
Abstract
The acetyl-CoA carboxylase isoform ACC2 expressed in the liver generates malonyl-CoA, which primarily regulates fatty acid oxidation through inhibition of the mitochondrial carrier carnitine palmitoyl-CoA transferase-I. Activity is initiated by sterol regulatory element-binding protein-1 (SREBP-1) binding to steroid response elements SRE in ACACB gene promoter P-II. We proposed that sequence variation in the promoter might affect expression. We investigated the effect of a single-nucleotide polymorphism −368 C/T (rs16939972) in ACACB P-II on activity in transfected HepG2 hepatoma cells. The T-allele construct showed significantly lower activity than the C-allele (p = 0.016) but only in the presence of SREBP-1a overexpression. Electrophoretic mobility shift assays showed that HepG2 nuclear proteins bound specifically to both allele probes, but with higher affinity to the T-allele. We tested competition for nuclear protein binding between the T-allele probe and unlabeled probes containing consensus sequences for six candidate transcription factors plus SREBP-1a. The SREBP-1a competitor probe had no effect on the shifted complex. GATA, c-Myb, and GR competitor probes abolished the complex; however, these proteins were undetectable in mass spectrometry of gel extracts from shifted bands. In conclusion, the −368 C/T single-nucleotide polymorphism in ACACB P-II binds HepG2 nuclear proteins that affect promoter activity in an allele-specific fashion.
Introduction
The ACC isoforms are encoded by separate genes, ACACA and ACACB, with different tissue distribution (Abu-Elheiga et al., 1995, 1997; Ha et al., 1996). ACC1 is highly expressed in liver and adipose tissues where lipogenesis is active, generating malonyl-CoA for de novo fatty acid synthesis (Oh et al., 2003). ACC2 is expressed in heart, skeletal muscle, and liver (Zhang and Kim, 1996), where the malonyl-CoA formed acts primarily as a regulator of fatty acid oxidation through carnitine palmitoyl-CoA transferase-I inhibition (Tong and Harwood, 2006). Transcription of the gene for ACC2, ACACB, is under the control of two promoters, P-I and P-II. In liver, P-I is inactive (Oh et al., 2005), but sterol regulatory element-binding protein-1 (SREBP-1) regulates expression through binding to steroid response elements (SRE) in P-II (Oh et al., 2003) in response to feeding status (McGarry, 1995).
We proposed that sequence variation in the ACACB gene promoter might affect expression in liver, influencing net levels of malonyl-CoA, with possible implications for energy balance dependent on genotype. We investigated the impact of a single-nucleotide polymorphism (SNP) −368 C/T (rs16939972) on P-II promoter activity in transfected HepG2 hepatoma cells. rs16939972 is located in a conserved region that we identified by alignment with rat and mouse sequences. It is the closest common SNP to the P-II transcription start site in the conserved region (minor allele frequency 0.29) (NCBI dbSNP CEU HapMap Phase 3, 2008) and is sited ∼300 bp upstream of the SREs (Oh et al., 2003). We investigated affinity of C- and T-allele probes for HepG2 nuclear proteins using electrophoretic mobility shift assays (EMSAs) and tested competition between the T-allele probe and probes containing consensus sequences for six candidate transcription factors plus SREBP-1a, for binding nuclear proteins. Finally, we attempted to confirm the identities of bound proteins using mass spectroscopy.
Materials and Methods
Cell culture
HepG2 human hepatoma cells were cultured in minimum essential medium supplemented with 10% (v/v) fetal bovine serum, 1% nonessential amino acids, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were maintained at 37°C with 5% CO2 and subcultured once a week. All reagents were from Sigma.
Sequence alignments
Alignment of human, mouse, and rat promoter sequences to identify SNPs in conserved sequences was performed by BlastZ in conjunction with the Ensembl Genome Browser (Schwartz et al., 2003).
SNP genotyping
The ACACB −368 C/T SNP was genotyped by Pyrosequencing (Qiagen) in 94 random subjects from TwinsUK (Spector and Williams, 2006) to identify homozygous subjects as DNA sources for reporter constructs. Template primers were as follows: forward, 5′-biotin-GGCGTCTGAC AGATGAACCC-3′; reverse, 5′-GCCCTACTTCACTGCTCA TGCA-3′. Sequencing primer (reverse strand): 5′-GGCTTTA AACAGTCCTGTA-3′.
Promoter-reporter constructs
To measure ACACB promoter II activity, a region from −870 to +37 relative to the transcriptional start site in ACACB exon 1b was amplified in a −368 C/C and a T/T subject. Primers were as follows: forward, 5′-GGTACCGTCCCAGTCTTTGGCTCTGTGG-3′; reverse, 5′-CTGGAGTCTTGAGTGAGGCTGACAGGCG-3′. Amplifications were performed for 30 cycles at 95°C for 30 s, 59°C for 30 s, and 72°C for 2 min in a buffer containing 1× Pfu buffer, 10 mM dNTP, 10 μM primers, and 0.4 U Pfu polymerase. All products were run on a 1% agarose gel in 1× TBE buffer, the bands were excised, and DNA was extracted and purified using the QIAquick gel extraction kit (Qiagen).
Cloning was performed using the TOPO TA Cloning kit (Invitrogen) according to manufacturer's instructions. Two microliter of the TOPO cloning reaction was used to transform TOP10 competent cells (Invitrogen) and plated on agar containing 100 μg/mL of ampicillin (Sigma-Aldrich) and incubated at 37°C overnight. The colonies were screened by polymerase chain reaction (PCR) for the presence of the insert using the primers shown above. Positive colonies were cultured in 10 mL lysogeny broth medium (Fisher BioReagents) containing 100 μg/mL of ampicillin and incubated at 37°C overnight. Plasmids were isolated using QIAprep miniprep kit (Qiagen) according to manufacturer's instructions and plasmid DNA was quantified using the NanoDrop ND-1000. Plasmid sequences were verified by Geneservice (
Transient transfection and luciferase reporter assay
Forty-eight hours before transfection, cells were set up for experiments at 2.5 × 105 cells per well on a 12-well plate. HepG2 cells were transfected with 500 ng constructs using FuGENE 6 (Roche Diagnostics). About 425 ng experimental DNA was co-transfected with 25 ng pRL-TK and either 50 ng of pcDNA3 or pcDNA-SREBP-1a. All experiments were performed with a positive control (pSyn-SRE) and a negative control (pGL3-basic) plasmid. The pSyn-SRE plasmid contains two SRE elements from the hamster HMG CoA synthase promoter (Dooley et al., 1998). pSyn-SRE and overexpression plasmid pcDNA-SREBP1a (Toth et al., 2004) were gifts from Tim Osborne. About 24 h after transfection, cells were washed once with phosphate-buffered saline (Oxoid) and lysed in 250 μL passive lysis buffer (Promega). Activities for SREBP-1 overexpression experiments were normalized to the amount of total protein because expression of Renilla pRL-TK was suppressed by overexpression of SREBP-1a. Total proteins in the lysate were determined using Bio-Rad Bradford protein assay (Bio-Rad) with bovine serum albumin (Promega) as standard. Three independent experiments were performed. In each experiment, transfection and luciferase assays were carried out in triplicate for each construct.
Electrophoretic mobility shift assay
Nuclear extracts were prepared from HepG2 cells transfected with pcDNA-SREBP-1a using the CelLytic Nuclear Extraction Kit (Sigma). The protein concentration was determined by the Bradford method (as above) and the nuclear extracts were stored at −80°C until used. Complementary oligonucleotides containing the alleles of ACACB −368 C/T are shown in Supplemental Table 1 (available online at
The forward primers were labeled with Cy3 at the 5′ end (
To investigate putative transcription factors binding to the ACACB −368 C/T SNP site (see below), unlabeled oligonucleotide probes containing a consensus binding motif were used in competition with the labeled T-probe. Supplemental Table 2 (available online at
The reaction mixtures were resolved on a native 6% Tris-borate-EDTA polyacrylamide gel in 0.5× TBE buffer. The gel was observed using the Ettan™ DIGE (GE Healthcare) and band intensities were measured using ImageQuant™ TL software (GE Healthcare).
Transcription factor binding site searches
To search for putative transcription factor binding sites at the ACACB −368 C/T SNP, a region comprising the SNP plus 10 bp 5′ and 3′ was analyzed with a variety of tools (Supplemental Table 3, available online at
Isolation of DNA-binding proteins
To isolate nuclear proteins binding at the SNP, the EMSA was repeated with several tracks containing samples of the ACACB T-allele binding reactions. The shifted band from each track was excised from the gel and pooled. In-gel reduction, alkylation, and digestion with trypsin were performed before subsequent analysis by mass spectrometry as described by Shevchenko et al. (1996).
Protein identification by mass spectrometry liquid chromatography–tandem mass spectrometry
Chromatographic separations were performed using an ultimate liquid chromatography system (Dionex). Peptides were resolved by reversed phase chromatography on a 75 μm C18 PepMap column. A 30 min gradient of acetonitrile in 0.05% formic acid was delivered to elute the peptides at a flow rate of 200 nL/min. Peptides were ionized by electrospray ionization using a Z-spray source fitted to a QTof-micro (Waters Corp.). The instrument was set to run in automated switching mode, selecting the two most intense precursor ions for sequencing by collision-induced fragmentation. The tandem mass spectrometry (MS/MS) analyses were conducted using collision energy profiles that were chosen based on the mass-to-charge ratio (m/z) and the charge state of the peptide.
Mass spectrometry data analysis
Tandem mass spectra were extracted by Masslynx version 4.0 and Protein Lynx Global Server 2.2.5. Charge state deconvolution and deisotoping were not performed. All MS/MS samples were analyzed using Mascot (Matrix Science; version Mascot). Mascot was set up to search mammalian entries of the sprot_56_8_090210 (SwissProtein) database (410518 entries) assuming the digestion enzyme trypsin. Mascot was searched with a fragment ion mass tolerance of 0.60 Da and a parent ion tolerance of 1.2 Da. Oxidation of methionine and iodoacetamide derivative of cysteine were specified in Mascot as variable modifications.
Scaffold (version Scaffold_2_02_03; Proteome Software Inc.) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95.0% probability as specified by the Peptide Prophet algorithm (Keller et al., 2002). Protein identifications were accepted if they could be established at >20.0% probability and contained at least one identified peptide. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al., 2003). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Results
Location of −368 C/T in conserved region
BlastZ was used to align 1 kb upstream of the human ACACB PII promoter with the mouse and rat counterparts (Fig. 1). The region from −1000 to −793 showed no conservation between the three species. The region from −792 to −582 showed moderate conservation between human and mouse only. From −581 to the transcription start site, mouse and rat sequences were highly conserved with stretches of human sequence (regions −520 to −493, −217 to −196, −169 to −157, −123 to −112, and −79 to −74). rs16939972 positioned at −368 and rs7305012 at −884 from the transcription start site were the only SNPs with reported frequencies >0.05 (respectively, 0.17 and 0.29) in the 1 kb aligned region. As rs16939972 lies in a region with a moderate degree of conservation, whereas rs7305012 lies in a region with no homology between human, mouse and rat, rs16939972 was chosen for analysis.

Alignment of human ACACB promoter with mouse and rat. Bases conserved between human and mouse and/or rat are highlighted in gray. Bases in known regulatory elements are underlined. Single-nucleotide polymorphisms are shown in bold. rs16939972 at −368 and rs7305012 at −884 were the only single-nucleotide polymorphisms with MAF >0.05. −368 C/T is shown in white, highlighted in black (T-allele shown).
Effects of ACACB −368 C/T on promoter activity in HepG2 cells
HepG2 cells were transiently transfected with the ACACB −368 C- or T-allele promoter constructs or the empty construct (pGL3-basic). In HepG2 cells, luciferase activity was minimal and only approximately five times higher than the empty construct. There was no significant difference in activity between constructs carrying the C- and T-alleles. We then tested the effect of overexpression of SREBP-1a on the C- and T-allele reporter construct activities. Figure 2 shows that luciferase activity in both constructs was stimulated by SREBP-1a. In overexpression experiments, promoter activity for the −368 C-allele construct was 63.5-fold higher than that for pGL3 and promoter activity for the −368 T-allele construct was 47.2-fold higher. Activity of the −368 C-allele construct was 1.35-fold higher than that of the −368 T-allele construct (p = 0.016).
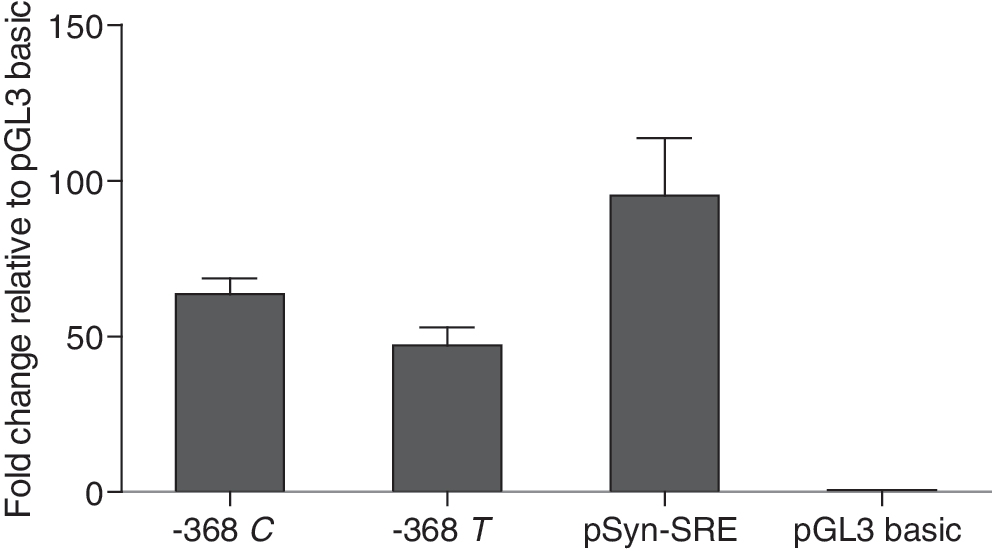
Activity of ACACB promoter-reporter constructs carrying alleles of −368 C/T. Each reporter construct was co-transfected into HepG2 cells with the overexpression plasmid pcDNA-SREBP-1a or pcDNA3 control. The reporter activities are shown as fold change relative to pGL3. The data represent the mean ± SD of three independent experiments performed in triplicate. The difference in activities of the −368 C and −368 T constructs is significant (p = 0.016). SREBP-1, sterol regulatory element-binding protein-1.
ACACB −368 C/T allele-specific binding of HepG2 nuclear proteins
Binding of nuclear protein extracts from HepG2 cells to double-stranded oligonucleotide probes carrying the ACACB −368 C- or T-allele was investigated by EMSA. Both C- and T-allele probes produced two shifted complexes, A and B (Fig. 3), but the T-allele probe in both complexes showed increased labeling intensity compared to the C-allele probe. The shifted complexes are specific to the −368 C/T SNP, as they were abolished by 25-fold excess unlabeled oligonucleotides.
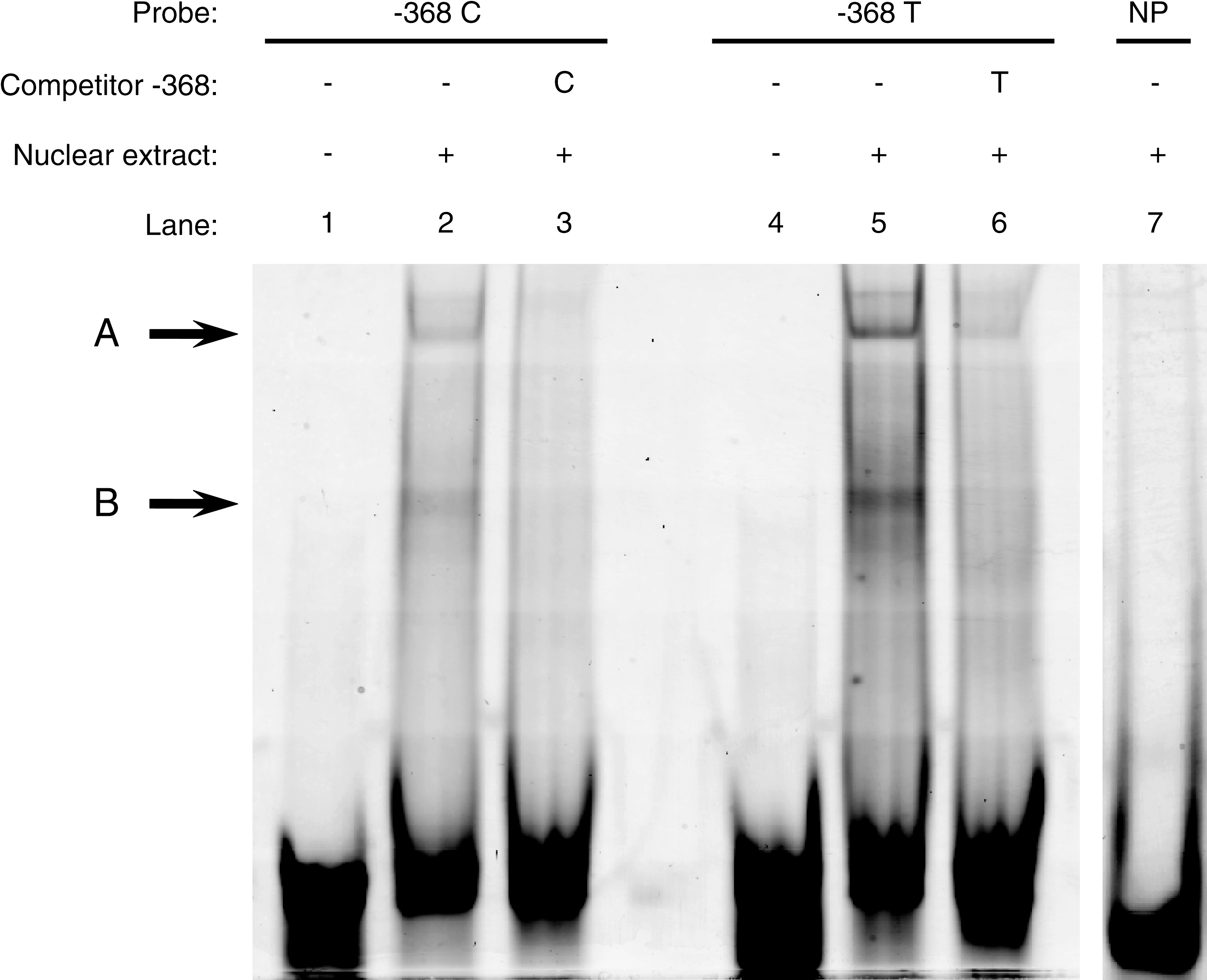
Electrophoretic mobility shift assay of the ACACB −368 C/T polymorphism. C- and T-allele probes produced two shifted complexes, A and B. Lanes 1 and 4: Negative reactions with labeled probes −368 C (1) and −368 T (4) in absence of nuclear extracts. Lanes 2 and 5: Shifts with labeled probes −368 C (2) and −368 T (5) and 6 μL of nuclear extract from HepG2 transfected with SREBP-1a plasmid. Lanes 3 and 6: Specific competitor reaction with same components as lanes 2 and 5 plus added 25-fold excess of unlabeled probes −368 C (3) and −368 T (6). Lane 7: Nonrelated probe (NP) with same components as lanes 2 and 5.
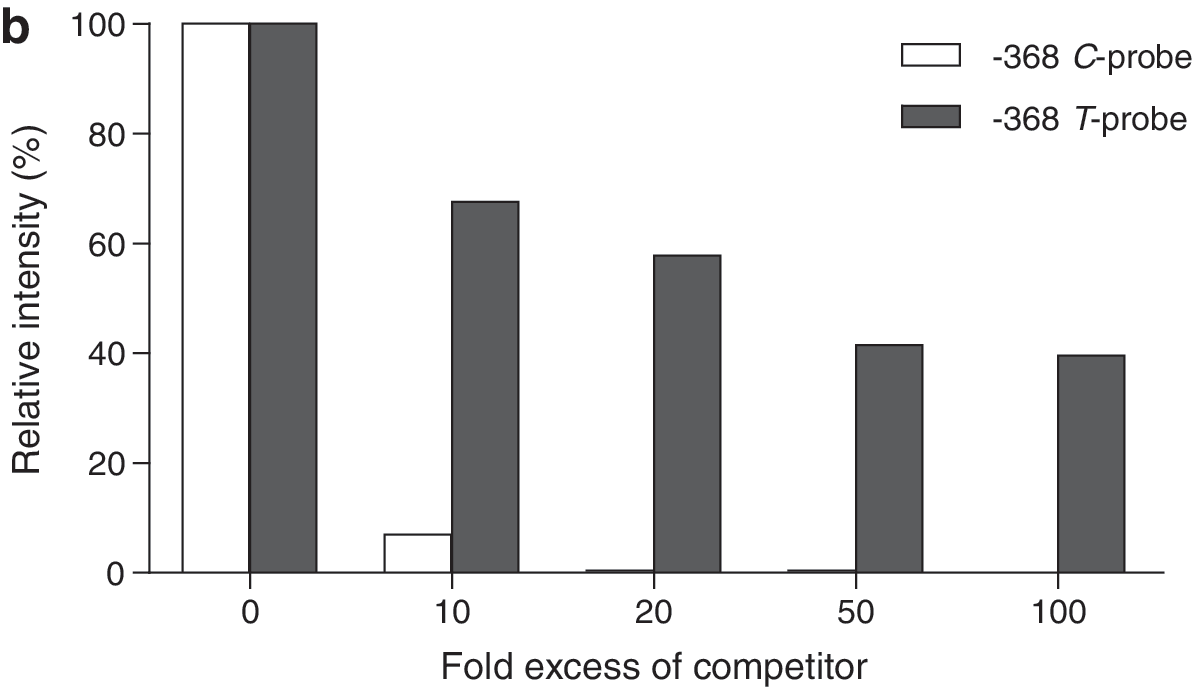
An allele-specific difference in protein binding affinity was shown by increasing amounts of unlabeled T-allele probe in competition with labeled C-probe and vice versa. Figure 4a shows that the unlabeled C-probe did not compete as efficiently as the unlabeled T-probe. Figure 4b shows that the unlabeled T-probe completely abolished complex B, whereas unlabeled C-probe reduced the intensity of labeled complex B to ∼40%, suggesting that the T-allele has a higher affinity for the nuclear proteins. Intensities of complex A (not shown) appeared almost identical to that for complex B shown in Figure 4b.
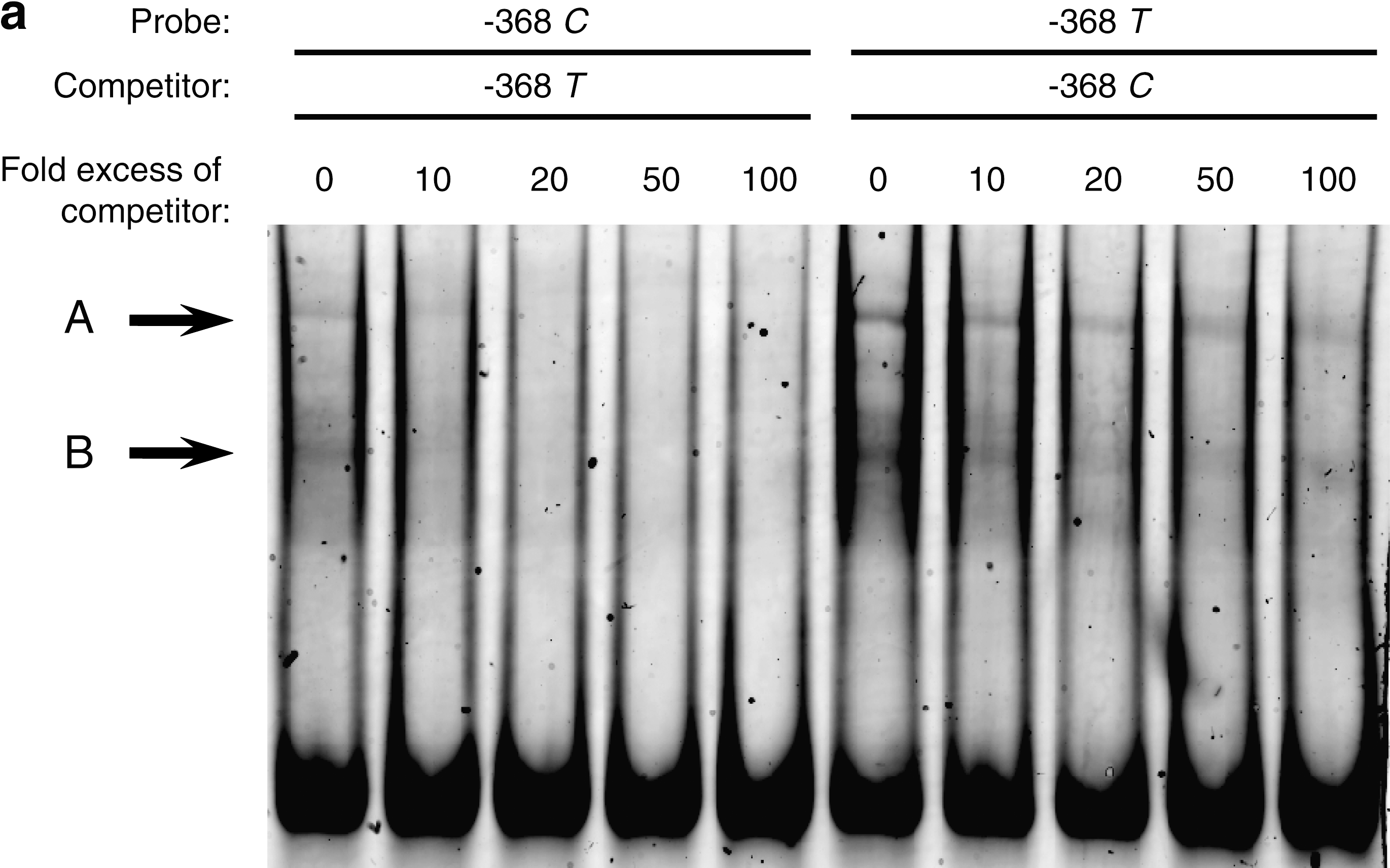
Effect of increasing competitor allele probe excess. A and B are two shifted complexes. (
Putative TF binding sites located in the ACACB promoter
In all the TF search programs the T-allele returned a higher number of putative transcription factors than the C-allele (Supplemental Table 3). We chose candidates based on the number of times they appeared in the programs, their core sequence location on the SNP, and their specificity for one of the alleles (Supplemental Table 4). Using MatInspector, sterol regulatory element binding protein (SREB) was found on the T-allele only. In TESS, GATA proteins (I00109, J00037, and R03442) and Gfi (J00038) were located on the T-allele only. In TESS, Oct-1 was found on the C-allele only, and in TF search, only on the T-allele. A range of transcription factors specific for each allele were found using AliBaba 2.1 and P-match, including CREB-BP1 (for the C-allele), GATA, CRE-BP1, and GR (for the T-allele).
The EMSA had shown that C- and T-alleles bind to the same proteins (shifted complexes A and B) but that the T-allele bound more strongly (Fig. 4a). Therefore, to determine whether putative transcription factors from the bioinformatics search may bind to the −368 C/T region, unlabeled double-stranded oligonucleotide probes containing consensus sequences for CRE-BP1, C-Myb, GATA, Gfi, GR, Oct-1, and SREBP-1 were used in competition with labeled T-probes in binding to HepG2 nuclear proteins. Although SREBP-1 was not indicated as a TF potentially binding at the SNP site in any searches and known SREs lie remote from the SNP, an SREBP-1a competitor probe was included to rule out any possibility of an allele-specific effect on activity based on affinity for SREBP-1a. Figure 5 shows that none of the unlabeled probes had an effect on complex A. However, unlabeled C-Myb, GATA, and GR completely abolished complex B. CRE-BP1, Gfi, Oct-1, and SREBP-1a competitor probes had no effect on either of the shifted complexes.
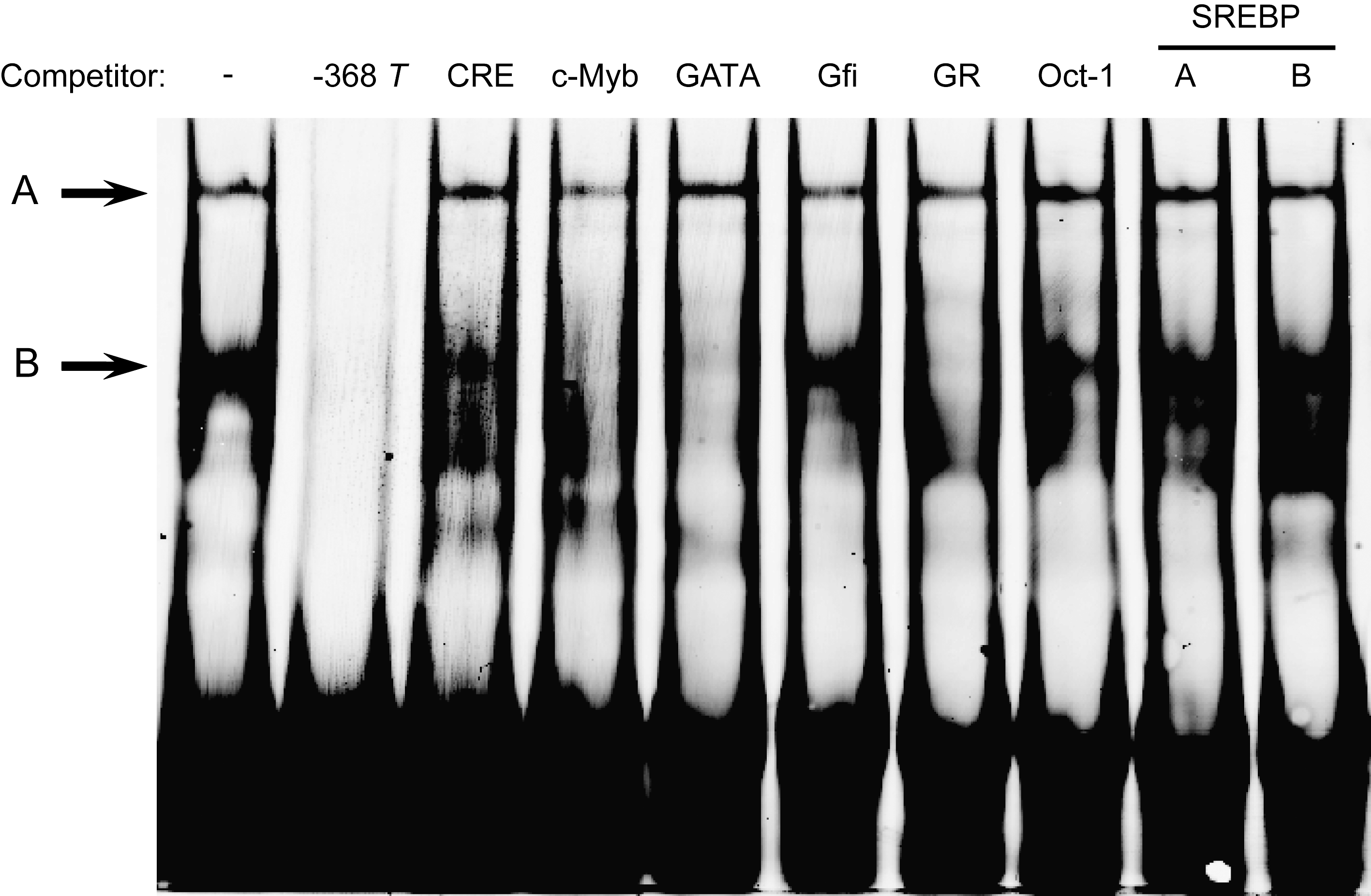
Effect of unlabeled probes containing transcription factor binding motifs. Lane 1: Binding reaction with ACACB −368 T-allele probe without competitor. Lanes 2–8: Binding reaction with competitor containing binding motifs for transcription factors shown above each lane. Lanes 9 and 10: Binding reaction with competitor containing binding motifs for SREBP-1 from A (Reed et al., 2008) and B (Ai et al., 2006).
Mass spectrometry of nuclear proteins bound to ACACB −368 C/T
In an attempt to confirm the identities of proteins bound to the ACACB T-allele probe, shifted complexes were excised from an EMSA gel and extracted proteins were analyzed by mass spectrometry. Mass spectral data were searched against all mammalian entries in the SwissProt database according to the criteria described in the Materials and Methods section. Summary results and validation by Scaffold are given in Table 1. Many peptides were assigned to protein orthologues from multiple species and the relevant accession names are provided for each case in the full spectrum report available upon request. However, in view of the experimental conditions, human and bovine are anticipated to be the most relevant protein assignments.
Results based on all mammalian entries in the SwissProt database. Criteria thresholds are 20% for protein identification, minimum 1 peptide per protein and 95% probability per peptide.
Results also show accession numbers from other species.
To assess false discovery rate and increase the confidence of protein assignments to the mass spectral results, the data for each sample were searched against a SwissProt decoy database using Mascot. This contains randomized sequences, and therefore any peptides matching will be highlighted as a false-positive. No false-positives were assigned within the dataset, indicating a zero false discovery rate for peptides above Mascot's identity threshold and thus provided high confidence of assignments. None of the putative transcription factors c-Myb, GATA, and GR were present in the protein complex B bound to the ACACB T-allele probe, but one transcription factor (splicing factor, proline- and glutamine-rich [SFPQ]), also known as polypyrimidine tract-binding protein-associated splicing factor, was identified (Table 1).
Discussion
In this study we have shown an SNP allele-specific effect on human ACACB gene promoter activity. ACC is one of the enzymes influencing cellular levels of malonyl-CoA, a key physiological regulator of cellular fatty acid oxidation and lipid partitioning. In liver and skeletal muscle, AMPK coordinates the changes in the activity of the enzyme through phosphorylation, regulating the partitioning of fatty acid between oxidative and biosynthetic pathways according to demand for ATP (Saha et al., 1994). The metabolic changes in the liver in response to nutrient availability are not as rapid as those in muscle. Here, transcriptional regulation may be more important than posttranslational modification of enzymes in accounting for slower changes (Oh et al., 2005). ACC2 activity in liver is initiated by SREBP-1 binding to SRE in P-II (Oh et al., 2003). Activity is decreased during starvation and restored on feeding (McGarry, 1995), in line with the role of malonyl CoA in regulating fatty acid oxidation in response to changing energy demands.
We were only able to demonstrate allele-specific differences in ACACB promoter activity in the presence of overexpression of SREBP-1a in transfected HepG2 cells, compatible with the reported role of SREBP-1a in the control of ACACB transcription (Oh et al., 2003). Without SREBP-1a, the constructs showed minimal promoter activity, but still 5 times higher than the empty construct. Recent work has highlighted the importance of SREBP-1a rather than the −1c isoform in liver ACC2 expression. ACACB in liver was downregulated in mice with specific inactivation of SREBP-1a, even though SREBP-1c is present at 10-fold higher levels. SREBP-1c could not substitute because transcriptional coactivators were recruited more efficiently to the more potent SREBP-1a activation domain (Im et al., 2009). The lower promoter activity associated with the −368 T-allele implies a reduction in ACC2 expression and malonyl-CoA production, inferring increased fatty acid β-oxidation and reduced levels of intracellular fatty acids, as seen in the Acc2 −/− mouse (Abu-Elheiga et al., 2001, 2003).
Nuclear proteins binding to ACACB −368 C- and T-allele-specific probes resulted in two shifted complexes in EMSAs. The T-allele probe showed higher affinity for proteins in both bands, suggesting that at least one bound protein may be acting as a repressor. SREBP-1a competitor probes had no effect on either of the shifted complexes A or B, which rules out any allele-specific effect on activity based on affinity for SREBP-1a. Binding at the SNP site was not indicated in any of the TF searches and the known SREs lie 306 bp downstream of the SNP (Oh et al., 2003). Complex B was completely abolished when competed with c-Myb, GATA, or GR unlabeled probes, but complex A was not affected. The unlabeled GATA oligonucleotides produced the clearest abolition of shifted complex B and the GATA consensus sequence 5′-TGATAA-3′ showed only one base pair difference to the −368 C/T region (5′-C/TGATTA-3′). Verification of the identities of bound proteins might have been possible by testing supershift of complex B with antibodies directed against each putative factor. Also, transient transfection of HepG2 cells with expression plasmids and allele-specific ACACB reporter gene constructs could confirm whether the TF was responsible for differential inhibition of ACACB promoter activity with respect to −368 T- or C-allele. However, the GATA family of transcription factors comprises six members (Ko and Engel, 1993). Even restricting candidature to GATA2, 3, 4, and 6 shown to be expressed in human Hep3B hepatocytes (Dame, 2004), together with c-Myb and GR, comprehensive investigation using these approaches would be a lengthy undertaking. We therefore attempted to identify proteins bound to the T-allele probe using mass spectrometry. None of the c-Myb, GATA, or GR proteins was identified. The proteins needed to survive gel electrophoresis, possible deterioration in the excised gel stored at −20°C, and the trypsin treatment before mass spectrometry. It is possible that the bound proteins may have been lost or they may have originally been present in the band at levels too low for identification. Although not confirmed, the results of the EMSA competition indicate that involvement of c-myb, GATA, and GR in modulating promoter activity cannot be ruled out.
One transcription factor, SFPQ, (defined in results) was identified with a probability of 91.8%. However, SFPQ was represented by only 2.12% of sequence coverage and 0.50% of the total spectra. It is unlikely that this protein is the transcription factor binding at the −368 T/C site. At present, the identities of the bound nuclear proteins associated with differential level of promoter activity remain unknown.
In conclusion, considerable evidence supports an important role for malonyl-CoA in the regulation of appetite and lipid homeostasis. In Acc2 −/− knockout mice, absence of malonyl-CoA promoted higher fatty acid oxidation in muscle and less fat deposition in adipose tissue (Abu-Elheiga et al., 1995, 1997) and higher energy expenditure (Choi et al., 2007) compared to wild-type mice. We have shown that rs16939972 is a regulatory SNP binding nuclear proteins that affect promoter activity in an allele-specific fashion. Lower levels of cellular malonyl-CoA in human ACACB T-allele carriers could result in reduced lipogenesis and promotion of fatty acid oxidation. There are no reported human studies of ACACB SNP associations with obesity or insulin sensitivity variables, but our results suggest that carriage of the variant T-allele might confer protective effects.
Footnotes
Acknowledgments
The authors thank the following for their kind gifts: Tim Osborne (University of California, Berkeley, CA) for pSyn-SRE and pcDNA-SREBP-1a plasmids, and Tim Spector (Twin Research and Genetic Epidemiology Unit, King's College London, United Kingdom) for DNA samples from the Twins UK cohort. This work was supported by a Medical Research Council Doctoral training account awarded to AKL.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.