones for which Sanger sequencing confirmed bi-allelic edits (100% editing frequency) at on-target sites (Supplementary Fig. S3A) were then subjected to WGS to identify OT mutations and analyzed by the BEIDOU toolkit on a genome-wide scale (Supplementary Fig. S4A).
We found the numbers of OT base substitutions in PE3-treated (Fig. 1D; p = 0.13, 0.08, and 0.02 for RNF2, FANCF, and SEC61B, respectively) and Cas9-treated single-cell clones (Fig. 1D; p = 0.24, 0.02, and 0.14 for RNF2, FANCF, and SEC61B, respectively) were similar to or even smaller than those in eGFP-treated single-cell clones, suggesting that PE3 and Cas9 induced no observable genome-wide OT base substitution.
Interestingly, PE3 and Cas9 induced significantly less genome-wide base substitutions than the eGFP control (Fig. 1D) in some samples, which is possibly due to the variation of the endogenous mutation background in different clones. In contrast, hA3A-BE3 induced more OT base substitutions than eGFP control (Fig. 1D; p = 0.004, 0.13, and 4 × 10–5 for RNF2, FANCF, and SEC61B, respectively), which is in line with previous reports showing that BE3 introduces substantial levels of gRNA-independent OT mutations in mouse embryos and plants.6,7
We further analyzed the types of base substitutions and found significantly more C-to-T or G-to-A substitutions than other subtypes of base substitutions (Fig. 1E and Supplementary Fig. S3B) induced by hA3A-BE3, while C-to-T or G-to-A substitutions were not enriched in cells treated with the other genome editors (Fig. 1E).
We also analyzed the sequence contexts of base substitutions induced by hA3A-BE3 and found that the OT mutation sites by WGS have little sequence similarity to on-target sites or the potential sgRNA/pegRNA-dependent or nicking sgRNA-dependent OT sites predicted by Cas-OFFinder 28 (Supplementary Fig. S5A and B). This finding is in line with previous studies6,7,29,30 and indicates that these OT mutations are gRNA-independent and randomly induced by the cytidine deaminase moiety of hA3A-BE3.10,11
Moreover, we used Sanger sequencing to confirm the gRNA-independent OT mutations we observed for hA3A-BE3 at selected loci that are associated with human diseases, and indeed we found mutations at these pathogenic sites in single-cell clones (Supplementary Fig. S5C). Other than base substitutions, we also analyzed whether PE3 induced genome-wide OT indels (Supplementary Fig. S4B) when generating on-target base substitutions. It was shown that PE3-treated single-cell clones only had background numbers of genome-wide indels compared to Cas9- or hA3A-BE3-treated clones or eGFP control (Fig. 1F).
To test whether the expression level or duration of treatment with PE3 has an effect on these pegRNA-independent OT mutations, we transfected different amounts of plasmids to express PE, pegRNA, and nicking sgRNA (Fig. 2A) or altered the duration of the editing time prior to collecting cells for analysis (Fig. 2D). We observed time and dose dependence of on-target editing efficiency (Fig. 2A and D), but in all tested conditions, PE3 did not generate pegRNA-independent OT mutations compared to the eGFP control (Fig. 2B, C, E, and F). These results together demonstrated that PE3 induced no detectable pegRNA-independent OT base substitutions or indels when applied to introducing single-base changes in 293FT cells.
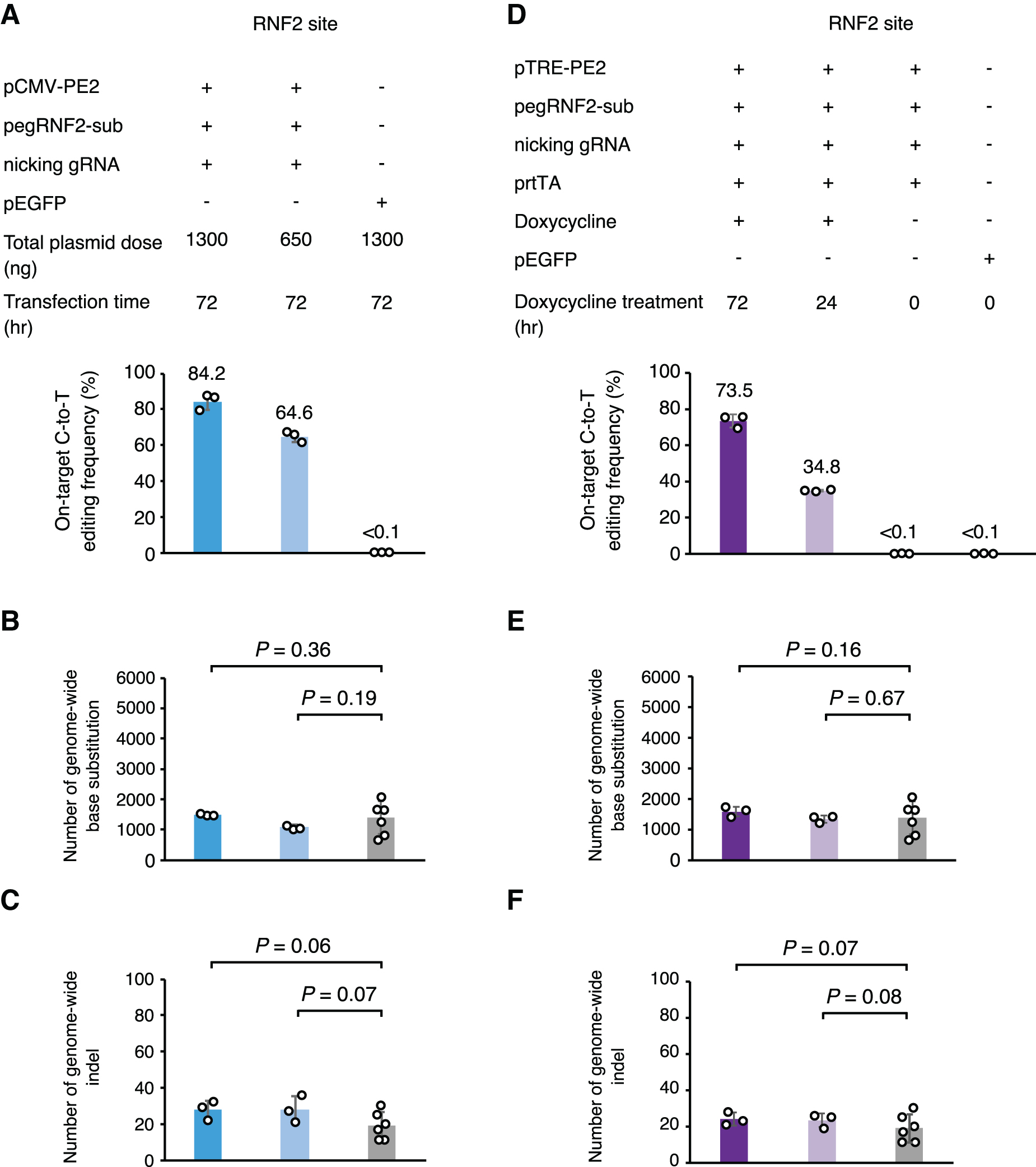
PE3 induced no observable pegRNA-independent OT mutations when expressed at a range of different doses or time periods.
No evidence for pegRNA-independent OT mutations in genomic DNA with PE3 introducing small indels
An advantage of applying PE3 for genome editing compared to other effectors is to introduce precise small indels at targeted sites.1,3 Therefore, we tempted to investigate pegRNA-independent OT effects when applying PE3 for generating specific, pegRNA-directed indels at target sites. Following the same strategy shown in Figure 1A, we transfected 293FT A3–/– cells with PE3, Cas9, or eGFP. With three pairs of pegRNA and nicking sgRNA with optimized editing efficiencies 1 (Supplementary Fig. S6A and B), PE3 was used to generate 3 bp deletions at EMX1, HEK1, and LSP1 sites (Fig. 3A), and deep-sequencing results showed that PE3 yielded ∼40–70% intended deletion frequencies in a bulk transfection setting (Fig. 3B).
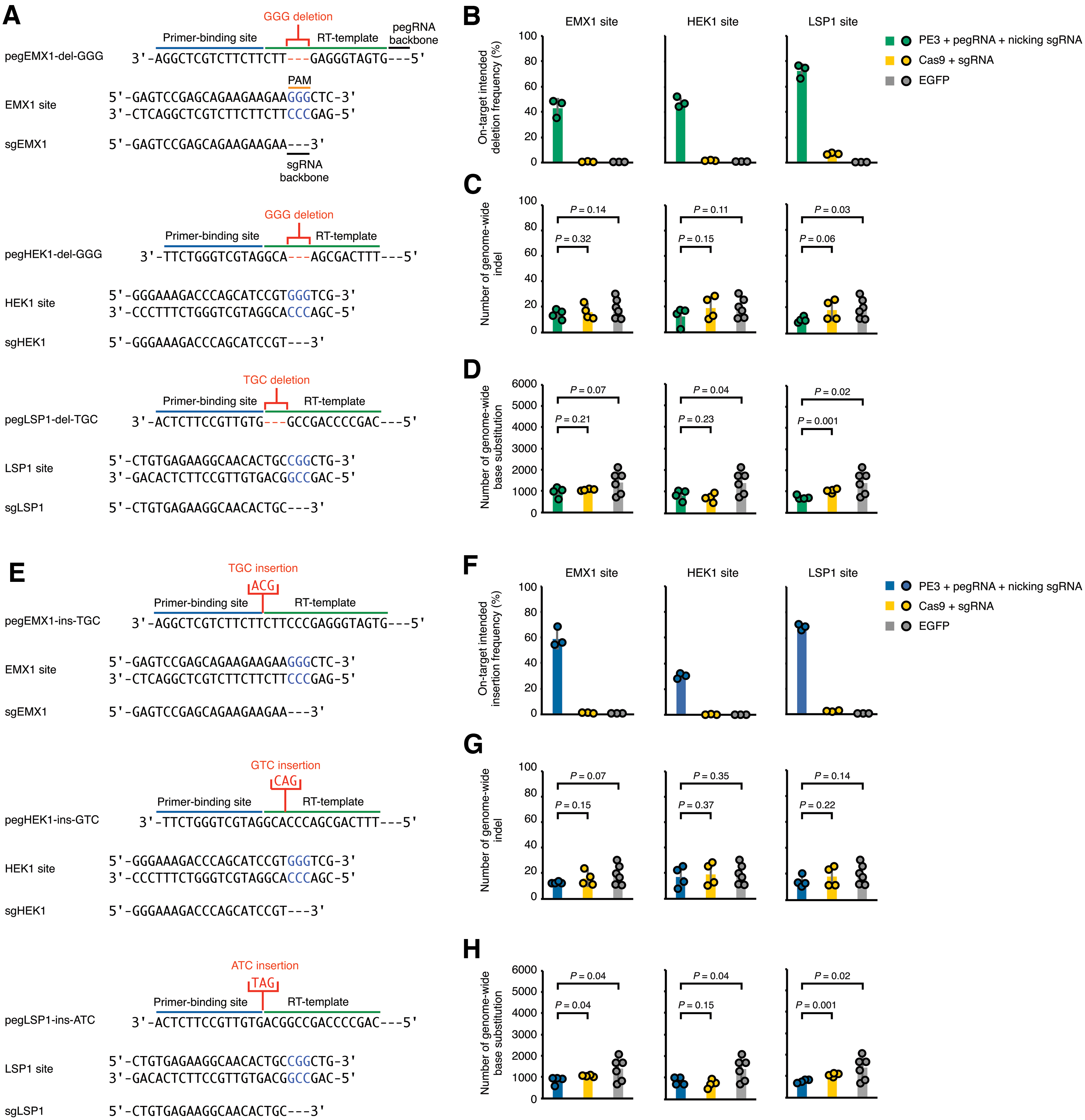
PE3 induced no observable pegRNA-independent OT mutations in genomic DNA when generating small indels.
Next, the whole genomes of edited single-cell clones with biallelic 3 bp deletions (Supplementary Fig. S7A) were sequenced and then analyzed by the BEIDOU toolkit. WGS results showed that PE3 induced similar or slightly fewer OT indels compared to Cas9 (Fig. 3C; p = 0.32, 0.15, and 0.06 for EMX1, HEK1, and LSP1, respectively) or eGFP control (Fig. 3C; p = 0.14, 0.11, and 0.03 for EMX1, HEK1, and LSP1, respectively).
Meanwhile, the numbers of base substitutions induced by PE3 were similar to or slightly smaller than Cas9 (Fig. 3D; p = 0.21, 0.23, and 0.001 for EMX1, HEK1, and LSP1, respectively) and eGFP control (Fig. 3D; p = 0.07, 0.04, and 0.02 for EMX1, HEK1 and LSP1, respectively), although various numbers of base substitutions were found in different single-cell clones treated with the eGFP control, possibly due to underlying subtle genetic differences in different single-cell clones.
Next, we used PE3 and three pairs of pegRNA and nicking sgRNA with optimal editing efficiencies 1 (Supplementary Fig. S6C and D) to generate 3 bp insertions at EMX1, HEK1, and LSP1 genomic sites (Fig. 3E). Deep sequencing showed that PE3 induced ∼30–70% intended insertion frequencies (Fig. 3F) at the on-target sites. WGS of the PE3-edited single-cell clones (Supplementary Fig. S7B) showed that PE3 induced similar or fewer indels (Fig. 3G) and base substitutions (Fig. 3H) compared to Cas9 or eGFP control.
PE3 did not affect telomeric regions or endogenous retroelements in the genomes of edited cells
As the effector moiety of PE3 is a RTase and reverse transcription is correlated to telomere maintenance and endogenous retroelements activity, we asked whether PE3 affected telomere integrity or endogenous retroelements in PE3-treated single-cell clones. We first determined the length of regions that contain telomere repeats in the genomes of edited single-cell clones in which PE3 mediated successful base substitution at on-target sites, and we found that the length of telomeric regions in PE3-edited cells was similar to those in hA3A-BE3-, Cas9-, or eGFP-edited cells (Fig. 4A). In addition, the length of telomeric regions was not affected when PE3 induced indels at on-target sites (Fig. 4C).
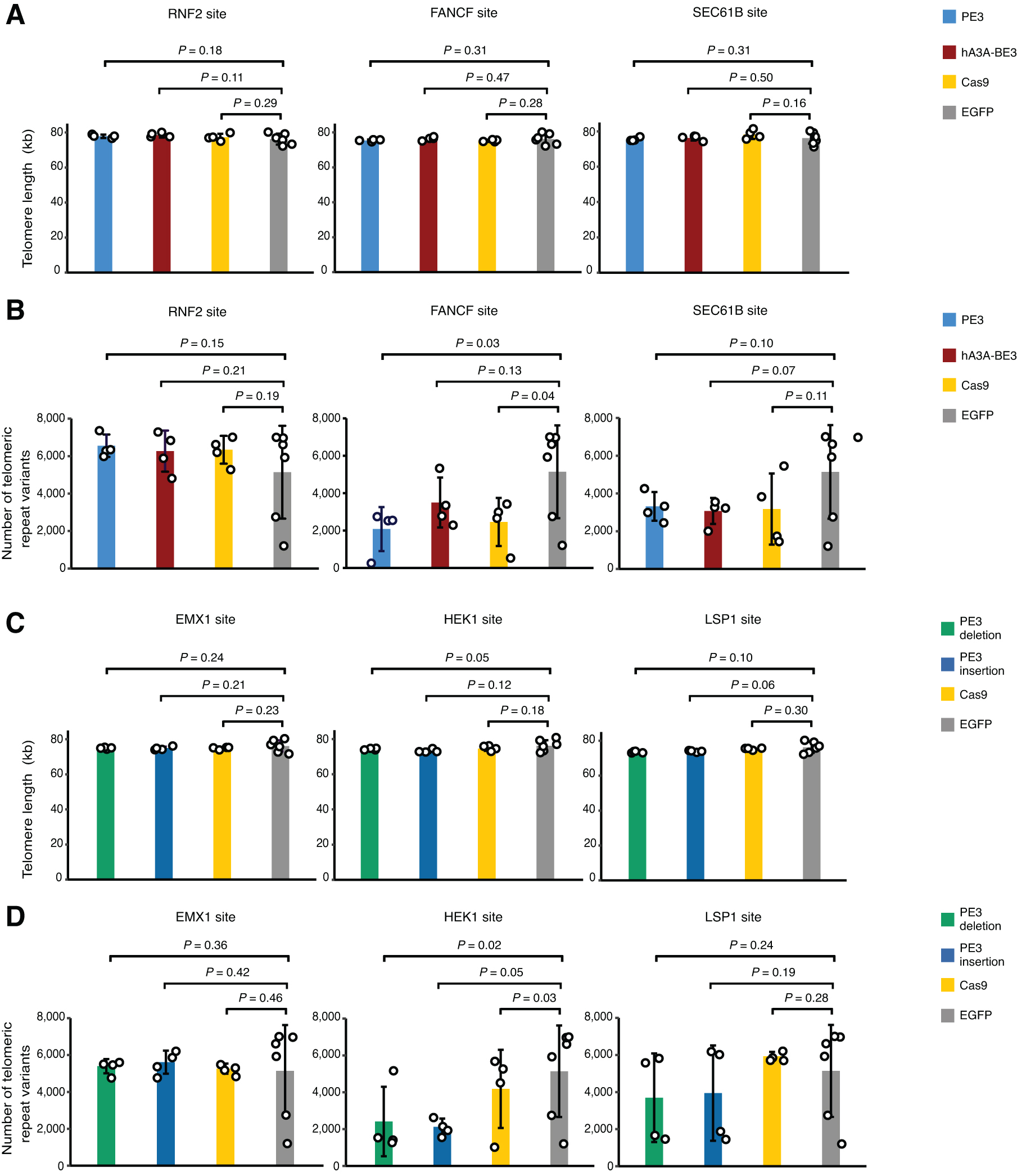
Telomere integrity was not affected by PE3. Telomere lengths
Furthermore, we also examined whether the base variants in telomeric repeat (TTAGGG) were altered by PE treatment. Comparing to BE3, Cas9, or eGFP treatment, the PE3 treatment did not significantly change the number of base variants in telomeric repeats (Fig. 4B and D). Moreover, we analyzed the effect of PE3-mediated editing on the copy numbers of two types of typical endogenous retrotransposons (Alu and LINE-1 [L1]) and found that the copy numbers of Alu and L1 were not significantly altered by the treatment of PE3 (Fig. 5). These data suggested that in spite of containing a MMLV RTase as the effector moiety, PE expression in human cells did not interfere with endogenous telomerase or endogenous retrotransposons.
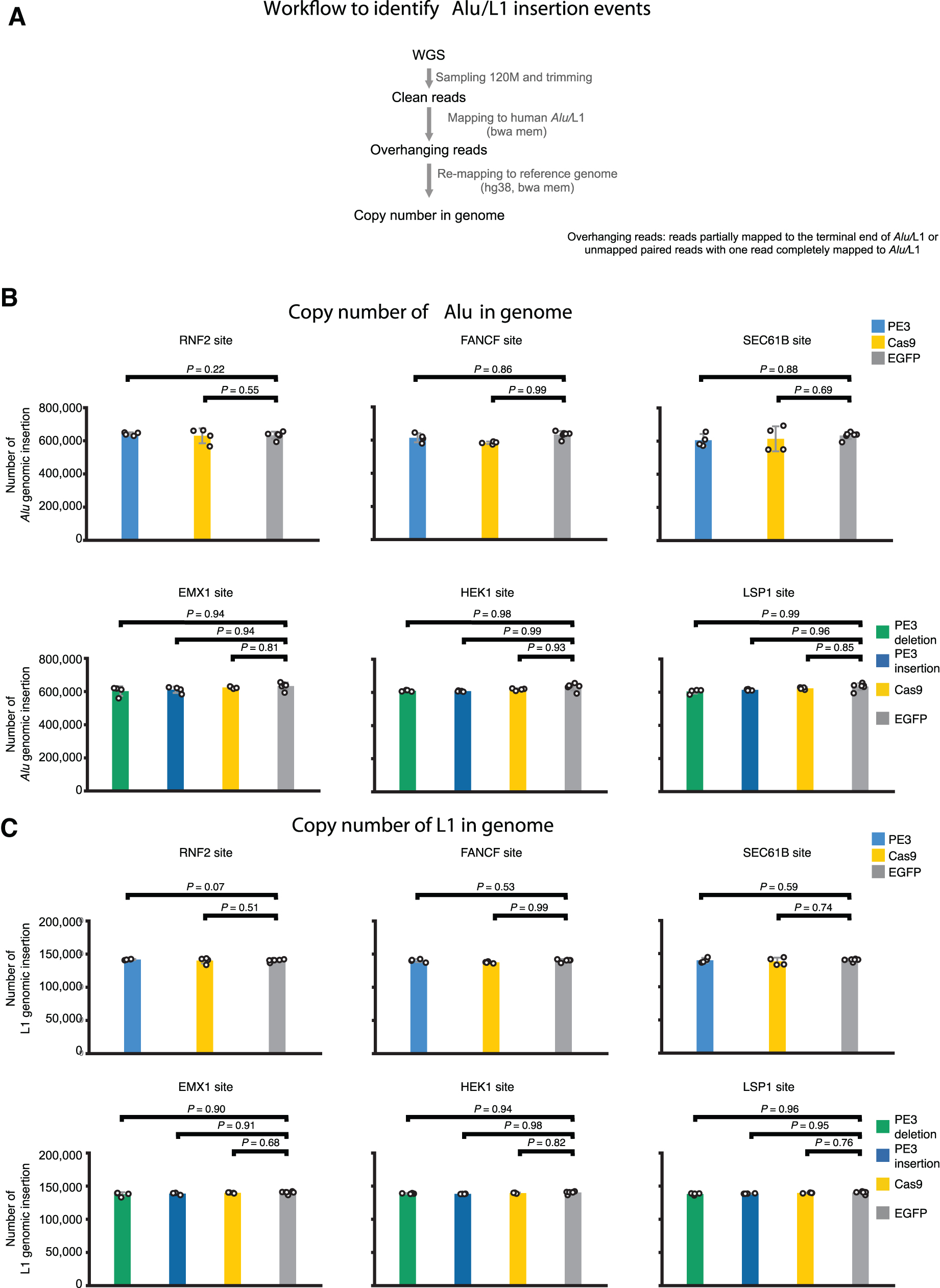
Endogenous retrotransposon copy numbers were not affected by PE3.
No evidence for pegRNA copying into the genome of PE3-edited cells
Previous studies reported instances in which PE3 editing can result in the pegRNA scaffold sequence being copied into the genome at some on-target sites.1,3 We searched for evidence of pegRNA copying into the genome at other sites genome-wide within our WGS data sets from PE3-edited single-cell clones, and we found ∼20 reads containing the pegRNA sequence in PE3-treated genomes (Supplementary Fig. S8A). However, all the pegRNA sequence–containing reads can be fully mapped to the pegRNA-expressing plasmids (Supplementary Fig. S8B), suggesting that these reads originate from the plasmid DNA that was not completely removed during the genomic DNA extraction process, and arguing against the presence of OT insertion of pegRNA-derived sequences.
PE3 induced no observable pegRNA-independent OT mutations in transcriptomic RNAs
In addition to gRNA-independent OT effects on genomic DNA, there have been concerns about possible gRNA-independent alterations to the transcriptome.8,9 Thus, we performed whole-transcriptome sequencing and used the RADAR pipeline 23 to detect whether PE3 induced pegRNA-independent OT mutations in transcriptomic RNA in edited 293FT cells (this time starting from WT cells rather than the A3–/– cells described above), again using hA3A-BE3, Cas9, and eGFP as controls, with the same pegRNAs or sgRNAs targeting the RNF2 site.
We first interrogated the introduction of C-to-U RNA mutations, revealing that there was no statistically significant enrichment for these mutations in PE3-edited cells compared to our negative controls expressing eGFP or Cas9 (Fig. 6A–C). In contrast, hA3A-BE3 induced a significantly higher level of C-to-U mutations in transcriptomic RNA (Fig. 6A–C), suggesting that these mutations were catalyzed by the hA3A deaminase moiety of hA3A-BE3 in a gRNA-independent manner. 15
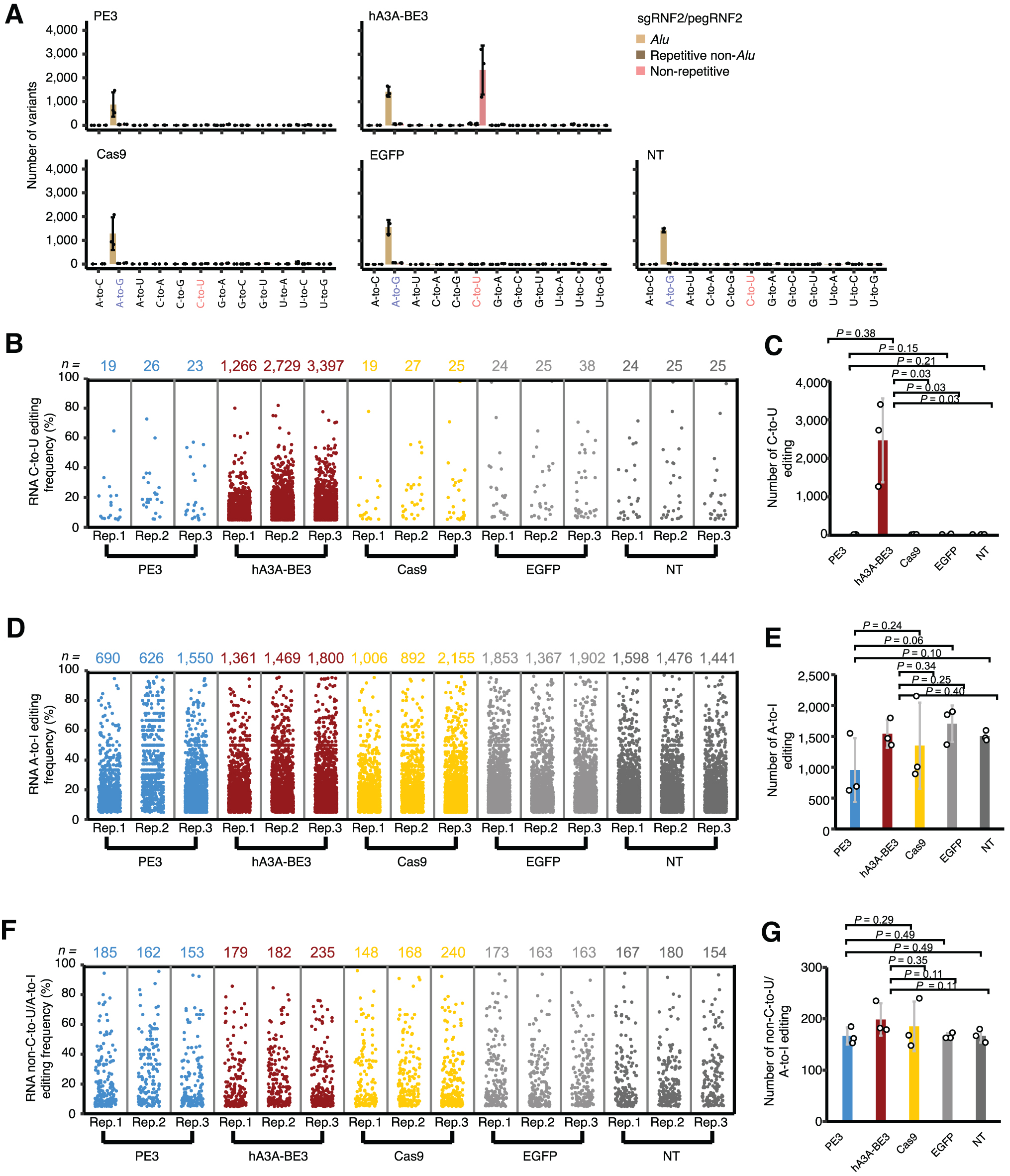
PE3 induced no observable RNA OT mutations transcriptome wide.
Moreover, we also examined other types of mutations in the transcriptomes of cells treated with our panel of genome editors. We found that the transcriptome-wide levels of A-to-I mutations—the major type of RNA editing in mammalian cells—were not significantly altered in cells treated with different genome editors (Fig. 6A, D, and E). We then compared non-C-to-U/A-to-I mutations in transcriptomic RNA and found that all tested genome editors did not significantly affect the levels of these types of mutations (Fig. 6A, F, and G). Altogether, our results indicate that PEs do not significantly alter the RNA mutational profile of the transcriptome above background levels.
PE3 does not alter splicing or gene expression patterns in edited cells
Finally, we examined whether PE3 could affect RNA splicing or gene expression in edited cells. We compared the alternative splicing events (including skipped exons, alternative 5′ splice sites, alternative 3′ splice site mutually exclusive exons, and retained introns) with rMATS 25 in cells treated with PE3, hA3A-BE3, Cas9, or eGFP, or untreated cells (Fig. 7A and B).
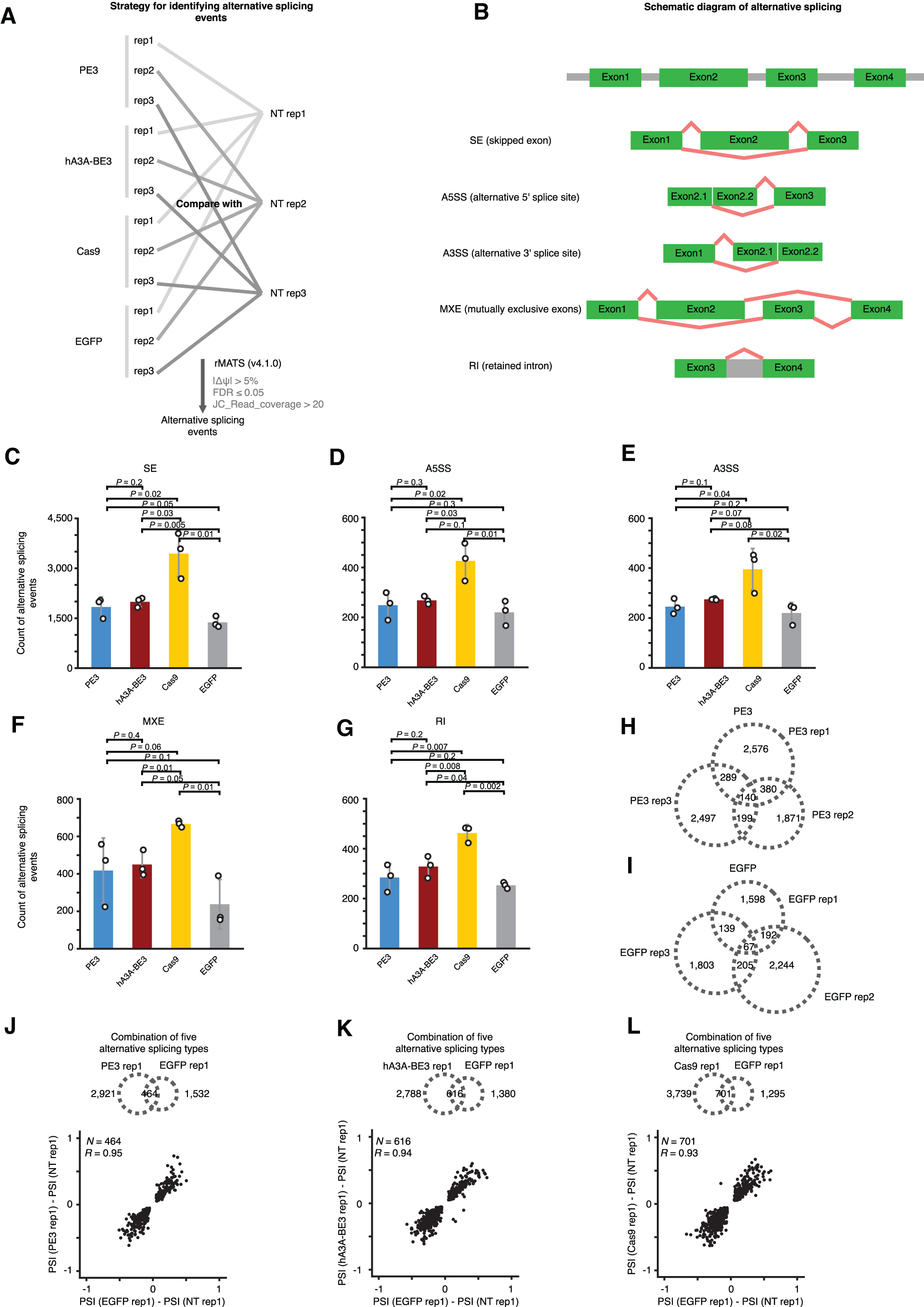
PE3 did not significantly alter the profile of alternative splicing events.
Alternative splicing events in PE3-treated cells were not significantly altered compared to those in eGFP-treated cells (Fig. 7C–G). Surprisingly, Cas9 triggered slightly increased events (around one fold) of alternative splicing, but the generalizability of these findings and the mechanisms by which this might occur remain unclear. We further evaluated the overlap in the profile of alternative splicing events detected between three biological replicates of cells treated with either PE3 or eGFP. Consistent with the high variability of alternative splicing reported previously, 25 only a few alternative splicing events were shared among three replicates of PE3 or eGFP (Fig. 7H and I).
Next, we compared the alternative splicing events between cells treated with PE3, hA3A-BE3, Cas9, or eGFP, and we found that only a few events were shared between different treatments as well. For the overlapping alternative splicing events detected in all of the conditions, the changes of exon-inclusion ratio (percent spliced in) relative to nontransfected cells for each treatment (i.e., PE3, hA3A-BE3, and Cas9) were highly correlated with that of eGFP (Fig. 7J–L; coefficients are 0.95, 0.94, and 0.93, respectively).
We also compared the gene expression profiles of untreated cells with cells treated with PE3, hA3A-BE3, Cas9, or eGFP (Fig. 8A) and did not detect significant alterations in gene expression (evaluated by FPKM) among experimental replicates or across different genome editors (Fig. 8B and C).
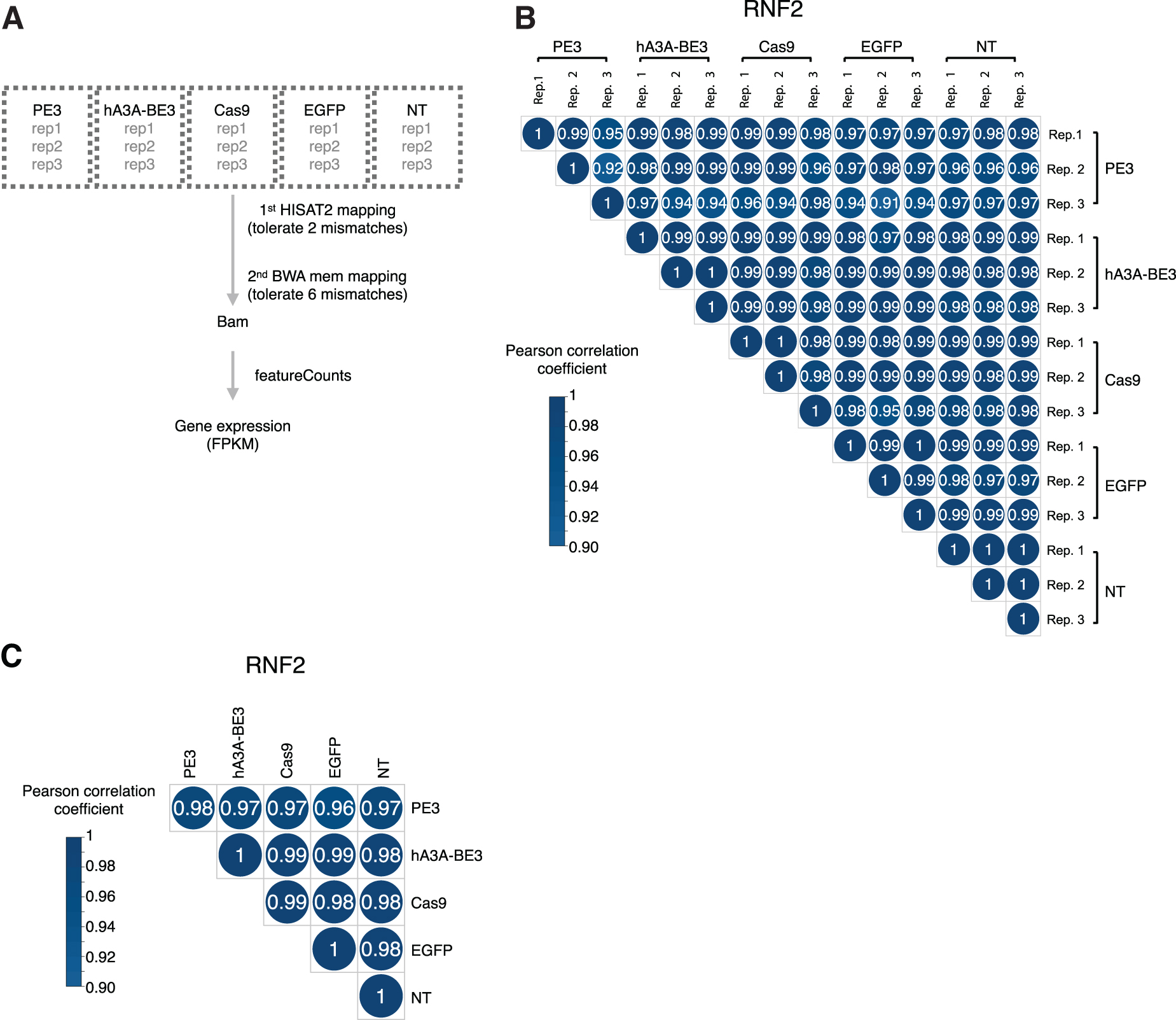
PE3 did not significantly alter cellular gene expression profiles.
Taken together, our results indicate that alternative splicing patterns and gene expression profiles are not disrupted in cells treated with PE3 or other tested genome editors.
Discussion
In this study, we evaluated pegRNA-independent OT effects of PE3 by performing WGS and whole-transcriptome sequencing. The WGS results showed that PE3 induced no observable genomic OT mutations in a pegRNA-independent manner, when generating three types of targeted edits: base substitutions, small deletions, and small insertions (Figs. 1–3). In addition, despite the RTase moiety of PEs, we did not detect any PE3-induced alterations in telomeric regions or endogenous retroelements (Fig. 4). Finally, whole-transcriptome sequencing showed that PE3 did not significantly introduce RNA mutations or changes in alternative splicing events or gene expression (Figs. 6–8).
As PEs have great potential in treating human genetic diseases, systematic investigation of any OT effects is of utmost importance. Although previous studies showed that PE can induce low levels of pegRNA-dependent mutations,4,5 the use of variant PEs comprising engineered Cas9 variants with high targeting specificity 11 could theoretically reduce these pegRNA-dependent OT mutations to acceptable levels.
However, any pegRNA-independent OT effects on cellular DNA or RNA would impact every application of PEs and significantly affect their safety profile. Considering the basal error rate of next-generation sequencing (NGS), gRNA-independent OT mutations—which by definition are not enriched at specific genomic or transcriptomic loci—are difficult to distinguish from the noise of NGS in heterogeneous pools of edited cells.
By sequencing expanded two-cell-stage mouse embryonic cells, 7 plant callus cells, 6 or human iPSCs, 26 previous studies were able to detect BE3-induced gRNA-independent OT mutations in genomic DNA. However, carrying out experiments in mouse embryos or iPSCs not only is costly but also requires special expertise, and findings in one cell type may not always be applicable to another cell type.
Thus, we sought to engineer a commonly used human 293FT cell line to reduce background mutation levels in a way that would allow sensitive detection of gRNA-independent OT mutations of various genome editors. Compared to WT 293FT cells, our engineered 293FT A3–/– cell line lacking the A3 cytidine deaminase gene cluster had a greatly reduced level of background base substitutions and indels (Supplementary Fig. S1), which therefore provides a useful platform to determine gRNA-independent OT mutations. We propose that our strategy could be similarly applied to other cell lines to investigate cell type-specific mutational profiles of PEs or other genome editing effectors further in the future.
Given the large size of the human genome, WGS with sufficient depth to capture rare mutational events is costly. However, taking advantage of the diploid nature of the human genome, random gRNA-independent OT mutations will have a frequency of at least 50% in a clonal population because they will occur at least at one allele (Supplementary Fig. S5C). Accordingly, by using a moderate depth of WGS (such as 12 × in this study), gRNA-independent OT mutations can be identified successfully in expanded single-cell clones (Fig. 1D and E). On the other hand, there are several sensitive methods to predict or detect gRNA-dependent OT mutations, which generally occur at a low frequency but at a fixed genomic locus (e.g., Cas-OFFinder, 28 GUIDE-seq, 31 Digenome-seq, 32 or CIRCLE-seq 33 ).
Conclusion
Our results demonstrate that CRISPR-based PE tools induced no observable genome- or transcriptome-wide pegRNA-independent OT effects in human cells, suggesting a high editing specificity of its RTase moiety. Therefore, we conclude that the main concern regarding OT mutagenesis during prime editing are those OT mutations that are pegRNA dependent (i.e., at other genomic sites bearing some similarity to the intended target site). These guide-dependent OT effects can likely be overcome by applying the insights gleaned from engineered Cas9 variants to reduce the binding of PE at OT sites, resulting in enhanced versions of PEs with improved editing efficiency and fidelity that will have great therapeutic promise.
Footnotes
Acknowledgments
We thank Molecular and Cell Biology Core Facility, School of Life Science and Technology, ShanghaiTech University for providing experimental service, and Haopeng Wang for providing the Tet-On 3G system.
Author Disclosure Statement
All the authors declare that they have no competing interests.
Funding Information
This work was supported by grants 2019YFA0802804 (L.Y.), 2018YFA0801401 (J.C.), and 2018YFC1004602 (J.C.) from MoST; 31925011 (L.Y.), 91940306 (L.Y.), 31822016 (J.C.), and 81872305 (J.C.) from NSFC; and 21JC1404600 (J.C.) from the Shanghai Municipal Science and Technology Commission.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.