
Abstract
With the advent of new genome editing technologies and the emphasis placed on their optimization, the genetic and phenotypic correction of a plethora of diseases sit on the horizon. Ideally, genome editing approaches would provide long-term solutions through permanent disease correction instead of simply treating patients symptomatically. Although various editing machinery options exist, the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas (CRISPR-associated protein) editing technique has emerged as the most popular due to its high editing efficiency, simplicity, and affordability. However, while CRISPR technology is gradually being perfected, optimization is futile without accessible, effective, and safe delivery to the desired cell or tissue. Therefore, it is important that scientists simultaneously focus on inventing and improving delivery modalities for editing machinery as well. In this review, we will discuss the critical details of viral and nonviral delivery systems, including payload, immunogenicity, efficacy in delivery, clinical application, and future directions.
Introduction
Since the development of the first successful transgenic organism in 1973, genetic engineering has become a critical tool in the world of research, and its optimization has become a focal point for many scientists around the world. 1 In light of ethical concerns surrounding germline genome engineering approaches, researchers have continued to pursue the refinement of these technologies for somatic cells due to their potential to treat and correct human disease. Recently, the introduction of site-specific double-stranded DNA break (DSB) inducing technologies, including zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)-Cas (CRISPR-associated protein), has paved the way to efficient genome modification in cells/living organisms.
These genome editing tools rely on endogenous cellular repair machinery. When DSBs naturally occur in the cell due to reasons like reactive oxygen species, there are two main avenues for DNA repair: nonhomologous end joining (NHEJ) and homology-directed repair (HDR) (Fig. 1). While NHEJ ligates two random strands together and can occur during any phase of the cell cycle, HDR relies on the presence of a sister chromatid during late S phase or G2 phase of the cell cycle. 2
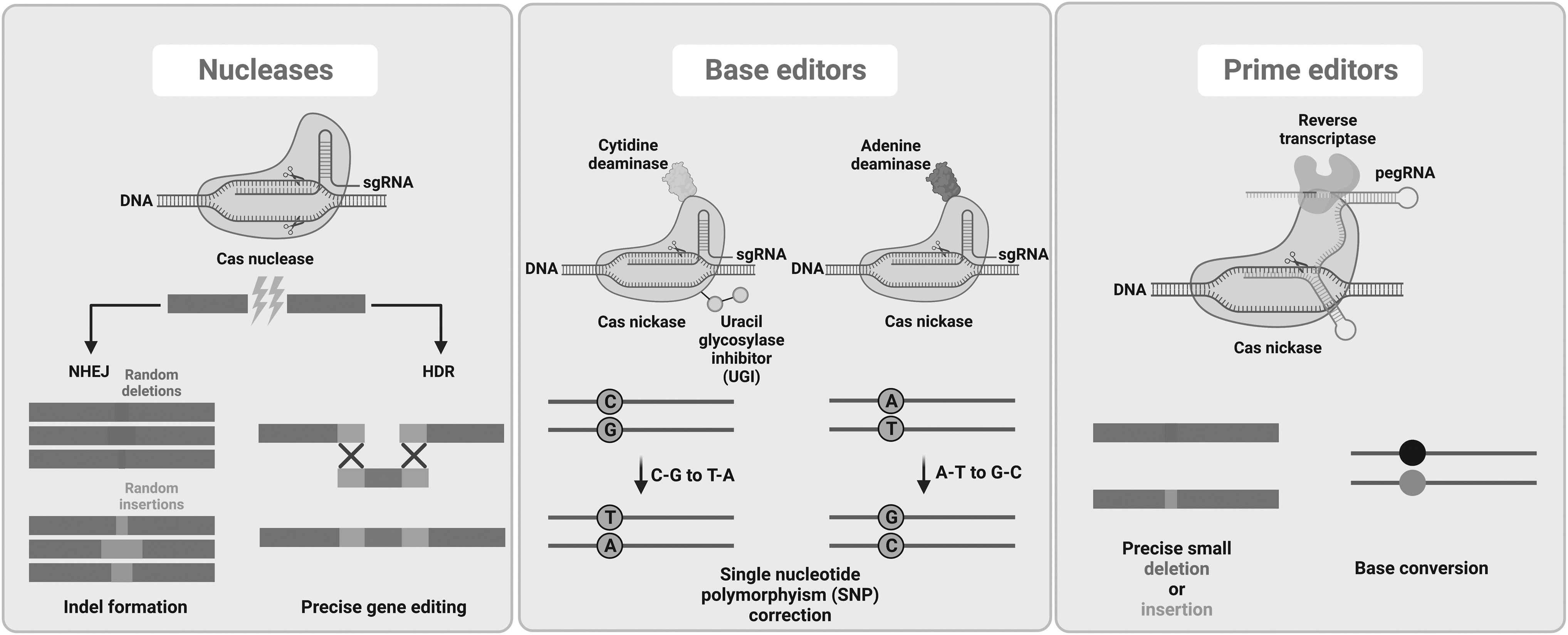
Overview of main genome editing approaches and potential outcomes. HDR, homology-directed repair; NHEJ, nonhomologous end joining; pegRNA, prime editing guide RNA; sgRNA, single-guide RNA.
Genome editing techniques utilize endonucleases to create DSBs (non-DSB approaches are discussed below), and then NHEJ or HDR takes place to modify the genome. Although NHEJ is active in both dividing and quiescent cells, it often introduces small insertions and deletions (INDELs) resulting in a nonfunctional DNA sequence. Therefore, it is mainly used for gene knockouts. In contrast, HDR, while mostly active in dividing cells, is less error prone and hence used for gene insertions/corrections when a homologous donor DNA template is provided. 2
Early in genome editing research, TALENS and ZFNs were the dominant genome editing techniques, relying on the FOK1 endonuclease, fused to a series of DNA-recognizing protein domains that required re-engineering for each site of interest.3,4 The CRISPR-Cas system has emerged as a simpler, quicker, cheaper, and more efficient alternative. The CRISPR-Cas system was initially discovered in Escherichia coli in 1987, 5 and was later determined to be part of the bacterial immune system. 6 Scientists have harnessed the sequence-specific cleavage of the RNA-Cas complex where a specific RNA sequence, guide RNA (gRNA), recognizes the target DNA region and directs the Cas nuclease to create targeted DSB and insert, correct, or silence genes. Since the CRISPR-Cas immune system can be found in 50% of bacteria and 90% of Archaea, 7 there is a diverse array of Cas genes and an extensive classification system, providing various options for genome editing (Cas classifications were reviewed elsewhere 8 ).
Many CRISPR-Cas-based therapeutic modalities are underway for various human diseases, including neurodegenerative diseases, cancer, HIV, inherited blood disorders (i.e., sickle cell disease [SCD] and β-thalassemia), and eye diseases. Recent clinical trial data displayed great promise in treating patients with hereditary transthyretin amyloidosis 9 or SCD and β-thalassemia 10 using in vivo CRISPR-Cas9 delivery or ex vivo CRISPR-Cas9-modified hematopoietic stem and progenitor cells (HSPCs).
Nonetheless, one of the main obstacles in the field of CRISPR-Cas editing approaches is efficient cellular delivery that determines the power of the application. CRISPR cargos can be introduced into cells in three modes: (i) DNA plasmid encoding CRISPR cargos, (ii) mRNA for Cas protein translation with a separate gRNA, or (iii) a ribonucleoprotein (RNP) consisting of a Cas protein and gRNA. Although RNP CRISPR-Cas delivery can edit target sequences with high efficiency, 11 the other systems have also been advanced. Of note, DNA plasmids and mRNA activate the innate immune system through multiple pathways. 12 As this could dramatically affect the overall toxicity of the genome editing approach, it must be taken into consideration when establishing in vivo CRISPR delivery.
Delivery systems can be classified into two major categories: viral systems, including lentiviruses (LVs), adenoviruses (AdVs), and adeno-associated viruses (AAVs), and nonviral delivery strategies, including physical and chemical delivery methods (Fig. 2). The viral delivery strategy uses viral vectors to traffic CRISPR cargos into cells. When a DNA template donor homologous to the DSB sites is delivered with CRISPR cargos, cells repair the break by “copying and pasting” the donor template using the cellular recombination machinery. In this sense, some hybrid delivery methods (i.e., combination of lipid nanoparticles [LNPs] with AAV vectors or electroporation methods with AAV vector) have been shown to be highly efficient in the correction of disease underlying mutations for tyrosinemia and SCD.13,14 In this review, we will discuss and assess the different nonviral and viral delivery systems, challenges, and future directions for CRISPR-Cas delivery systems.
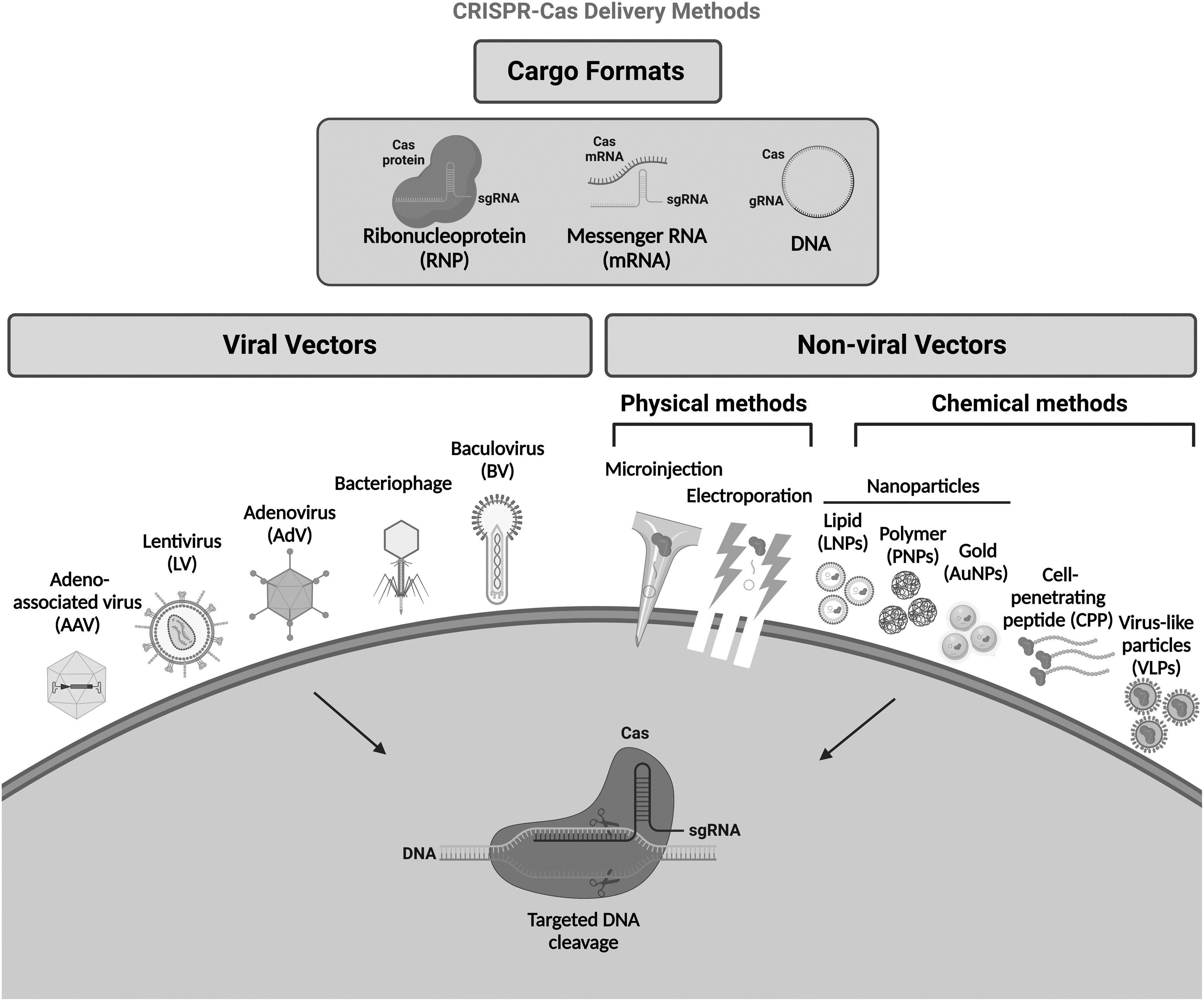
Major CRISPR-Cas delivery methods/formats.
Nonviral Delivery
Physical delivery methods
Physical transfer of CRISPR cargos into the cell relies on permeabilization of the cell membrane by externally applied physical force (electrical, thermal, mechanical, etc.) to allow genome editing tools to enter cells. Although most of these techniques are straightforward and provide efficient editing with almost no limit to the cargo size, some of them target a small number of cells; they might dramatically affect cell viability or produce tissue damage in some circumstances (Table 1). As a side note, most of the physical delivery methods rely on ex vivo cultured cells, including immune cells and HSPCs. Therefore, due to current toolbox limitations, they are generally not applicable in certain tissues such as the brain, lung, and spinal cord.
CRISPR-Cas, clustered regularly interspaced short palindromic repeats (CRISPR)-Cas (CRISPR-associated protein).
Microinjection
Microinjection can deliver all three modes of CRISPR cargos to individual cells (i.e., zygote) to create genetically modified organisms using a microneedle (0.5–5.0 μm) under a microscope. 15 For optimal results, microinjection ought to be performed by skilled personnel. Microinjection does not have limitations in CRISPR cargo size, has almost 100% efficiency without affecting surrounding tissues/cells, and guarantees that the cargos are delivered exactly to the targeted cells. This approach can transfer CRISPR cargo directly into the cytoplasm of target cells or the pronucleus of fertilized eggs to create in vivo animal models (i.e., zebrafish, Caenorhabditis elegans and mice—reviewed elsewhere 16 ). Because of its simplicity and accuracy, microinjection can also be used to investigate the genome editing efficacy of various plasmid-based CRISPR systems for the same target. 17 However, microinjection is only suitable for editing a small number of cells since a single cell is targeted in each injection cycle.
Electroporation
Electroporation is a long-standing physical method for delivering genome editing tools into a population of cells. All three CRISPR cargo modes can efficiently be introduced into host cells by electroporation. It has good scalability and can be applied to almost all types of mammalian cells with various efficacies. The major drawback of electroporation is low cell viability. Numerous efforts have been made to minimize voltage-induced cytotoxicity, including postpulse centrifugation 18 and an enhanced electroporation cell density. 19 Although clinically irrelevant, the inclusion of serum in the electroporation buffer improves cell viability with no negative effect on gene transfer and expression in mammalian cells. 20
Optimization of postelectroporation recovery also decreases the cytotoxicity for primary stem cells. 21 Moreover, the addition of 2–8% glycerol to the electroporation buffer improves the cell viability of HSPCs without loss of editing efficiency and engraftment potential. 22 Optimization of the pulse strength, doses of CRISPR cargos, and cell preparation could ensure high editing efficiency with minimal cytotoxicity. However, low cell viability after electroporation still remains a significant obstacle for patients with limited stem and progenitor cells. In addition, electroporation and genome editing also affect long-term repopulating HSPCs. It is a well-known phenomenon that after DSB formation, the P53-dependent apoptotic pathway is activated.
However, it was recently discovered that the average expression of known P53 target genes in stem cell-enriched fractions was higher compared to the relatively differentiated fraction when nuclease-mediated DSBs were created. 23 In addition, while a single electroporation dosage does not seem to affect engraftment potential, 24 second rounds reduced human cell chimerism in an immunocompromised mouse model, indicating that long-term repopulating cells would be more vulnerable to electroporation-dependent toxicity.
Electroporation has been widely studied in ex vivo genome editing research, including chimeric antigen receptors (CAR)-T cell-based immunotherapy studies to (i) insert the CAR cassette into a specific genomic location, (ii) disrupt inhibitory molecules and signaling axis, (iii) reduce the occurrence of cytokine release syndrome and graft-versus-host disease, (iv) establish allogeneic universal CAR-T cells, or (v) provide resistance to TGF-β's suppressive effect. 25 Other than the ex vivo electroporation approach, in utero electroporation of mouse embryos at embryonic day 15 was also successfully applied to tag GluA2 gene in vivo using plasmids coding for Cas9 protein, gRNA, and donor DNA template. 26 Last but not least, as a highly efficient method, it has good clinical potential. Electroporation-based CRISPR cargo delivery provided robust ex vivo BCL11A gene editing in patient-derived HSPCs (74–86%) and long-term engraftment of edited cells after infusion, and reversed the SCD and β-thalassemia phenotypes. 10
Hydrodynamic delivery
Hydrodynamic delivery is a quick injection of fluid carrying the gene editing machinery into an animal's bloodstream. The volume is about 8–10% of the animal's body weight (i.e., ∼20–25 mL for rats).27,28 It ruptures the endothelium and cell membranes to allow CRISPR cargos to enter parenchymal cells. Studies have reported a genetic correction rate of about 1/250 in mouse liver cells 27 and approximately partial (2.6%) liver genome modification targeting PTEN. 28 Hydrodynamic delivery is not currently considered for clinical applications because it increases blood pressure and induces temporary cardiac dysfunction, liver expansion, and even animal death.29,30
Microfluidic cell deformation
Mechanical cell deformation with a microfluidic device using electric pulse, hydrodynamic force, and optical energy induces cell membrane permeability for cellular uptake of macromolecules. 31 Microfluidic-based mechanoporation strategies are reviewed at length elsewhere. 32 It is an efficient way to deliver CRISPR cargos into hard-to-transfect cells. Han et al 33 used this method to introduce DNA plasmids encoding CRISPR-Cas components into breast cancer (MDA-MB-231) and lymphoma (SU-DHL-1) cells, and demonstrated 90% and 70% knockout efficiency of enhanced green fluorescent protein (EGFP), respectively. In addition, delivery of RNP to breast cancer cells (SK-BR-3) by this method resulted in roughly 90% editing efficiency with 70% cell viability. 34 Although its capacity is limited and it is only suitable for a small number of cells, this high efficiency delivery method could be very useful in the future for research and clinical applications.
Chemical delivery methods
Large RNP complexes and negatively charged naked nucleic acid molecules cannot efficiently pass through the cell membrane. Electrostatic interactions between nucleic acids and positively charged delivery agents, or encapsulation of the RNP complex by these materials allows efficient cell entry through endocytosis and protects cargos from being degraded (Table 1). Usually, the size of the genome editing cargo is not a consideration for chemical delivery methods. These types of delivery approaches mostly provide short-term CRISPR cargo expression that reduces off-target activity. All chemical delivery agents are cleared by liver; thus, delivery efficiency to non-liver tissues remains poor. However, the optimization of vehicle structure (i.e., size, composition, addition of aptamers/antibodies) has proven to increase targeted delivery. 35 This will hopefully expand delivery to non-liver tissues such as the central nervous system, eye, and lung.
Lipid nanoparticles
LNPs are used to deliver DNA, RNA, or protein CRISPR components to cells for therapeutic purposes or to establish knockout/knock-in animal models. An association between LNPs and CRISPR components is achieved through either encapsulation or complexation. LNPs can protect payloads from nuclease degradation or immunological response 36 and their compositions can be adjusted for an effective in vivo delivery. 37 However, small particle size, good uniformity, high stability, and targeted delivery are essential for clinical use of LNP in vivo.
A landmark study reported by Gillmore et al demonstrated the use of a safe and efficacious CRISPR components agent (NTLA-2001), an LNP product encapsulating mRNA for Cas9 protein combined with a chemically modified sgRNA targeting the transthyretin (TTR) gene in liver cells. It led to substantial reductions in serum TTR protein concentrations (47% to 96%) in six patients with hereditary transthyretin amyloidosis treated through intravenous administration. 9 In another experiment, single infusion of LNPs carrying CRISPR base editors provided near-complete knockdown of PCSK9 in the liver, and robust reductions in PCSK9 (90%) and low-density lipoprotein (LDL) cholesterol (60%) levels in the blood of cynomolgus monkeys. 35
While targeting liver diseases using LNP-delivered CRISPR components is highly efficient, systemically administered component delivery to non-liver tissues remains a challenge. To address this problem, Sago et al established a high-throughput in vivo screening approach where they identified an LNP formulation (7C3) that could deliver Cas9 mRNA and sgRNA to splenic endothelial cells as efficiently as hepatocytes. 38
Commercially available lipid-based particles such as Lipofectamine (ThermoFisher Scientific) are widely used to deliver DNA plasmid encoding CRISPR components, Cas9 mRNA/sgRNA mixtures, and RNPs to tissues and various cell lines such as HEK293FT, U2OS, mouse embryonic stem cells, N2A, and A549, for genome editing.11,39 Among these particles, lipofectamine CRISPRMAX displayed 40% and 15% higher genome editing efficiency in various mammalian cell lines than Lipofectamine 3000 and RNAiMAX, respectively. 40 The lipofectamine LNP (RNAiMAX) can support efficient multiplexed genome editing. It results in 8–11% editing efficiency through HDR ex vivo and 20% genome editing efficiency in vivo (i.e., mouse inner and ear hair cells). 39
A generalizable engineering approach is to formulate LNPs by adding permanently cationic lipids that preserve RNP integrity, mediate RNP encapsulation, and redirect CRISPR components to targeted tissues following low-dose intravenous injection. Along with efficient ex vivo genome editing, these LNPs delivered RNPs in vivo to effectively edit tissues, including muscle, brain, liver, and lungs, suggesting the broad applicability of this approach.41,42 Similarly, a recent study demonstrated tissue-specific delivery of Cas9 mRNA and sgRNA using LNPs engineered by changing the internal charge with additional lipid compositions, which allowed efficient editing in targeted tissues, including the liver, lung, and spleen. 43 The specificity of bioreducible LNPs for in vivo targeting was screened by using firefly luciferase mRNA, 44 which highlighted the potential of LNP-directed delivery as a specific, effective, and safe platform for CRISPR-based therapeutics.
Polymer nanoparticles
Polymer nanoparticles (PNPs), due to their small size, high volume-surface area ratio, and tunable pores, have been extensively used for the delivery of drugs and nucleic acids, including plasmid DNA and mRNA. Association of CRISPR components with PNPs can be achieved by noncovalent encapsulation or covalent derivatization. While covalent conjugation of Cas9 to the polymer does not impair endonuclease activity and editing efficacy, noncovalent encapsulation may better protect CRISPR components from immune responses and nuclease degradation in vivo. 45 However, noncovalent encapsulation has a lower loading capacity and packaging efficiency, and requires administration at high dosages leading to significant cytotoxicity.46,47 Cationic polymers for PNPs can be functionalized with active targeting components that specifically bind to surface proteins of the target cell type for in vivo delivery. For instance, an amphiphilic block polymer was shown to be able to deliver CRISPR materials to macrophages. 48
Recently, a multistage delivery polymer nanocarrier (MDNP) was able to efficiently deliver CRISPR-dead(d)Cas9–miR-524, inhibiting tumor growth in mice and providing a feasible approach for the development of CRISPR-based cancer gene therapies. 49 MDNP exhibits corresponding surface properties at different delivery stages. When circulating in the bloodstream, MDNP maintains the main structure. However, upon entering tumor tissues, the acidic microenvironment triggers the decomposition of DMMA groups in the polymer shell. This leads to its rapid conversion from an anionic to a cationic polymer, causing the polymer shell to detach from the MDNP core.
PNPs can also be modified for the delivery of RNP. In a recent study, the negatively charged red fluorescent protein was first anchored to the shell of chitosan nanoparticles, which can simultaneously deliver Cas9 RNPs with a poly-glutamate tag and single-strand DNA template. 50 Such nanoparticles were shown to have about 77.6% transfection efficiency and 12.5 ± 3.0% HDR frequency, which is comparable to that of Lipofectamine CRISPRMAX (14.5 ± 3.5%).
Gold nanoparticles
Gold nanoparticles (AuNPs) are gold cores coated with synthetic or biological compounds that can be modified to covalently or noncovalently conjugate biomacromolecules. 51 Attaching a negatively charged glutamate peptide to the Cas9 protein enabled efficient assembly with cationic arginine AuNPs. This led to ∼90% intracellular delivery efficiency, which resulted in up to 30% editing rate in Hela cells. 52 These AuNPs have high cytoplasmic/nuclear delivery (∼90%) and genome editing efficiencies (23–30%).52,53 AuNPs can be successfully applied to in vivo CRISPR cargo delivery approaches. In a mouse model of fragile X syndrome, AuNP-CRISPR efficiently delivered Cas9 and Cas12a RNPs in the brain after an intracranial injection that inhibited 40–50% of the autism causal gene, Grm5, and rescued mice from increased repetitive behaviors. 54 AuNPs can also be expanded to treat polygenic diseases such as Huntington's disease by simultaneously using two or more sgRNAs. 55
AuNPs are relatively nontoxic and, therefore, have great potential for the delivery of CRISPR cargos to nonmalignant dividing somatic cells such as HSPCs.56,57 Shahbazi et al designed a simple AuNP-based CRISPR nanoformulation with layer-by-layer conjugation of RNP on the surface of AuNPs with or without the single-strand DNA donor to target primary human blood progenitors. It demonstrated 17.6% total editing with 13.4% HDR at the CCR5 locus, and 12.1% total editing with 8.8% HDR at the γ-globin promoter locus. 58 This indicates that HSPCs could be efficiently targeted using AuNP-delivered CRISPR components.
An AuNP-condensed, lipid-encapsulated, and laser-controlled delivery system was recently developed, providing a versatile method for high-efficiency CRISPR cargo delivery with broad clinical applications. 59 In this system, the plasmid encoding CRISPR cargos (PC) was condensed on TAT peptide-modified AuNPs through electrostatic interactions. Then it was coated with lipids to form lipid-encapsulated, AuNP-condensed PC (LAPC). Once inside tumor cells, PC was released from LAPC into the cytosol by laser-triggered thermo-effects of AuNPs, enabling effective knockout of a target gene in the tumor.
DNA nanoparticles
The DNA nanoparticle is a novel delivery vehicle for CRISPR cargos based on a yarn-like DNA nanoclew (NC) for both ex vivo and in vivo applications. 60 The DNA NCs are synthesized by rolling circle amplification with palindromic sequences encoded to drive the self-assembly of synthesized DNA through Watson-Crick base pairing. DNA NC is designed to contain sequences that are partially complementary to the gRNA. As a result, the synthesized DNA NCs and RNPs can quickly associate into complexes, which are then coated with the cationic polymer, polyethyleneimine, to enhance cellular uptake and endosomal escape.
The genome editing efficiency using this delivery method was ∼28%. 60 DNA nanoparticles have a large loading capacity and good biocompatibility, and can be modified to target specific membrane receptors for effective cellular uptake. DNA nanostructures can also be designed to encode DNA aptamers for specific tumor cell targeting 61 and miRNA-responsive structures for Cas9/sgRNA release by toehold-mediated strand displacement to achieve cell type-specific targeting. 62
Induced transduction by osmocytosis and propanebetaine
Induced transduction by osmocytosis and propanebetaine (iTOP) is a relatively new approach for RNP delivery.62,63 It uses salt and propanebetaine to cause hyperosmolality, which triggers macropinocytotic uptake and intracellular release of extracellularly applied macromolecules. Kholosy et al optimized the iTOP method, achieving highly efficient genome editing in diverse human cells, including the difficult-to-transduce human induced pluripotent stem cells (iPSCs) and primary T cells. 64 The editing efficiency was 40–95% depending on the cell type with high cell viability (70–95%). In addition, it has similar gene editing efficiencies to the electroporation method in Jurkat, ARPE-19, and HEK 293 cells, but achieved twice as high cell viability (80% vs. 40%). This suggests that the iTOP delivery strategy is a competitive alternative.
Cell-penetrating peptide
Cell-penetrating peptides (CPPs), peptides fewer than 30 amino acids, can translocate into cells through direct penetration, endocytosis, or the formation of micelles. Conjugation of Cas9 proteins with CPPs enhanced RNP delivery and achieved editing rates of 2.3–16% in different cell lines, including HeLa cells, HEK293T cells, dermal fibroblasts, embryonic cells, and embryonic stem cells.65,66 In addition, Cas9 and Cas12a RNPs were efficiently delivered to stem cells and hard-to-transfect cells, including cancer cell lines and natural killer cells, using a cationic helical amphiphilic peptide.
Interestingly, Cas9 and Cas12a RNP delivery using an engineered amphiphilic peptide system provided clinically relevant genome editing in mouse airway epithelia without any detectable short-term toxicity. 67 Although conjugation of Cas9 protein with CPP may increase transport across the cell membrane, it does not protect from protease degradation and cell type-specific targeting. Thus, to successfully employ CPPs, they may require encapsulation or combination with other delivery methods.
Virus-like particles
Virus-like particles (VLPs) are self-assembled nanoparticles that consist of one or more viral structural capsids and envelope proteins, but lack the viral genome. Therefore, they remain replication incompetent and insertion deficient. This property of VLPs makes them attractive as a relatively safer viral delivery method. In addition, having a transient expression of Cas proteins reduces potential off-target activity, which renders them even more relevant. Although production of VLPs requires cell culture and is relatively costly, the production is simple and straightforward; thus, they have been extensively used as nanocarriers for drugs and vaccines. 68
Due to these advantages, scientists are currently focusing on the development and optimization of VLPs as CRISPR cargo delivery agents. They efficiently delivered CRISPR cargos, including donor DNA templates for HDR both ex vivo and in vivo.69–71 Initially, VSV-G and Gag/Pol plasmids were used with Cas9 fused to the N-terminus of the Gag gene. 69 During the viral maturation step, a protease cleaved the HIV cleavage sequence site between the fusion proteins releasing Cas9 protein for delivery. This VLP system effectively edited the CCR5 gene in primary CD4+ cells with a much lower off-target rate compared to lentiviral transduction, which leads to continuous Cas9 expression. 69 To have sgRNA in vesicles, a VLP approach, VEsicCas, was established in which VLPs were synthesized using a producer cell line through co-expression of VSV-G, sgRNA (T7 RNA polymerase-driven transcription system), and Cas9 protein. 71
VEsicCas with passively encapsulated Cas9 RNPs was shown to efficiently edit genomes in various cell lines, exerted minimal cell toxicity, and provided around 30% editing rate in the injection area of cardiac muscle of a mouse model. All-in-one VLPs, including donor DNA template for HDR, Nanoblades (murine leukemia VLPs), were synthesized in the gesicle producer 293T cell line by transfection of wild-type Gag-Pol, Gag::Cas9, VSV-G, sgRNA, and Baboon Endogenous retrovirus Rless glycoprotein (BaEVRless) plasmids. 70 Along with achieving high editing rates (50–76%) in tested cell lines and primary cells, including macrophages, human hematopoietic progenitors, and primary hepatocytes, more than 50% knock-in efficiency was obtained in HEK293T cells when Nanoblades were coupled with donor DNA templates.
More interestingly, using a tail injection route, Nanoblades provided significant genome editing ratios (7–13%) in adult mouse hepatocytes. 70 This moderate level of in vivo editing was improved by David Liu's group by optimizing the cleave linker sequence in the fusion sequence and Gag-Cargo fusion/wild-type Gag-Pol stoichiometry, and adding nuclear export signals to the fusion sequence. Along with robust editing ratios in cell lines and primary cells, engineered VLP administration led to 63% hepatocyte editing that reduced serum Pcsk9 levels by 78% and partially restored visual ability in a mouse model of genetic blindness. 72 Targeted delivery is one of the most important research areas for CRISPR-reliant scientists. In this sense, a recent article showed the proof-of-principle for cell-type specific delivery of Cas9 RNPs using surface glycoprotein-engineered VLPs. 73
In this work, HIV-1 envelope-targeted particles were able to preferentially transduce CD4+ cells, while co-cultured CD8+ cell transduction/editing rates were limited. In a word, VLP-based CRISPR cargo delivery has comparable editing efficiency to other efficient delivery methods (such as electroporation or AAV delivery) with transient Cas expression, targeted and multiplexed editing potentials, low off-target rates, and high cell viability. Currently, there is an ongoing clinical trial study (NCT04560790) using VLP (BD111) targeting herpetic stromal keratitis in Shanghai, China.
Viral Delivery
Although physical delivery systems (i.e., microinjection and electroporation) for CRISPR-Cas tools are popular, these approaches are often not relevant in vivo and induce high levels of cytotoxicity. Thus, viral delivery techniques have gained prominence as an alternative method due to their wide applicability for various cell types, high transfection rate, and minimal immunogenicity and cytotoxicity. Viral-based delivery systems have also been modified to display safer profiles and enhance study applicability (Table 2). As a result, not only have these techniques been successfully implemented in vitro, ex vivo, and in vivo 74 but also have been employed in many preclinical and clinical gene addition trials in the past 20 years.75–77
AAV, adeno-associated virus; AdV, adenovirus; LV, lentivirus.
Adeno-associated virus
Various viral vectors have been successfully used to deliver CRISPR-Cas tools, which are summarized in Table 2. AAVs are among the most popular viral delivery platforms. They are composed of an icosahedral capsid and a single stranded DNA genome, which can be sense (+) or anti-sense (−). Since AAVs induce negligible cytotoxicity, are replication defective, can infect quiescent and dividing cells, can integrate at a well-defined locus on chromosome 19 (AAVS1) without requiring a DSB, and have various serotypes allowing for infection of targeted tissues, they have become a dominant player in the world of gene therapy. In addition, their safety profile has led to their approval for clinical use.74,78–81 AAVs deliver DNA encoding for Cas, gRNAs, and/or donor DNA template into cells by transduction. This approach avoids the toxicity of electroporation or potential mutagenesis of vectors that require integration.77,82
However, as AAVs have a limited payload (<4.7 kb), it makes it challenging to package the commonly used Streptococcus pyogenes (Sp)Cas9 and a gRNA together (∼4.2 kb). Thus, scientists have either modified the AAV delivery approach, using dual or triple AAV systems, or have utilized smaller Cas9 orthologs, such as Staphylococcus aureus (Sa)Cas9, to circumvent its limited packaging capacity.83–85 One such approach was able to correct the disease phenotype of adult phenylalanine hydroxylase in mice. Using an intein-split system, they split a base editor, a CRISPR system allowing the introducing of point mutations to the DNA sequence without creating DSBs, into two separate recombinant AAVs, each of which expressed a portion of the desired protein.
Following injection of the vectors, the approach leads to the creation of one functional protein. 86 In addition to CRISPR tool delivery, AAVs are also being used in combination with RNPs to transfer donor DNA templates for gene correction through the HDR pathway. This has been shown to be the most effective gene knock-in technique compared to other nonviral delivery approaches. 87 These findings suggest the applicability and efficiency of AAV as a vehicle to deliver genome editing tools.
AAVs tend to integrate into CRISPR-induced DSBs (up to 47%). 88 That would lead to continuous expression of Cas9 posing a safety concern by augmenting cytotoxicity, immune response, and off-target activity. To address this hurdle, a self-deleting AAV-CRISPR system, which creates INDELs in the AAV genome, was introduced. 89 Using this approach, high on-target activity, robust reduction in Cas9 protein levels (>79%), and no detectable off-target activity were reported. Nonetheless, despite its general effectivity, some individuals have a pre-existing immunity to AAVs, which may hamper the efficacy of the technique.90,91 Thus, other viral delivery techniques may be preferable in these cases. 92
Although several preclinical and clinical studies have been completed using AAVs without any significant safety problem, in 2020, a gene replacement therapy using AAV8 aimed at treating X-linked myotubular myopathy (XLMTM) (NCT03199469) led to death of three children, which was attributed to high-dose AAV vector-dependent fatal liver dysfunction. 93 This study highlights the importance of clinical trial designs, prescreening process, and carefully conducted preclinical studies for safety and efficacy assessment.
Lentivirus
Another popular viral delivery method is the use of LVs. It is a retrovirus composed of a single-stranded RNA genome that can be used for CRISPR-Cas delivery in vitro and ex vivo.94,95 LV vectors are capable of transducing both dividing and nondividing cells and have relatively big cloning capacities (<8 kb), which enable all CRISPR components to be carried in one LV backbone. 96 Moreover, LVs maintain stable expression as they get randomly integrated into host cell genomes. Nonetheless, LVs still come with some risk of insertional mutagenesis.97,98 To avoid random LV integration and sustained Cas expression, researchers have developed alternative approaches such as integration-defective lentiviruses (IDLVs).
In IDLVs, a single point mutation is introduced into the integrase to prevent LV integration and increase the safety profile of using the LV system. 99 Nonetheless, IDLVs show lower transgene expression levels. These lower levels can be improved by the introduction of the IS2 element into the vector.100,101 Another technology that improves the safety of LVs is the utilization of transiently expressed, self-inactivating Cas9. This system utilizes two different gRNAs, one targeting the gene of interest, while the second targets the Cas9 gene to prevent Cas9 re-expression. This reduces the off-target effects caused by sustained Cas9 expression. 102
Adenovirus
The AdV is a nonenveloped and transcriptionally active virus. It contains double-stranded DNA (dsDNA) and can infect both dividing and quiescent cells. Due to its large cloning capacity (>35 kb), it can easily hold CRISPR-Cas machinery. In addition, it does not integrate into the host genome, reducing the potential for off-target effects, making it one of the most commonly used vectors in clinical trials.103,104 While the first AdVs produced acute and chronic immune responses, they have since been optimized to be safer for gene delivery. 105
The latest generation of AdVs, known as helper-dependent AdVs (HDAds), completely removes all viral genes. Not only does this reduce chronic toxicity, while maintaining high levels of transduction, but also increases cloning capacity to 37 kb.106,107 Despite AdV advancements, it has yet to become a universally effective treatment method. Many individuals are exposed to AdVs throughout their lifetimes and develop neutralizing antibodies that elicit acute immune reactions.104,108 A recent study injected baboons intravenously with an HDAd expressing human alpha1-antitrypsin (hAAT), but noticed a gradual decline in transgene expression. This was attributed to prior AdV exposure. 109 Nonetheless, AdV has still been successfully applied as an editing tool ex vivo and in vivo. For example, in Duchenne muscular dystrophy, which is caused by genetic mutations in the dystrophin-encoding DMD gene, AdV-mediated CRISPR-Cas9 delivery successfully corrected the mutation in patient-derived muscle progenitor cells and in the liver of mdx mice.75,110,111
This technology has oncological therapeutic potential as well. It was able to disrupt tumor growth and enhance tumor shrinking in a xenograft mouse model of human lung cancer by disrupting epidermal growth factor (EGFR) expression, a key player in tumor progression. Other studies that used adenoviral-mediated CRISPR-Cas9 delivery include the following: one that targeted PCSK9 in mouse livers to reduce LDL and cholesterol levels and another that disrupted CCR5 expression in CD4+ T cells, a key receptor in HIV infection.112,113
Bacteriophage
Bacteriophages, or phages, are a class of viruses that have the ability to infect bacteria. They are made up of different virus families, each with its own structure and target organism. 114 With the rise of antibiotic resistance, phage therapy has emerged as a promising treatment option to fight antimicrobial-resistant (AMR) bacteria. In the last few years, several studies have been published on phage therapy and the successful application of this approach.115,116 In addition, the CRISPR-Cas9 system has been repurposed as a novel therapeutic strategy for AMR bacteria through targeting of multidrug-resistant genes in the bacterial genome. 117 However, delivering the CRISPR-Cas9 tool kit into targeted bacteria remains a significant obstacle. Engineered phage-based vectors offer an effective delivery method for the CRISPR-Cas9 system into microorganisms.
There are two major delivery forms: phagemids and the phage genome. 118 As plasmids, phagemids can hold the CRISPR-Cas9 system and can access different bacterial strains, such as E. coli and S. aureus. Two studies recently and successfully employed this antimicrobial delivery technique to target AMR genes, lowering AMR bacterial survival rates.119,120 CRISPR-Cas9-dependent antimicrobials can also be introduced by taking advantage of the phage genome, an even better alternative to phagemids. 121 Recent studies using the engineered phage-derived delivery system have showed re-sensitization of E. coli to antimicrobials and have reduced the infectivity of S. aureus.122–124 However, phage use as a delivery method for the CRISPR-Cas9 system into bacteria has a number of drawbacks, including a limited host range, microbial resistance, and safety concerns. 125
Baculovirus
As summarized above, the CRISPR tool kit can be efficiently delivered through viral vectors. However, viral capsids have limited packaging capacities. This restricts their ability to deliver all the necessary editing machinery (SpCas9, gRNA expression cassette, selection markers, HDR templates, etc.), which is critical to achieving maximal editing efficiency. 126 Baculovirus (BV), namely the Autographa californica multiple nucleopolyhedrovirus (AcMNPV), is a large circular dsDNA enveloped insect virus with a genome of ∼134 kb. It contains more than 100 genes and has the capacity to harbor large DNA segments. 127 It has been identified as an efficient vector for gene delivery into a variety of human cells and is widely used for high-level expression of recombinant proteins in insect cells.128,129
The BV is capable of transducing various mammalian cells and expressing the desired genes under the control of mammalian cell promotors without viral replication. It is associated with low cytotoxicity compared to mammalian virus-based vectors. The integration frequency of baculoviral DNA in mammalian cells is extremely rare due to its restriction to insect cells. Most importantly, it has a high transgene capacity and can deliver large strands of foreign DNA. Transferring a 38 kb size of DNA insert was successfully conducted without interfering with the viral titer. 130 Due to these advantages, especially its huge DNA packaging capacity, BV-mediated delivery of CRISPR-Cas is becoming a promising delivery approach that ensures codelivery of all CRISPR-Cas components and HDR templates into the target cell. For instance, EGFP knock-in in the HMGA1 locus of human umbilical vein endothelial cells (HUVECs) (4.5%) and iPSCs (3%) was obtained using BV-mediated CRISPR cargo transfer. 131
With a similar approach, CDCA8 gene was efficiently edited (15–61%) in various human cell lines and GSG2 gene was tagged with yellow fluorescent protein when sgRNA, Cas9, and donor DNA encoding BV transduction was used. 126 More recently, high efficiency (up to 30%) of whole-exon replacement in the intronic β-actin locus was achieved with a single baculoviral vector encoding Cas9, sgRNA, and donor DNA template. 132 However, there are a few potential drawbacks of using BV as a delivery method, including having a time-consuming production process, and its sensitivity to centrifugal stress during purification, which in turn can reduce viral vector titration. 133
Conclusion and Future Perspective
CRISPR-Cas systems have already proven efficient in treating and correcting human diseases in preclinical and clinical studies. However, the development of newer technologies with the goal of optimizing specificity and efficacy are already underway. Among these evolved editing systems, base editors (dCas9 or nickase Cas9 [nCas9] fused with a deaminase domain) and prime editors (prime editing gRNA [pegRNA] and Cas9 nickase [nCas9] fused with Moloney Murine Leukemia Virus Reverse Transcriptase) do not require classical DSB-based repair mechanisms or donor DNA templates to modify genomes (reviewed elsewhere 134 ) (Fig. 1).
In addition to editing genomic sequences, dCas9 fused with epigenetic modifiers (epigenome editors) can be used to alter the epigenomic status of a particular locus, either increasing or decreasing gene expression. 135 These new systems along with classical CRISPR-Cas tools offer promising treatment options to a plethora of genetic/epigenetic diseases. However, to unlock their full potential and become clinically applicable, several obstacles need to be addressed.
Irrespective of CRISPR-Cas application, safe and efficient delivery of editing tools without significant immunogenicity, genotoxicity, and cytotoxicity are critical for their future clinical application. For example, a less cytotoxic delivery strategy is vital for ex vivo genome editing for patients with low numbers of target cells (i.e., limited HSPCs in patients with SCD 136 ). Most of the delivery systems are useful for efficient genome editing in ex vivo conditions (although off-target activity [reviewed elsewhere137,138] is still a major concern). Ex vivo editing relies on the collection of the target cells (generally restricted to HSPCs, and immune cells, including HSPCs, T cells, and natural killer cells) followed by ex vivo genome modification and infusion to the patients.
This approach has mostly been tested on patients with severe combined immunodeficiency, β-thalassemia, SCD, or cancer in a number of ongoing clinical trials (Table 3, ClinicalTrials.gov). To reduce off-target activity, along with sequence optimization and backbone modification of the gRNA, duration of the Cas expression in the target cells should be limited. 137 Therefore, traditional plasmids or integrating nonmodified viral delivery systems are poor options to reduce off-target effects due to their long-term expression profiles. In this sense, nonviral delivery methods providing transient cargo expression remains an attractive alternative to reduce the off-target risk.
ATTR, amyloid transthyretin; CAR, chimeric antigen receptor; HSPCs, hematopoietic stem and progenitor cells.
In addition to the nonviral delivery methods, RNP mode of delivery for CRISPR tools is generally preferred for preclinical and clinical ex vivo applications as off-target activity is lower, and it has no risk for insertional mutagenesis and has reduced immunogenic response. 139 However, RNP delivery mostly relies on electroporation, which dramatically reduces infusion cell number and generates a lot of stress on the electroporated cells. In this manner, less cytotoxic electroporation conditions and other ex vivo delivery methods are still being investigated for optimal delivery of genome editing tools.
Ex vivo cell handling also has several drawbacks/risks such as contamination, reduction in cell function, and cell loss during manipulation. Most importantly, the majority of genetic diseases can only be targeted using in vivo editing. Therefore, direct intravenous/local administration of the genome editing tools, providing cell-/tissue-targeted and high-efficiency genome editing, while bypassing costly ex vivo manipulation techniques and cell culture-related risks, is desirable.
However, several aspects should be optimized and taken into consideration to achieve successful in vivo editing. First, delivery of genome editing tools should be highly specific to avoid editing of untargeted cells in the body that can cause malignancy. Second, the editing efficiency in the target population should be high enough to provide the desired therapeutic effect. For example, in an allogeneic HSPC transplantation setting, at least 20% healthy donor cell chimerism alleviated the clinical symptoms and severity of SCD.140,141 In another work, 38–82% pathogenic mutation correction in human airway epithelial cells provided functional correction of CFTR-dependent anion channel activity. 142 Although the threshold for different diseases varies, higher levels of editing theoretically provide better outcomes. Third, the delivery system should be biocompatible and mechanically stable, so as to not cause any immunogenicity and protect the CRISPR tools until their targeted delivery.
Immune response to delivery agents or CRISPR cargo, particularly in in vivo approaches, would hamper the efficiency of the treatment and raise safety concerns. For instance, the capsid proteins of AdVs stimulate an immune response in a dose-dependent manner and more than 50% of individuals have preexisting humoral neutralizing antibodies to type 2 AdV. This interferes with transduction, activates the complement system, and induces an inflammatory response in the presence of AdVs. 143
As a nonviral delivery approach, LNPs have great potential for in vivo CRISPR delivery. However, they might also provoke innate and adaptive immune responses based on their sizes, shapes, surface charges, and compositions. 144 For instance, cationic lipids stimulate dendric cells, lipoplexes cause systemic inflammation, and grafted polyethylene glycol (PEG) polymers in lipid-based systems induce the synthesis of anti-PEG antibodies.144,145 Therefore, in vivo delivery of some CRISPR cargos may require co-administration of immunosuppressants to achieve relatively safer and more efficient therapeutic modality.
To date, various nonviral and viral in vivo delivery systems have been established (Table 4). Since creating INDELs through NHEJ is relatively easier than HDR-based modifications, most of the research has been focused on gene/sequence disruptions. Aside from delivery to the liver, a current drawback of in vivo delivery method is low editing frequencies at the targeted site, which likely provide little therapeutic benefit. 146 The other significant challenge for in vivo delivery systems is the immunogenicity of the CRISPR-Cas components or carriers (viral or nonviral particles).
AML, acute myeloid leukemia; RNP, ribonucleoprotein.
Recent studies have shown the presence of anti-Cas9 antibodies and antigen-specific T cells in human plasma.147,148 Given preexisting immunity to SaCas9 in a mouse model completely eliminated genome-edited liver cells, 149 this immunological rejection of Cas machinery and/or delivery vehicles (i.e., AAV 150 or nanoparticles 151 ) could hamper the efficacy of this editing approach. This could prove to be particularly problematic for repeated treatment modalities, and may present safety issues.
In summary, CRISPR-Cas editing has the potential to cure a number of devastating diseases. However, without optimized delivery methods, this technology remains largely inaccessible and inapplicable in a number of critical settings. Despite all the progress that has been made to increase efficiency and reduce immunogenicity and cytotoxicity, further optimization is still required. Currently, as there is no truly dominant delivery technique, researchers should assess which of the methods discussed above best fits their project's objective. Ideally, dominant methods will soon emerge and their applicability will reliably and thoroughly encompass the needs of scientists and clinicians, hoping to use CRISPR-Cas genome editing technologies.
Footnotes
Authors' Contributions
S.D. and J.F.T. designed the content. S.D., K.E., P.G., X.L., W.H., and J.F.T. wrote the article. S.D., K.E., P.G., and W.H. drew the figures. S.D., P.G., and J.F.T. edited the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The authors received no financial support for publication of this article.