
Abstract
Antiviral innate immune responses and apoptosis are the two major factors limiting viral infections. Successful viral infection requires the virus to take advantage of the cellular machinery to bypass cellular defenses. Accumulated evidences show that autophagy plays a crucial role in cell-to-virus interaction. Here, we focus on how viruses subvert mitophagy to favor viral replication by mitigating innate immune responses and apoptotic signaling.
Autophagy and Viral Infections
A
Autophagy and/or autophagy-related proteins can be both proviral and antiviral factors depending on viral subtypes (Kudchodkar and Levine, 2009). Recent works show that autophagy is involved in the regulation of inflammation and constitutes an essential innate responsive mechanism defending pathogens in primitive eukaryotic cells (Kudchodkar and Levine, 2009; Deretic, 2012). Autophagy can engulf bacteria and viruses for lysosomal degradation (Levine, 2005; Knodler and Celli, 2011; Wild et al., 2011). Moreover, autophagy is also required for presenting viral antigens to major histocompatibility complex class II, leading to the activation of adaptive immunity (Hayward and Dinesh-Kumar, 2010). Interestingly, some viruses have evolved countermeasures to escape the antiviral properties of autophagy. For instance, matrix protein 2 of influenza A virus and Nef protein of human immunodeficiency virus inhibit the maturation of autophagosomes into autolysosomes by targeting beclin1 (Gannage et al., 2009; Kyei et al., 2009; Rossman and Lamb, 2009). Herpes simplex virus 1 restrains early stage of autophagic process through its Us 11 protein interacting with the protein kinase PKR (Lussignol et al., 2013).
In contrast, some viruses can benefit from autophagy for their infections. Several families of RNA viruses may subvert autophagy for their replication by targeting immunity-associated GTPase family M (IRGM) (Gregoire et al., 2011). In addition, some viruses utilize autophagic mechanisms for enhanced viral transmission and replication to be even more devastating, such as dengue virus (Heaton and Randall, 2011), hepatitis C virus (Shrivastava et al., 2011), poliovirus (Taylor and Kirkegaard, 2007), coronaviruses (Maier and Britton, 2012), chikungunya virus (Joubert et al., 2012), and adenovirus (Rodriguez-Rocha et al., 2011).
Mitophagy Promotes Viral Replication by Mitigating Antiviral Immune Responses
Mitophagy is known as a process of the selective engulfment of mitochondria by autophagosomes and their subsequent degradation by lysosomes (Kim et al., 2007), which functions as selective removal of damaged mitochondria (Jin and Youle, 2012). Mitophagy occurs after recognition of altered mitochondria by some autophagic receptors, such as SQSTM1, ATG32, or NIX (Zhang and Ney, 2009; Geisler et al., 2010; Aoki et al., 2011). The crucial role of mitophagy in manipulation of viral infections is now getting clarified. Previous studies have observed mitophagy in cells infected by seasonal influenza A virus (Lupfer et al., 2013), hepatitis C virus (Kim et al., 2013b, 2014), and hepatitis B virus (Kim et al., 2013a). Following seasonal influenza A virus infection, receptor interacting protein kinase 2 (RIPK2)-mediated mitophagy reduces immunopathology by negatively regulating activation of NOD-like receptor family pyrin-containing domain protein 3 (NLRP3) signaling and inflammation (Lupfer et al., 2013). We have recently reported that measles virus Edmonston vaccine strain (MV-Edm) infection induces mitophagy in nonsmall cell lung cancer (NSCLC) cells leading to enhanced viral replication (Xia et al., 2014a). While MV-Edm infection induces innate immune responses by means of activation of DDX58/MAVS, it in parallel stimulates SQSTM1-mediated mitophagy to degrade mitochondrion-tethered mitochondrial antiviral signaling protein (MAVS), a key adaptor protein of DDX58/IFIH1 signaling, and thus attenuates antiviral immune responses (Fig. 1). In this process, SQSTM1 plays a critical role in tipping the balance of antiviral or proviral roles of autophagy following MV-Edm infection. Of note, many previous studies show that SQSTM1 is upregulated in many cancers (Thompson et al., 2003; Duran et al., 2008; Mathew et al., 2009), which might explain why oncolytic viruses preferentially replicate in cancers. The induction of mitophagy by MV-Edm remains to be determined. It is possible that MV-Edm causes mitochondrial dysfunction by not yet well-defined mechanisms, thereby targeting mitochondria toward autophagic degradation.
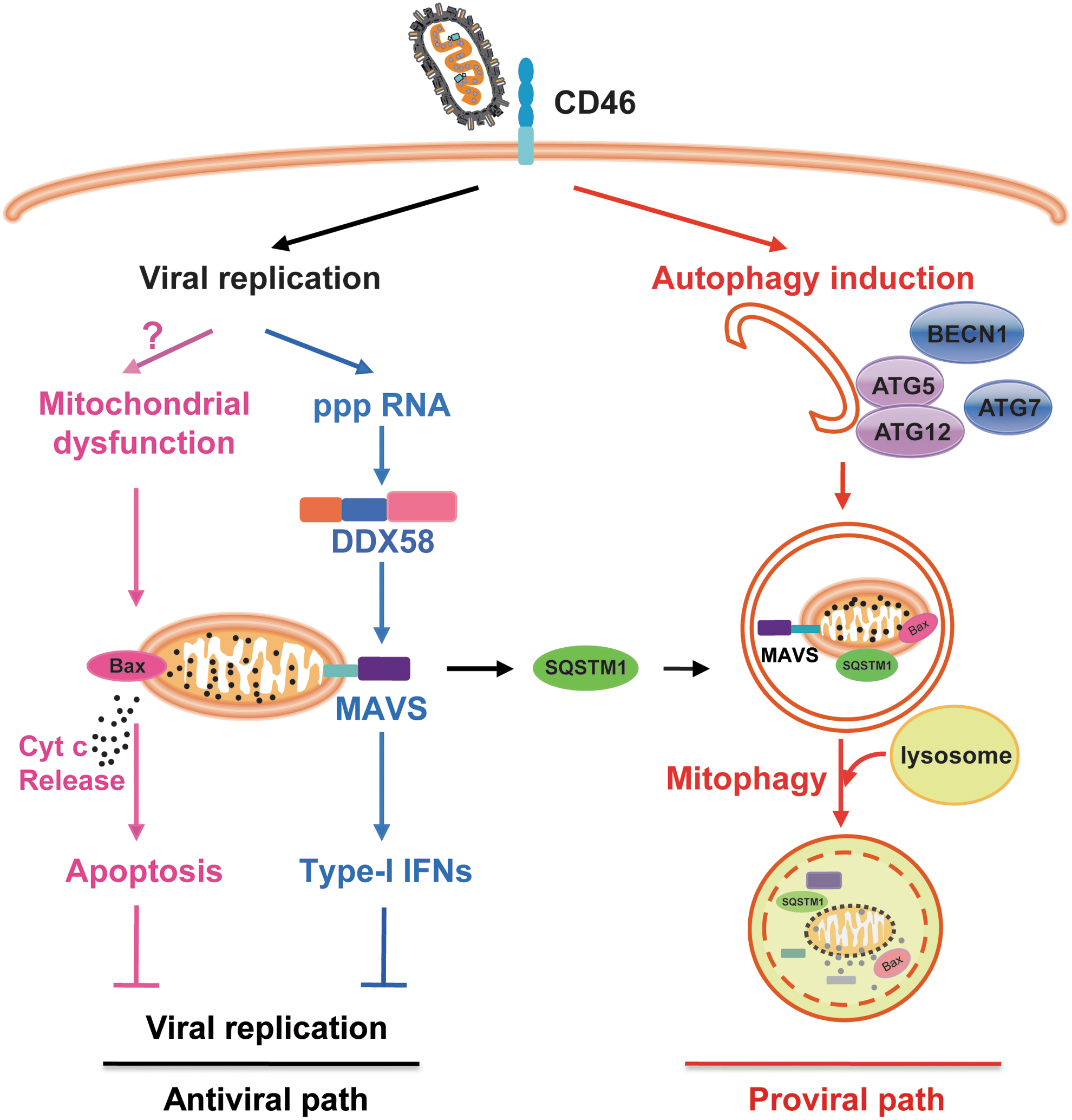
Measles virus Edmonston vaccine strain (MV-Edm) subverts mitophagy to attenuate both innate immune responses and apoptosis. In this model, antiviral innate immunity and apoptosis are countermeasures limiting MV-Edm infections (antiviral path). MV-Edm subverts mitophagy to bypass these restrictions by two different mechanisms: mitophagy-mediated reduction of mitochondrion-tethered mitochondrial antiviral signaling protein (MAVS) and mitophagy-mediated reduction of cytochrome c release (proviral path). SQSTM1 may play a crucial role in tipping the balance of antiviral or proviral functions of autophagy enhancing replication of MV-Edm, as it mediates mitophagy.
Mitochondrial dynamics also regulate antiviral innate immune responses. Mitochondrial fusion may increase the interaction of key factors such as MAVS in signaling of immune responses, and mitochondrial fission may mitigate these pathways (Castanier et al., 2010; West et al., 2011; Lartigue and Faustin, 2013). Given that IRGM contributes to proviral autophagy following measles virus infection (Gregoire et al., 2011) and that IRGM is implicated in the regulation of mitochondrial fission (Singh et al., 2010), it remains to be clarified whether IRGM primes infected cells for mitophagy by disrupting mitochondria dynamics.
Mitophagy Favors Viral Replication by Attenuation of Apoptosis
Apoptosis is another crucial mechanism by which hosts defend themselves against viral infection. Since self-destruction may restrict viral replication and spread. However, viruses evolve various mechanisms to delay or prevent apoptosis.
Recent studies unveil various cross talks between autophagy and apoptosis. It has been shown that autophagy inhibits apoptosis by preventing the activation of Bid or degrading active caspase-8 by Beclin 1 (Djavaheri-Mergny et al., 2010). Cleaved ATG5 by calpains translocates to mitochondria and interacts Bcl-xL leading to controlled cytochrome c release (Giansanti et al., 2011). Bcl-2 or Bcl-xL inhibits the proautophagic activity of Beclin 1 by binding to Beclin 1 (Maiuri et al., 2007). In line with these, autophagy can be utilized by viruses to counteract apoptosis. For instance, autophagy induced by chikungunya virus delays caspase-dependent cell death by the IRE1α–XBP-1 pathway interaction with ROS-mediated mTOR inhibition (Joubert et al., 2012), and autophagy induced by flavivirus NS4A protein protects epithelial cells from cell death (McLean et al., 2011).
As mitochondria are regarded to function as the central executioner in apoptotic pathways (Estaquier et al., 2012), it is plausible that mitophagy may participate in the modulation of apoptosis. Indeed, Bnip3-induced mitophagy negatively regulates cytochrome c release capacity by mitophagic degradation of mitochondria before dysfunction (i.e., mitochondrial membrane permeability transition, depolarization, and release of cytochrome c), which results in decreased apoptosis (Zhu et al., 2013). Another study shows that mitophagy eliminates damaged mitochondria and thus reduces cytochrome c release to cytoplasm under heat shock (Yang et al., 2010). Recent studies also unveil the role of mitophagy in controlling apoptosis following viral infections. We find that mitophagy induced by MV-Edm switches cell death from apoptosis to necrosis in NSCLCs (Xia et al., 2014b). In addition to attenuation of antiviral immunity, mitophagy is also utilized by MV-Edm to eliminate defective mitochondria before cytochrome c is released, which results in decreased apoptosis following viral infection and therefore sustains viral replication in NSCLCs (Fig. 1). Several other studies show that HBV and HCV induce Drp1-mediated mitochondrial fission (mitochondrial fragmentation) and Parkin-dependent mitophagy, which attenuate apoptosis by reducing cytochrome c release and may have significant contribution in persisting viral infection (Kim et al., 2013a, 2013b, 2014).
An Introduction of Measles Virus Edmonston Vaccine Strain
Measles virus is a negative-sense single-strand RNA paramyxovirus. The attenuated strains of the Edmonston vaccine lineage (MV-Edm) have been used for vaccination over 50 years with excellent safety records (Blechacz et al., 2006). In the past decades, MV-Edm and its genetic engineered substrains has been proved as promising oncolytic viruses against a number of tumor types, including ovarian cancer, glioma, myeloma, mesothelioma, adult brain tumor, and lymphoma (Russell et al., 2012). As an excellent oncolytic virus with safety records, MV-Edm has been investigated in several clinical trials and holds promising for advanced cancer patients (Russell and Peng, 2009; Russell et al., 2012). MV-Edm enters cells via its cognate membrane-bound receptor CD46, which has been shown overexpressed on tumor cells (Msaouel et al., 2009). It has also been shown that measles virus could promote proviral autophagy by means of IRGM-dependent pathway (Gregoire et al., 2011). Autophagosomal formation can be triggered by MV-Edm at very early stage during infection through a CD46-Cyt-1/GOPC pathway and matures into autolysosomes (Meiffren et al., 2010). At the late stage following infection, MV-Edm protein C contributes to sustain autophagy for viral infectivity (Richetta et al., 2013).
Perspectives
Mitophagy favors measles virus replication by two different ways: mitophagy-mediated reduction of mitochondrion-tethered MAVS and mitophagy-mediated reduction of cytochrome c release. It deserves further intensive investigations to clarify if such mechanisms are also commonly subverted by other viruses. This will be of great interest not only for designing antiviral strategies but also for oncolytic virotherapies.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81071860 and 81172143), Jiangsu Special Program for Clinical Medical Science and Technology (BL2014054), and the State Key Laboratory of Pharmaceutical Biotechnology (KF-GN-201301).
Disclosure Statement
The authors have no conflicts of interest.