
Abstract
Pulmonary arterial smooth muscle cell (PASMC) phenotype switching, which is characterized by changes in smooth muscle (SM)-specific gene expression, contributes to vascular remodeling in pulmonary hypertension. In addition, it has been shown that the transcription of SM-specific genes is modulated by cytoskeleton rearrangement. However, the intracellular mechanisms and signaling pathways that regulate these relationships are largely unknown. In the present study, we aimed to investigate the roles that phosphoinositide 3-kinase (PI3K) and protein kinase B (PKB), also known as AKT, play in modulating the cytoskeleton and phenotype of rat PASMCs. To observe the downstream effects of inhibiting or enhancing PI3K/AKT pathway activity, we used various approaches to manipulate protein function and gene expression. Treatment of PASMCs with platelet-derived growth factor (PDGF)-BB or PIK3CA-adenovirus induced cytoskeleton rearrangements and downregulated SM22α and α-SM actin gene expression. Inhibition of PI3K led to blocking of AKT phosphorylation and attenuated the PDGF-BB-induced downregulation of F-actin and SM-specific genes, the downstream effector of PI3K. The decrease in SM22α and α-SM actin mRNA levels induced by PDGF-BB was markedly and reproducibly blocked by LY294002. PI3K/AKT pathway plays a vital role in the modulation of PASMCs cytoskeleton rearrangement and phenotype switching.
Introduction
P
It is well established that VSMCs, in contrast to myocardial and skeletal muscle cells, are not terminally differentiated and exhibit prominent plasticity (Owens et al., 2004; Bochaton-Piallat et al., 2005). Based on variances in their proliferation, synthetic activities, and contractile protein expression levels, VSMCs can demonstrate either a contractile phenotype (differentiated state) or a synthetic phenotype (dedifferentiated state) (Rensen et al., 2007). The former type of VSMCs, present within mature animal blood vessels, typically proliferate at a relatively low rate and produce a repertoire of smooth muscle (SM)-specific contractile proteins. However, there is extensive evidence showing that these slow-proliferating VSMCs can reversibly switch to a dedifferentiated state under the control of certain critical environmental cues during embryonic development and pathophysiological processes (Owens and Wise, 1997). This phenotypic change is characterized by over-proliferation; increased ability of migration; secretion of collagen, elastin, and proteoglycans into the extracellular matrix; and low expression of contractile proteins (Jeffery and Morrell, 2002; Mandegar et al., 2004).
The SM-specific contractile proteins are considered to be markers of VSMCs. The earliest known and most abundant VSMC marker, α-SM actin, and another marker discovered early on and involved in the contraction of VSMCs, SM22α, are currently the subject of wide study (Mack and Owens, 1999; Fu et al., 2000). Investigators have confirmed that the transcription of SM-specific genes is regulated by actin polymerization (Mack et al., 2001; Liu et al., 2003; Han et al., 2009). In this process, individual subunits of actin called globular actins (G-actins) assemble into long filamentous polymers and stress fibers known as F-actins. Actin filaments have been shown to be regulated by several intracellular signaling pathways, including Src, mitogen-activated protein kinase (MAPK), and major members of the Rho family of the small GTPases (i.e., Rho, Cdc42, and Rac) (Carpenter, 2000; Tang et al., 2008).
Several studies have demonstrated that phosphoinositide 3-kinase (PI3K)/AKT signaling is required for the platelet-derived growth factor (PDGF)-induced proliferation and migration of VSMCs (Goncharova et al., 2002; Choi et al., 2010), which are prominent features involved in the process of phenotype switching and the changes of cytoskeleton (Jefferyand Morrell, 2002; Mandegar et al., 2004). The PI3Ks constitute a family of enzymes involved in cell proliferation, differentiation, motility, survival, and intracellular trafficking (Katso et al., 2001; Vanhaesebroeck et al., 2001). These enzymes are activated by stimulation with an environmental cue, such as PDGF, which in turn recruits pleckstrin homology domain-containing signaling molecules such as protein kinase B (PKB), also known as AKT, and other protein kinases including PDK1 and CRTC2 to the cell membrane to result in AKT phosphorylation (Stokoe et al., 1997; Vanhaesebroeck et al., 2012). Phosphorylated AKT can then activate or deactivate a huge variety of substrates via its kinase activity and thereby affect multiple cellular processes that include apoptosis, proliferation, and transcription. The PI3K family is divided into Class I, II, and III members (Stokoe et al., 1997; Vanhaesebroeck et al., 2012). Class I PI3Ks are composed of a regulatory and a catalytic subunit. PIK3CA is a Class I PI3K catalytic subunit, and recent evidence has shown that the PIK3CA gene is mutated in a range of human cancers (Stokoe et al., 1997; Vanhaesebroeck et al., 2012).
However, the role of PI3K/AKT in pulmonary arterial smooth muscle cell (PASMC) cytoskeleton rearrangement and phenotype switching remains unclear. Thus, we inhibited or activated PI3K/AKT signaling to observe the downstream effects on these cellular processes, using multiple approaches to interfere with protein function and abundance and gene expression. We end by discussing how PI3K/AKT could be involved in pulmonary vascular remodeling during the development of pulmonary hypertension.
Materials and Methods
Reagents
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), L-glutamine, streptomycin/penicillin, and trypsin were purchased from HyClone. LY294002 was acquired from Sigma-Aldrich, and PDGF-BB was supplied by R&D Systems. The adenovirus that contains the coding sequences for PIK3CA (GenBank No. NM_133399) and green fluorescent protein (GFP) was constructed by the Chinese National Human Genome Center (China) and named PIK3CA-adenovirus. HiPerFect Transfection Reagent was purchased and the PIK3CA small interfering RNA (PIK3CA-siRNA) (target sequence: 5′-AACCCTTAAAGGGCATAATTA-3′) was custom-designed and synthesized by Qiagen using the rat sequence with accession number NM_133399. Rhodamine-phalloidin and 4′,6-diamidino-2-phenylindole (DAPI) were bought from Invitrogen. NP-40 lysis buffer, the SDS-PAGE Gel Preparation Kit, and the BCA Protein Quantitation Kit were provided by Beyotime. The polyvinylidene fluoride (PVDF) microporous membranes and ECL Western Blotting Detection System were obtained from Millipore. Primary antibodies against β-actin (cat No. ab8227), phosphorylated AKT (Ser473) (cat No. ab66138), total AKT (pan) (cat No. ab8805), α-SM actin (cat No. ab5694), and SM22α (cat No. ab14106) and the goat secondary antibodies against mouse, rat, and rabbit IgG were purchased from Abcam. SYBR Green and Titanium™ Taq DNA polymerase purchased from Invitrogen and Clontech, respectively.
Isolation and culture of PASMCs
All procedures were performed under the guidelines set forth by Sichuan University. Male Sprague–Dawley rats obtained from the Animal Breeding Center of Sichuan University were sacrificed by cervical dislocation. A segment of pulmonary artery just proximal to the lung entry point was collected under aseptic conditions and trimmed of connective and fat tissues. The media of the pulmonary artery was then dissected away from the adventitia and intima and cut into small blocks. These tissue blocks were cultured in DMEM containing 20% FBS, 100 U/mL penicillin, 100 mg/mL streptomycin, and 200 mM glutamine at 37.0° in a humidified 5% CO2 atmosphere. Primary PASMCs were subcultured to the third generation for experiments and were identified by α-SM actin staining (Supplementary Figure S1; Supplementary Data are available online at
Transduction of PIK3CA-adenoviruses into PASMCs
PIK3CA-adenoviruses or control adenoviruses containing only the GFP coding sequence were diluted in serum-free DMEM to the final concentration of 1×108 PFU/mL and added to the starved PASMCs. The medium was replaced 6 h later, and the culture of PASMCs continued. The efficiency of PIK3CA-adenovirus overexpressing PIK3CA gene was measured by real-time quantitative reverse transcription–polymerase chain reaction (RT-qPCR).
Transfection of PIK3CA-siRNA into PASMCs
PIK3CA-siRNA was used at a concentration of 120 nM, and a nonsilencing siRNA containing only the GFP coding sequence was used at 120 nM as a negative control. Cells were transfected with a 1:2 mixture of siRNA to HiPerFect™ Transfection Reagent according to the manufacturer's instructions. After 6-h incubation, the medium was replaced, and the culture of PASMCs continued. The efficiency of PIK3CA-siRNA silencing PIK3CA gene was measured by RT-qPCR.
Western blot analysis
PASMCs were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in cold NP-40 lysis buffer (50 mM Tris-HCl [pH 7.4], 1% Nonidet P-40, and 150 mM NaCl) for 30 min. Cell lysates were then collected by centrifuging at 12,000 g for 5 min at 4°C, and the protein concentration was determined using the BCA protein assay kit. Proteins (30 μg/lane) were separated by electrophoresis on 10% polyacrylamide sodium dodecyl sulphate gels and transferred onto PVDF microporous membranes. The membranes were incubated with primary antibodies against p-AKT, total AKT, SM22α, α-SM actin, or β-actin overnight, washed twice with PBS-Tween 20, and then incubated with secondary antibody for 45 min. The blots were developed using the ECL Western Blotting Detection System with a ChemiDoc™ MP imager (Bio-Rad). The densitometry values of the bands were measured using Image Pro-Plus software (Media Cybernetics).
Fluorescent RT-qPCR
Cells were washed with PBS and lysed using TRIzol reagent (Invitrogen), and total RNA was isolated according to the manufacturer's protocol. Total RNA (1 μg) was used as template for the reverse transcriptase (RT) reaction using MMLV, 2 μg random hexamers, 0.5 mM dNTPs, and 5 mM DTT (Life Technologies). RT-qPCR was performed in the iCycler iQ™ Detection System (Bio-Rad) using SYBR Green and Titanium™ Taq DNA polymerase according to the manufacturer's recommendations. The protocol consisted of 45 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 1 min with the primers for PIK3CA (NM_133399), TAGLN (SM22α, NM_031549), ACTA2 (α-SM actin, NM_031004), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, NM_17701). The following primer sequences were used for the amplification reactions: for PIK3CA, forward primer: 5′-GAA ACC TCA GGC TTG AAG AGT-3′, reverse primer: 5′-CTC ATT GTT CTG AAA CAG T-3′; for TAGLN, forward primer: 5′-AAC GCT ACT CTC CTT CCA G-3′, reverse primer: 5′-GCT CCT CAT CAT ACT TCT TCT C-3′; for ACTA2, forward primer: 5′-ATA TTC TGT CTG GAT CGG C-3′, reverse primer: 5′-GCT TCG TCA TAC TCC TGT TT-3′; and for GAPDH, forward primer: 5′-TTC AAC GGC ACA GTC AAG G-3′, reverse primer: 5′-CTC AGC ACC AGC AT C ACC-3′. The expected sizes of the amplified DNA fragments were 110 bp for PIK3CA, 145 bp for TAGLN, 149 bp for ACTA2, and 114 bp for GAPDH. The expression levels of the target genes were each normalized to the GAPDH level. The 2−ΔΔCt method was used to calculate the relative expression levels for each target.
Visualization of actin filaments under laser confocal microscopy
PASMCs were cultured on 18-mm glass coverslips before being treated, fixed with 4% cold formaldehyde for 20 min at 37°C, and then rinsed and permeabilized with 0.2% Triton for 10 min. After being washed thrice with PBS and incubated with rhodamine-phalloidin (1:100) for 2 h in the dark, the cells were again washed twice with PBS to remove unbound label. Next, the cells were incubated with DAPI for 10 min in the dark. Finally, actin filaments were visualized and imaged using a Zeiss laser confocal microscopy with a 20×objective lens.
Statistics
All results are expressed as mean±standard error. The significance of differences between groups was tested by one-way analysis of variance (ANOVA). Differences were considered statistically significant when p<0.05.
Results
AKT phosphorylation in PASMCs was induced by PDGF-BB and blocked by LY294002
Phosphorylated AKT levels in PASMCs increased in a time-dependent manner following stimulation of the cells with PDGF-BB (Fig. 1A, B). Our experiments showed that AKT phosphorylation was rapid and reached its maximal extent by 15 min post stimulation. Subsequently, the activation level decreased slightly, fluctuated, and then was sustained for over 3 h with no significant decrease. LY294002 is a specific inhibitor of PI3K that is soluble in DMSO (Kaplan-Albuquerque et al., 2003). Increasing concentrations of LY294002 (3.3, 10, 30 μM) blocked AKT phosphorylation by 23%, 100%, and 100%, respectively (Fig. 1C). These results showed that PDGF-BB activated PI3K/AKT signaling, resulting in the phosphorylation of AKT. This process can be blocked by the PI3K inhibitor, LY294002.
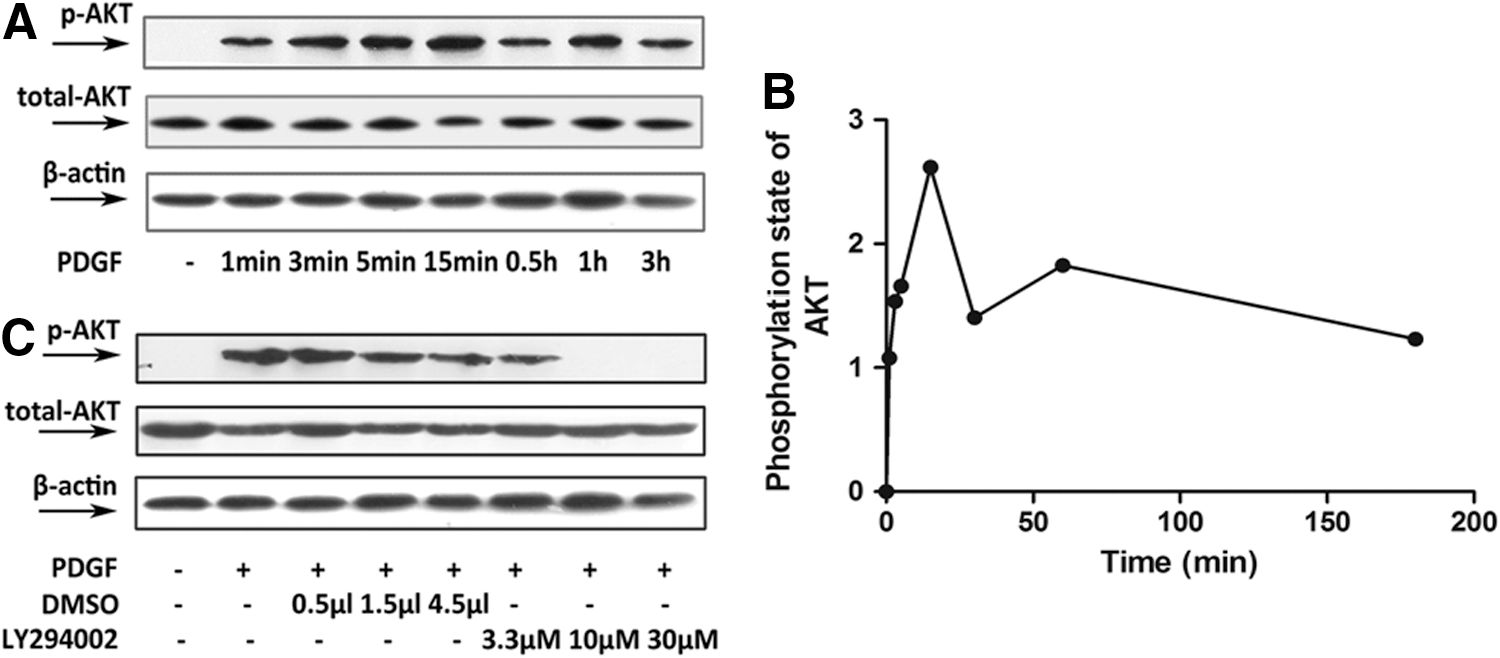
Platelet-derived growth factor (PDGF)-BB activated the phosphoinositide 3-kinase (PI3K)/AKT pathway in pulmonary arterial smooth muscle cells (PASMCs). In
Inhibition of the PI3K/AKT signaling pathway in rat PASMCs inhibited the PDGF-BB-induced reduction of VSMC marker gene expression
As detected by western blotting, the relative expression levels of SM22α and α-SM actin were markedly decreased in rat PASMCs incubated with PDGF-BB or with DMSO plus PDGF-BB (p<0.05) (Fig. 2). LY294002 significantly inhibited this PDGF-BB-induced downregulation of SM22α and α-SM actin (p<0.05) (Fig. 2C, D).
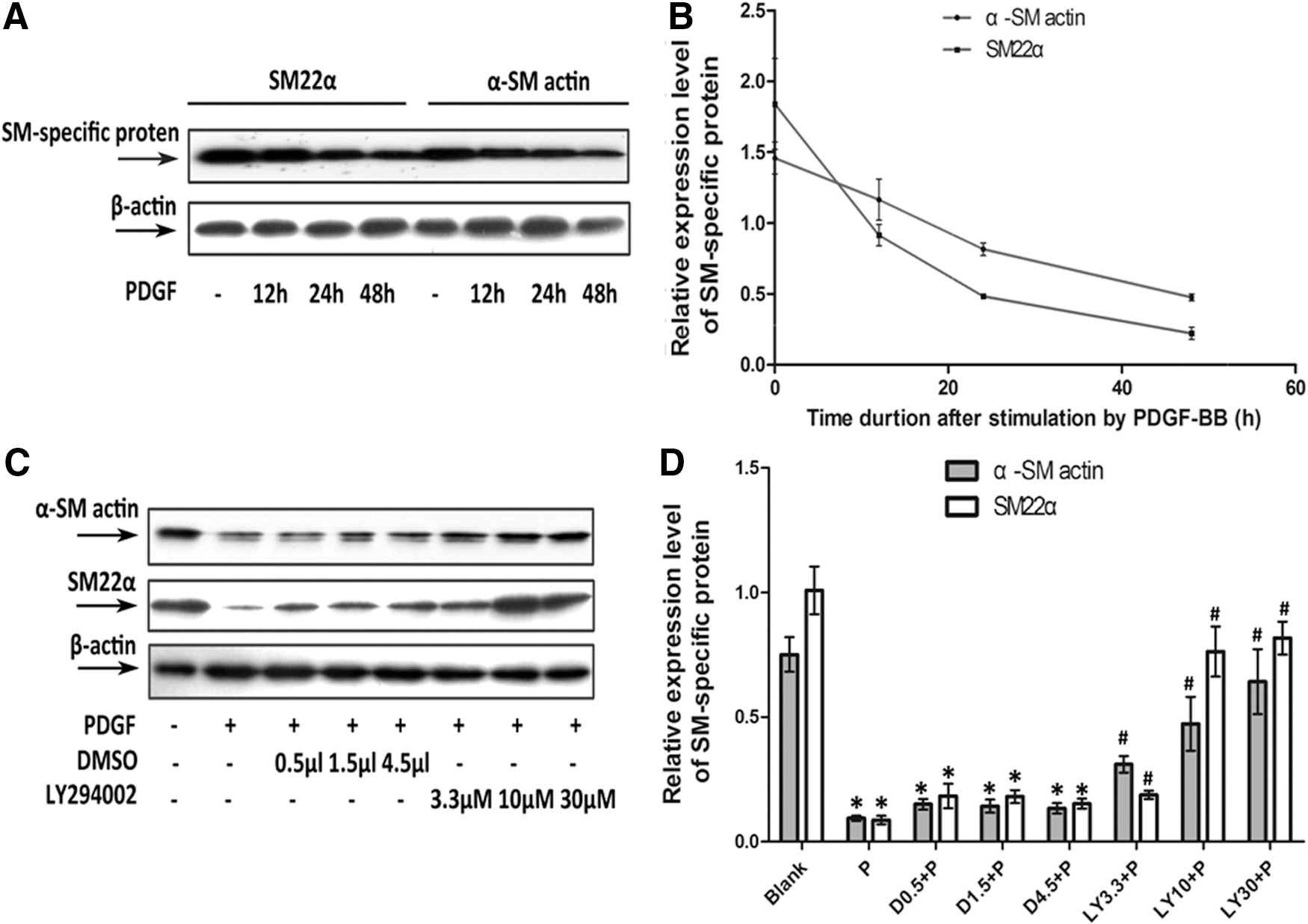
LY294002 blocked the PDGF-BB-induced downregulation of vascular smooth muscle cell (VSMC) marker gene expression at protein level. In
PASMCs transfected with PIK3CA-siRNA displayed a decrease in AKT phosphorylation (Fig. 3A), demonstrating that the target gene was successfully silenced. In PASMCs incubated with PDGF-BB or control siRNA plus PDGF-BB, we found low expression levels for α-SM actin and SM22α (p<0.05), whereas higher levels of these marker proteins were detected in PIK3CA-siRNA-transfected PASMCs (p<0.05) (Fig. 3B, C). The efficiency of PIK3CA-siRNA silencing PIK3CA gene was measured by RT-qPCR shown in the Table 1.
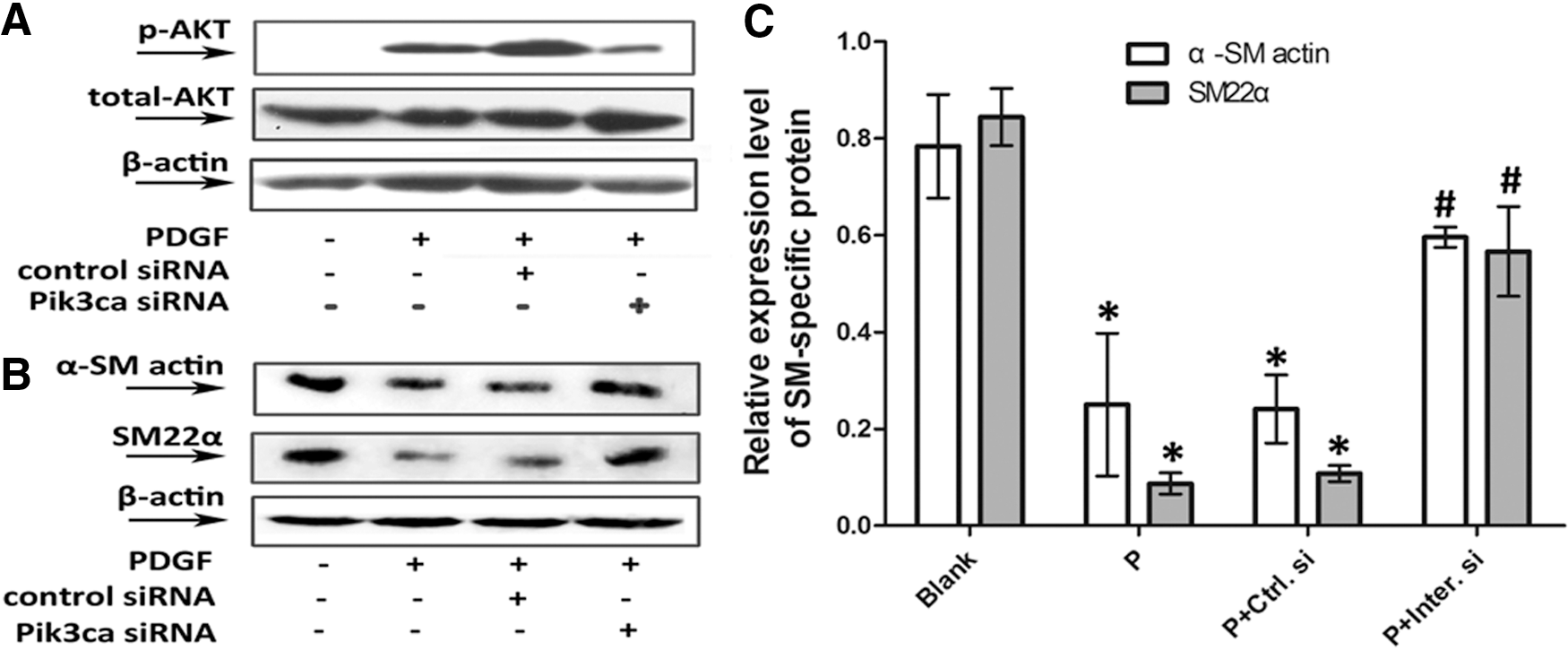
PIK3CA-siRNA attenuated the PDGF-BB-induced downregulation of VSMC marker gene expression at protein level. Starved PASMCs were left unstimulated (Blank group), were stimulated with 20 ng/mL PDGF-BB alone for 15 min in
Starved PASMCs were left untransfected (Blank group), were transfected with empty adenoviruses (Empty V. group), were transfected with PIK3CA-adenoviruses (PIK3CA V. group) and were pretreated with 120 nM control siRNA (P+Ctrl.si group) or PIK3CA-siRNA (P+Inter.si group) before stimulation by PDGF-BB for 48 h.
p<0.05, versus Blank group.
p<0.05, versus Empty V. group.
p<0.05, versus P+Ctrl.si group.
PASMCs, pulmonary arterial smooth muscle cell.
The observed PDGF-BB-dependent decline in α-SM actin and SM22α mRNA levels (p<0.05) was reproducibly and significantly prevented to a similar extent by LY294002 and PIK3CA-siRNA (p<0.05) (Fig. 4A).
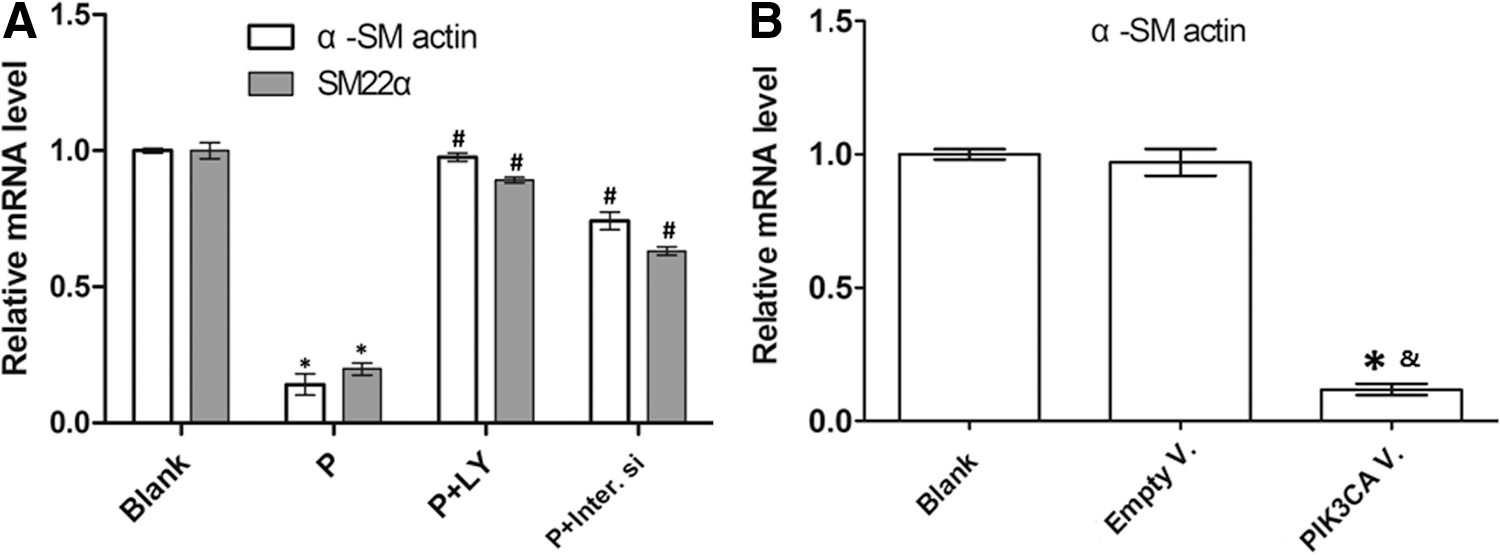
PI3K/AKT signaling pathway regulated VSMC marker gene expression at mRNA level. In
Enhancement of PI3K/AKT signaling pathway in rat PASMCs reduced VSMC marker gene expression
A high level of AKT phosphorylation was detected in PIK3CA-adenovirus-transfected PASMCs (Fig. 5A), indicating that PIK3CA-adenoviruses successfully activated the PI3K/AKT pathway. Further, the over-expression of PIK3CA led to an obvious downregulation of α-SM actin and SM22α expression (p<0.05) (Fig. 5B, C).
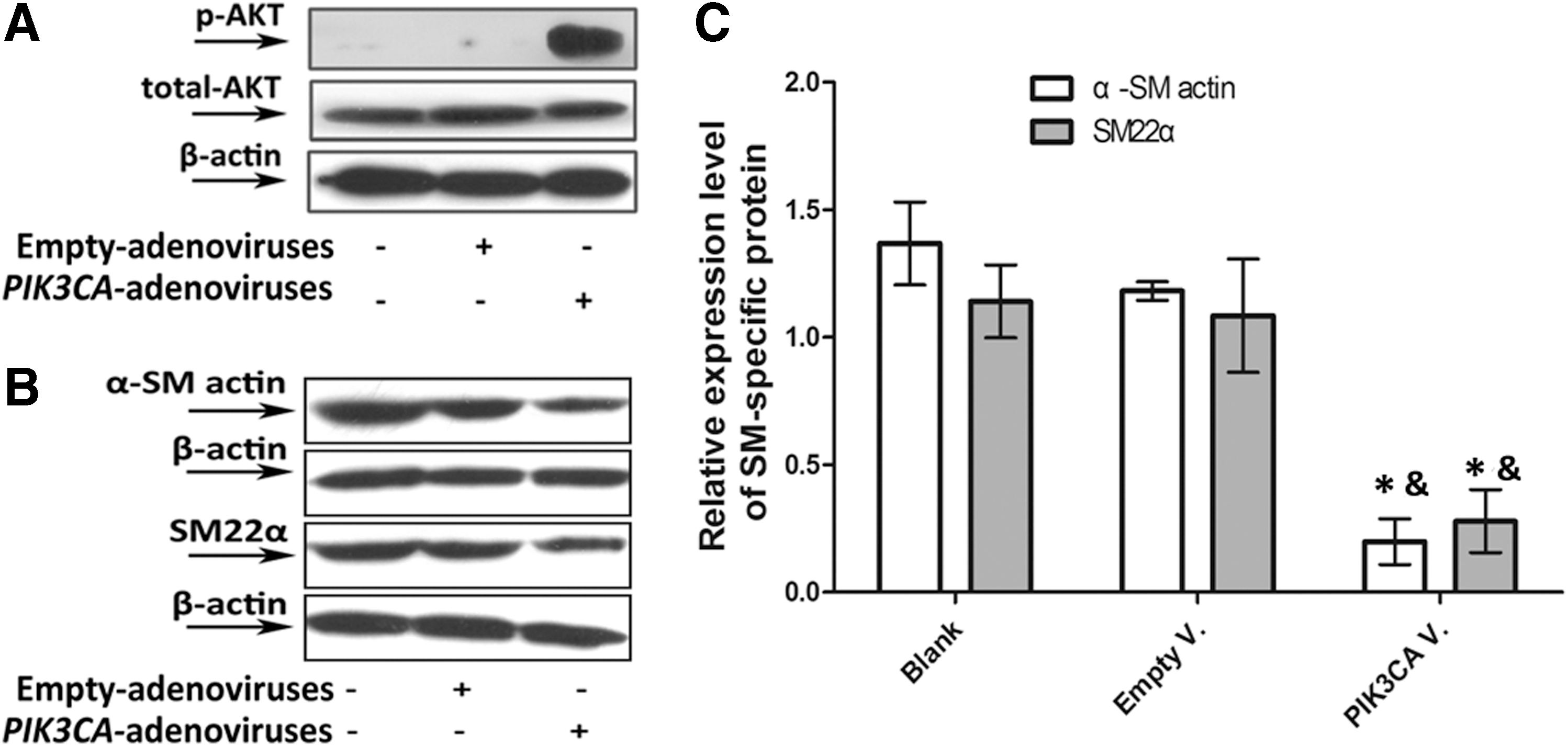
PIK3CA-adenoviruses induced downregulation of VSMC marker gene expression at protein level. Finally, starved PASMCs were left untransfected, were transfected with empty adenoviruses or PIK3CA-adenoviruses. High levels of AKT phosphorylation in
The observed PDGF-BB-dependent decline in α-SM actin and SM22α mRNA levels was also produced by overexpression of PIK3CA (p<0.05) (Fig. 4B). The efficiency of PIK3CA-adenovirus overexpressing PIK3CA gene was measured by RT-qPCR shown in the Table 1.
Inhibition of the PI3K/AKT pathway in rat PASMCs blocked the PDGF-BB-induced reduction of F-actin level
In untreated control PASMCs, we found that F-actins were well-developed, thick, and long fibers with high intensity of fluorescence (Fig. 6A). The fluorescence intensity of labeled F-actins in the PDGF-BB-treated PASMCs was significantly lower than that in the control cells (Fig. 6B, C). However, as shown in Figure 6D, this effect of PDGF-BB was dramatically blocked by LY294002 (30 μM), which demonstrated that PI3K/AKT mediated the PDGF-BB-dependent cytoskeleton rearrangements.
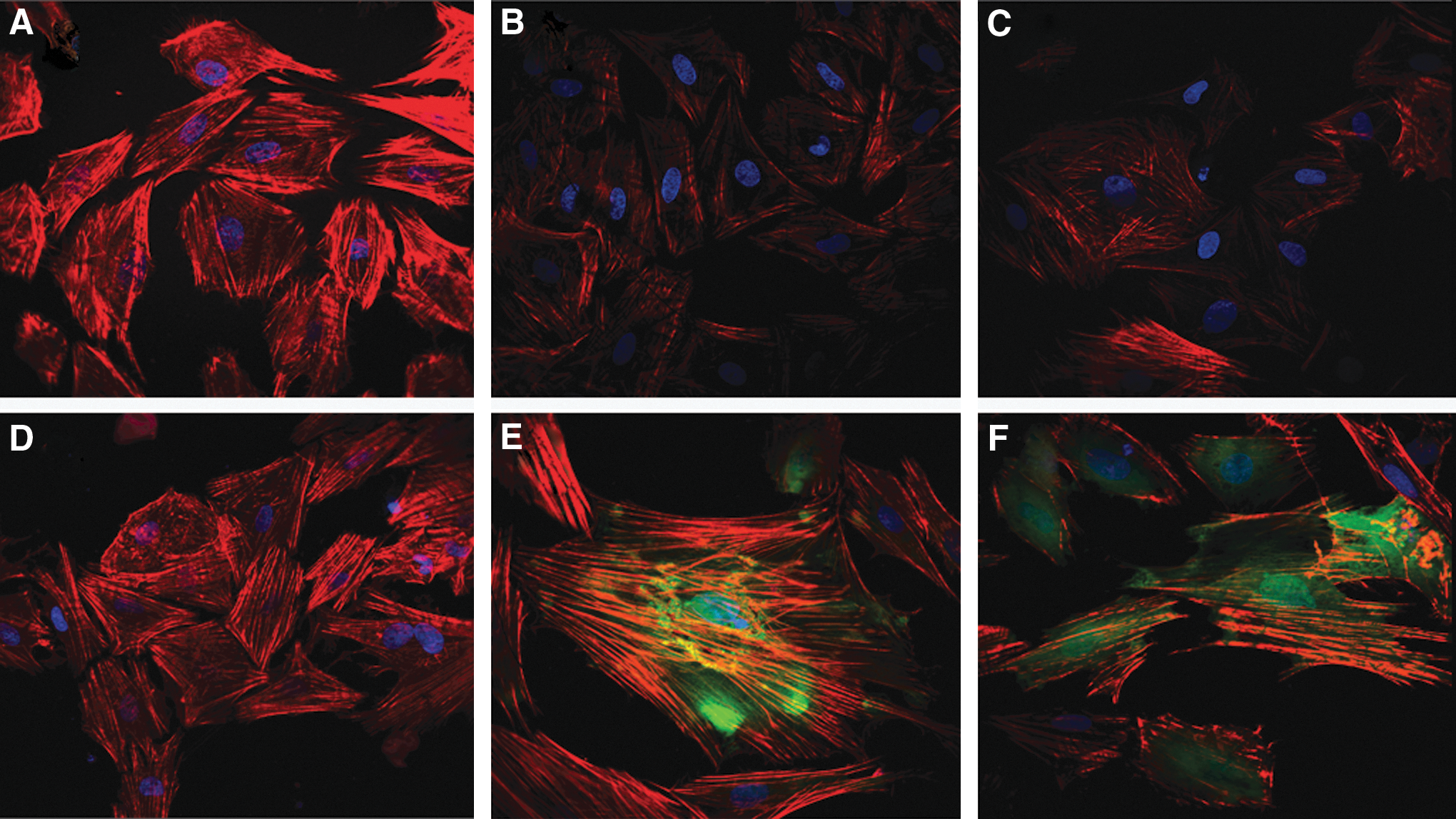
PI3K/AKT signaling pathway regulated F-actin level in rat PASMCs. PASMCs were left unstimulated
Enhancement of PI3K/AKT signaling in rat PASMCs reduced F-actin level
As expected, PASMCs transfected with empty control adenoviruses showed an intensity of cytoskeleton fluorescence similar to that of the untransfected PASMCs (Fig. 6A, E). Decreased intensity of F-actin fluorescence was observed in the PIK3CA-adenovirus-transfected PASMCs (Fig. 6F), indicating the involvement of PI3K/AKT signaling in cytoskeleton rearrangement.
Discussion
The idea that phenotype switching of PASMCs plays a vital role in the progression of pulmonary hypertension is well established (Humbert et al., 2004; Mandegar et al., 2004; Morrell et al., 2009; Schermuly et al., 2011). Experiments that manipulate relevant signaling pathways provide insight into possible interventional targets for the prevention and treatment of pulmonary hypertension. Blocking a single unique pathway or multiple pathways has become the latest new strategy. However, our understanding of the signaling pathways responsible for phenotype switching is inadequate. Recently, PI3K/AKT signaling was reported to be involved in VSMC proliferation and migration (Goncharova et al., 2002; Choi et al., 2010), processes that commonly accompany phenotype switching and cytoskeleton alterations. In the present study, we focused on the role of PI3K/AKT signaling in pulmonary remodeling as it affects phenotype switching and cytoskeleton rearrangement.
SM22α and α-SM actin expression levels have been known for many years as indicators of VSMCs phenotype switching. Our study demonstrated that PDGF-BB-induced downregulation of SM22α and α-SM actin expression in PASMCs was significantly blocked by the PI3K inhibitor LY294002 or by PIK3CA-siRNA. Additionally, adenovirus-mediated PIK3CA overexpression induced AKT phosphorylation and also led to decreased SM22α and α-SM actin levels. These findings suggested that PI3K/AKT signaling is responsible for controlling phenotype switching.
The polymerization of actin filaments in VSMCs was found to increase SM-specific gene promoter activity. In addition to providing cells with mechanical support, actin filaments may provide trafficking routes that support signal transduction throughout the cytoplasm and nucleus (Mack et al., 2001; Liu et al., 2003; Hellstrand et al., 2005; Han et al., 2009). Cytoskeleton rearrangements may change the trafficking routes along which serum responding factors (SRF) travel to reach their binding sites on CArG boxes in the promoters of SM-specific genes (Kaplan-Albuquerque et al., 2003); decreased distribution of SRF to the nucleus and suppression of SM-specific gene expression have been observed following cytoskeleton rearrangement. These findings from a previous study implied that PI3K/AKT-mediated phenotype switching may be regulated by actin filament rearrangement in PASMCs. Moreover, cytoskeleton alterations contribute to cell migration, and the PI3K/AKT pathway was previously shown to contribute to VSMC migration. We therefore investigated whether PI3K/AKT signaling is involved in cytoskeleton rearrangement.
In the present study, a PDGF-BB-induced reduction in the level of F-actin in PASMCs was blocked by LY294002. Like PDGF-BB treatment, transduction of cells with PIK3CA-adenoviruses induced a decrease in F-actin formation. These results indicated that the PI3K/AKT pathway was responsible for controlling cytoskeleton rearrangements. Previous studies have shown that the Rho family of GTPases regulates the actin filament cytoskeleton, cell polarity, migration, and gene transcription (Tapon and Hall, 1997; Aspenstrom, 1999). Further studies are therefore needed to determine whether PI3K/AKT acts upstream of Rho in the modulation of the cytoskeleton and cell phenotype.
Conclusion
To summarize, our study provided novel findings through the use of pharmaceutical inhibitors, gene silencing, and gene overexpression in PASMCs to show that the PI3K/AKT pathway modulated cytoskeleton rearrangements and phenotype switching. More specifically, PI3K/AKT appeared to regulate phenotype switching through changes to the cytoskeleton. The findings of our study clearly demonstrated that PI3K/AKT signaling pathway regulated the conversion of PASMCs between phenotypes with distinct proliferative and migratory characteristics. Our investigation of the PI3K/AKT pathway also produced a new potential target for therapeutic intervention in patients with pulmonary hypertension. In conclusion, given the importance of phenotype switching in the pathophysiological events of pulmonary remodeling, further exploration is critical to elucidating the downstream signaling pathways that drive the mechanisms responsible for pulmonary hypertension.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 30872783), the State Key Development Program for Basic Research of China (No. 2007CB511900), and the Program for Changjiang Scholars and Innovative Research Team in University (No. IRT0935).
Disclosure Statement
The authors declare no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.