
Abstract
The congenital cataract has been a clinically important cause of impaired vision development, making up about 10% of the cases of childhood blindness. Mutations of more than 40 genes have been identified causing congenital cataract with Mendelian inheritance, which indicated that it has an extremely high genetic heterogeneity. In this study, we recruited a large congenital cataract family and identified a missense mutation (c.143A>G: p.E48G) within gap junction protein alpha-3 (GJA3) gene in the proband using whole exome sequencing. Subsequent Sanger sequencing of this mutation in all family members revealed that this mutation cosegregated with the phenotype in the family with full penetrance. Our study identified a mutation in GJA3 that correlated with congenital cataract phenotype, which was not reported previously, and would be of benefit to the diagnosis of this genetic disorder. This finding expands the mutation spectrum of GJA3 and provides useful information for further study of the molecular pathogenesis of congenital cataract.
Introduction
T
It is revealed that congenital cataract has a high genetic heterogeneity. About one-third of the congenital cataract cases are familial, most of which are inherited with autosomal dominant model. By now, more than 40 genes have been linked to nonsyndromic congenital cataract. It has been identified that mutations of gene coding, gap junction protein alpha-3 (GJA3) and gap junction protein alpha-8 (GJA8), are associated with congenital cataract in humans (Addison et al., 2006). Furthermore, animal experiments have identified that targeted disruption of GJA3 or GJA8 would lead to cataract (Gong et al., 2007).
By using whole exome sequencing, we successfully identified a missense mutation (c.143A>G) within the GJA3 gene (NM_021954) in a five-generation Chinese family with congenital nuclear cataract. This mutation was not found in any of the unaffected family members and 500 normal controls. Our study suggested that exome sequencing was an effective way in the genetic diagnosis of Mendelian disorders with high genetic heterogeneity.
Materials and Methods
Clinical evaluations
A five-generation Chinese pedigree with bilateral congenital cataract was recruited. This family consists of 70 individuals, including 20 affected individuals. The proband was diagnosed with bilateral congenital cataract. Other members of the family took a physical and ophthalmologic examination, which included visual acuity testing, slit lamp examination, intraocular pressure measurement, and fundus examination with dilated pupils. Five hundred unrelated subjects were recruited as controls. The study protocol was approved by the Institutional Review Board of the Central South University, and informed consent was obtained from each patient and control.
Exome sequencing
The mutant gene for congenital cataract was not initially known, so the proband in the bilateral congenital cataract family was selected for exome sequencing. Exome library was constructed with 500 μg genomic DNA, which was exacted from the peripheral blood lymphocytes of the individual. The SureSelectXT Target Enrichment System Kit for Illumina Paired-End Sequencing Library (Agilent Technologies, Santa Clara, CA) and the Agilent SureSelect Human All Exon V5 Kit (Agilent Technologies) were used to capture and construct this library. We sequenced the library on the Illumina HiSeq2000 platform following the manufacturer's instructions. Two lanes of the flow cells were used for the sample to perform paired-end sequencing.
Bioinformatics analysis
We evaluated the sequence quality by using FastQC (
Lasergene MegAlign (DNASTAR, Madison, WI) software was used to analyze conservativeness of the identified mutation on the GJA3 protein in different species. The mutation amino acid structure and function were evaluated by using PolyPhen-2 software online (
Sanger sequencing analysis
A pair of locus-specific PCR primers was designed as 5′-ctcatggacgcttgcacttg-3′ and 5′-AGAAGCGGATGTGGGAGATG-3′ in accordance with the results of exome sequencing. Peripheral blood was collected and genomic DNA was extracted from blood lymphocytes and leukocytes by the standard phenol–chloroform method. Genomic DNA of all these available family members and 500 ethnically matched unrelated controls were amplified by PCR, which is performed by GeneAmp PCR System 2720 (ABI, Foster City, CA) in a 10 μL reaction volume, according to the manufacturer's instructions. Sanger sequencing of all the individuals was performed on an ABI 3100/3130 automated sequencer (ABI) following the standard procedures. Lasergene (version 12.2; DNASTAR) was used for analyzing the Sanger sequencing results, and the reference sequences in the database at the National Center for Biotechnology Information (NCBI; NM_021954) were used for comparing. The mutation was named in accordance with the nomenclature recommended by the Human Genomic Variation Society.
Results
Clinical evaluation
The proband is a 24-year-old male (IV:9), who had a cataract extraction, from a five-generation pedigree (Fig. 1A). All of the 14 affected individuals in the family were affected by bilateral cataract at 2 or 3 years of age and had no other oculopathy. Most of them had the cataract operation when they were young. None of the healthy family members had any other ocular or systemic abnormalities identified after the complete ophthalmologic examination.
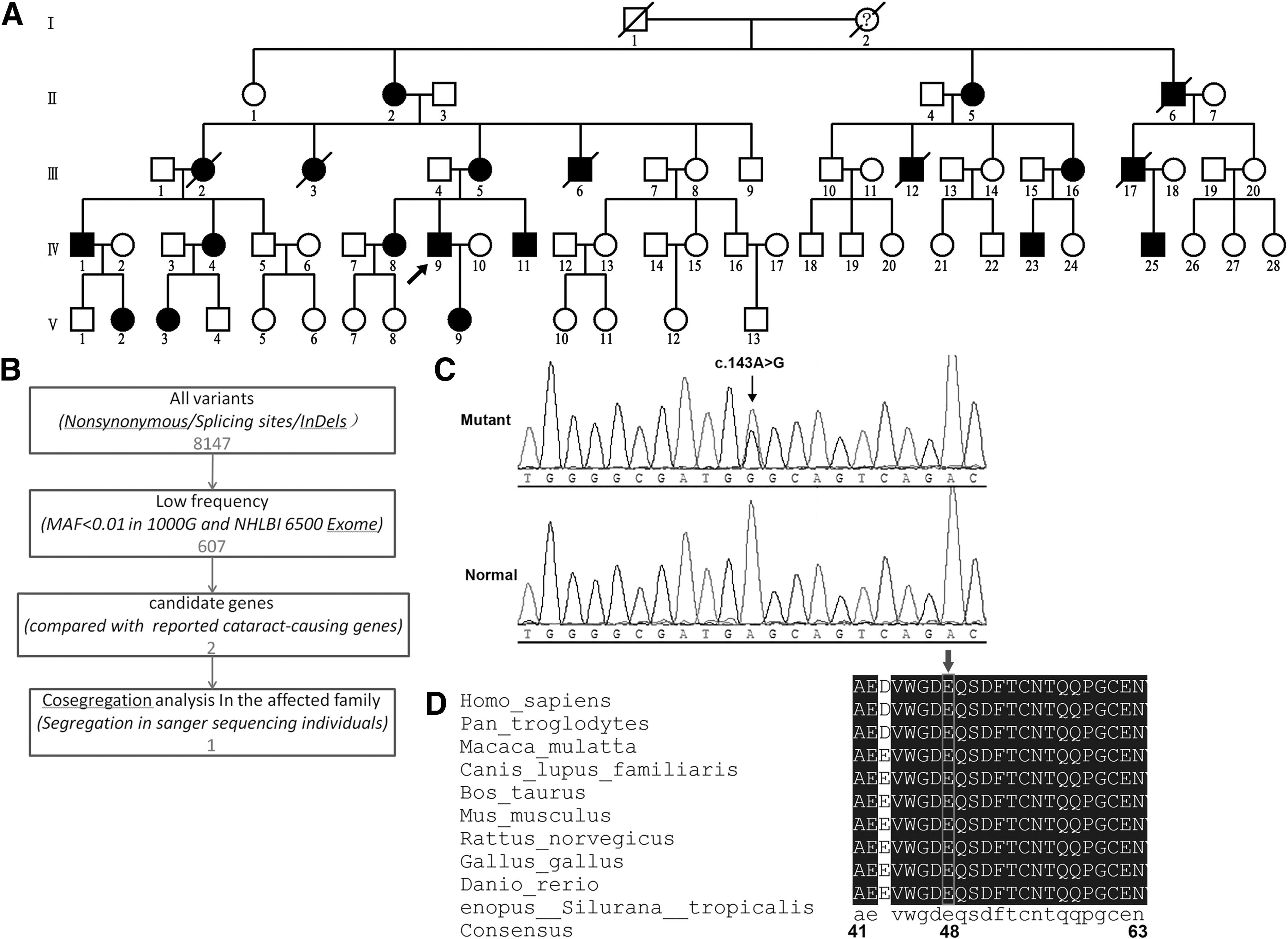
Whole exome sequencing and bioinformation analysis for the identification of a novel genetic variant for congenital cataract.
Mutation screening
By exome sequencing, we identified 20,992 (including 8147 nonsynonymous SNVs, splicing SNVs, or InDels) coding variants in the proband. Following the filtering protocol described in the Methods section, only one variant (c.143A>G within GJA3), which caused a substitution of glycine (G) for glutamate (E) at codon 48 (p.E48G), was left for further analysis (Fig. 1B). Results of further PCR and Sanger sequencing demonstrated that this mutation was detected and cosegregated with all affected individuals in the family and was not observed in other unaffected members in the family and 500 unrelated controls from the same ethnic background (Fig. 1C). No other changes were found in the coding sequence. Additionally, the identified mutation was not reported in dbSNP (
Bioinformatics analysis
The glutamate at the 48th amino acid in connexin46 (Cx46) is highly conserved, by aligning sequences with various species using Lasergene MegAlign (DNASTAR) (Fig. 1D). PolyPhen-2 software produced a score of 1000 and predicted to be probably damaging. J. Craig Venter Institute SIFT analyzed the variant (E48G) to give out a PROVEAN score of −6.705 and predicted it to be deleterious. MutationTaster showed a Disease-Causing prediction on the mutated connexin. I-TASSER Protein Structure and Function Prediction online and PyMol-1.5.x software predicted that compared with wild type, the conformation of mutant Cx46 had taken a great change (Fig. 2).
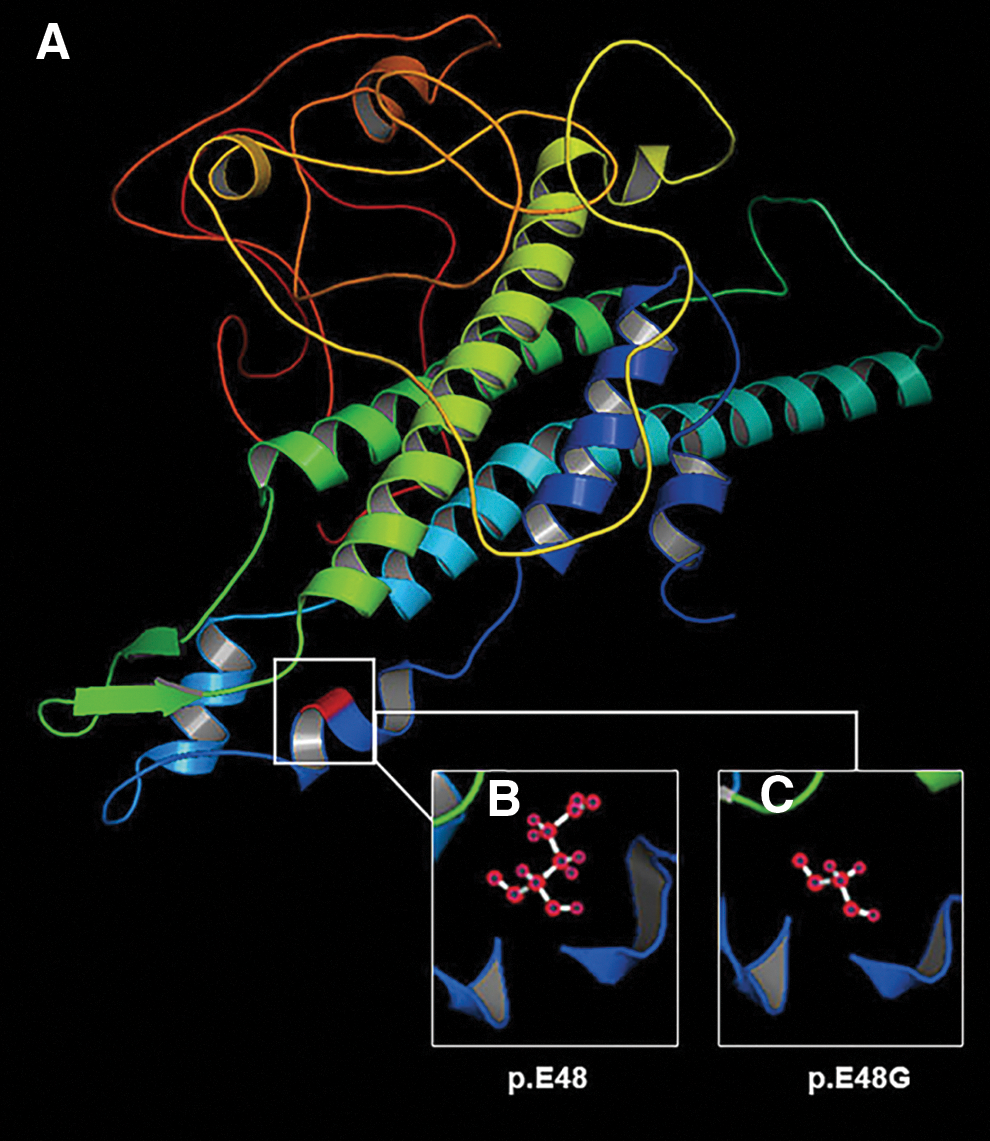
The predicted 3D conformation by software.
Discussion
The crystalline lens is composed of the lens fiber, lens epithelium, and lens capsule from inner to outside in proper order. The lens, known as an avascular structure, discarding all intracellular organelles during its development, highly depends on intercellular communication for their survival (White, 2002). Since the intercellular communication network is formed mainly by the gap junctions, working order of the gap junctions is vital for maintaining osmotic and metabolic homeostasis in lens fiber cells and ultimately maintains lens transparency (Goodenough, 1992). One gap junction channel that is working for exchanging small molecules (Mr ≤1 kDa), such as ions, metabolites, and second messengers from cell to cell, is composed of two hemichannels, respectively, from adjacent cells docking together (Paul et al., 1991; Goodenough et al., 1996; Guleria et al., 2007). Six connexins construct one hemichannel (Mese et al., 2007). It has been verified that Cx46 and connexin50 (Cx50) are expressed in the lens fiber cells of human and mouse eyes in previous studies (Guo et al., 2013). Both are important for the correct development of the crystalline lens, even if their levels are expressed low (Reddy et al., 2004; Sohl and Willecke, 2004).
Cx46, which is expressed by GJA3 gene on chromes 13q 12.11, is composed of 435 amino acids. It contains four transmembrane domains, referred to as M1, M2, M3, and M4, proceeding from N- to C-terminus. Three loops, including two extracellular loops and one cytoplasmic loop, are available for connecting the transmembrane domains. The first extracellular loop (E1) connects M1 and M2, and the second extracellular loop (E2) connects M3 and M4, then the sole cytoplasmic loop connects M2 and M3. Both the N-termini and C-termini stay in the cytoplasm (Milks et al., 1988; Yeager and Gilula, 1992; Yeager and Harris, 2007).
Gap junction takes a large degree on the lens homeostasis and the maintenance of transparency (Zhang et al., 2012). It has been identified that the E1 domain plays a very important role on the function of Cx46 channel in previous studies (Trexler et al., 2000). E1 domain consists of 35 amino acids, from Glu42 to Pro71, in proper order (Bennett et al., 2004). The mutations in E1 and M1 were associated with gap junction formation and pore structure/gating, respectively. So far, there have been at least 29 mutations on Cx46 reported and considered as resulting in cataract by various studies in the world (Sun et al., 2011; Beyer et al., 2013; Kumar et al., 2013; Ponnam et al., 2013; Zhou et al., 2013; Hu et al., 2014; Yang et al., 2014; Yuan et al., 2015) (Fig. 3).
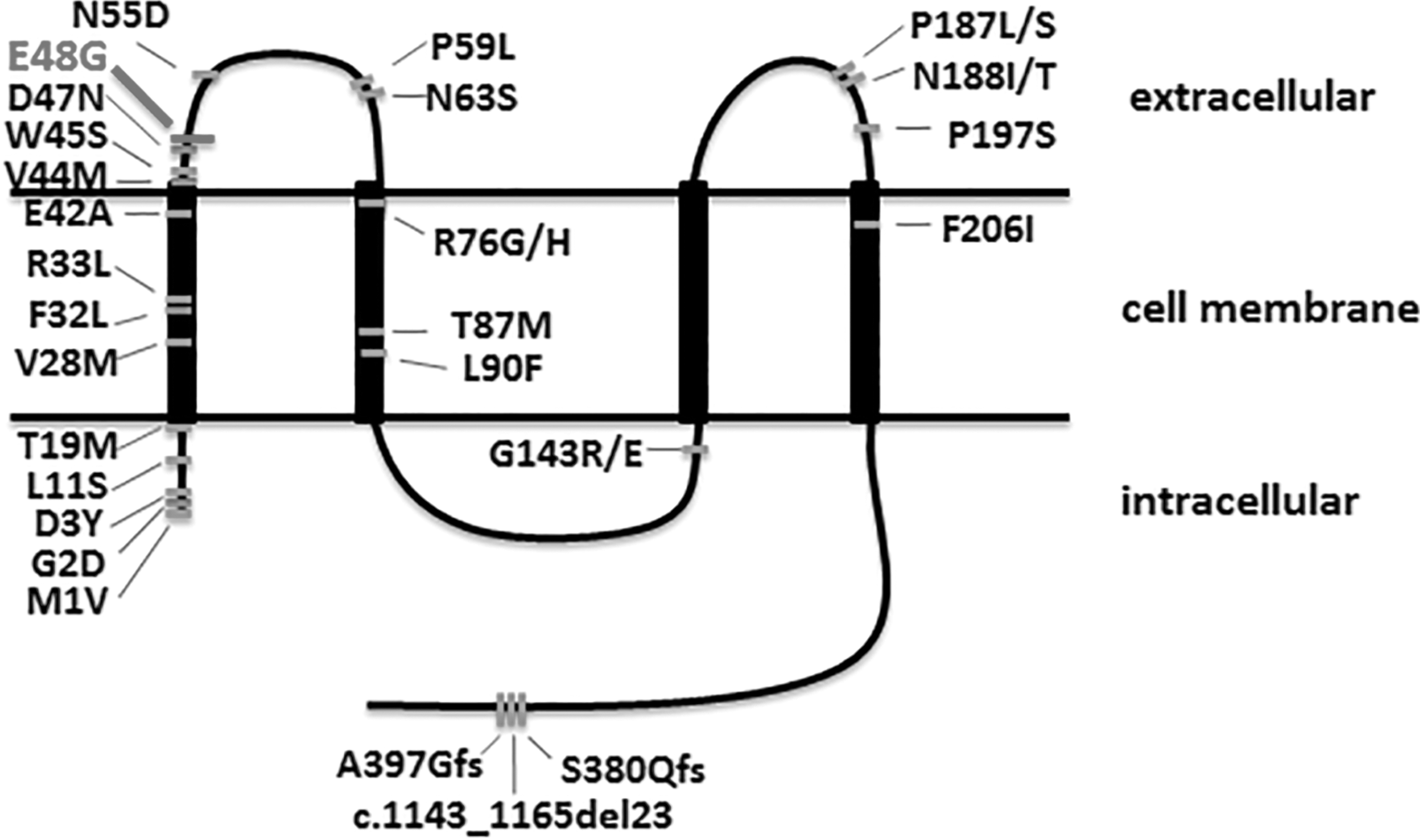
The diagram illustrates the topological structure of human Cx46 and the mutation locations that had been reported.
In this study, we have detected a c.A143G mutation in GJA3 gene, which is expressed as p.E48G mutation on Cx46, in a Chinese family affected with congenital cataract, and it is deduced to locate at the E1 domain exactly.
By aligning the Cx46 amino acid sequence with other Euteleostomi downloading from NCBI database, we observed that the glu48 was highly conserved. It suggested the p.E48G mutation may probably be morbigenous. All three kinds of function prediction software gave a disease-causing prediction consistently.
The p.E48G mutant protein might induce a cleaved form of γ-crystallin, and lead to a vital change in the 3D structure, resulting in abnormal interaction with other proteins (Gong et al., 1999). We predicted the protein secondary structures with I-TASSER (Iterative Threading ASSEmbly Refinement,
In summary, we found out a heterozygous p.E48G mutation in Cx46 in a five-generation Chinese family with autosomal dominant bilateral congenital cataract, using exome sequencing. This finding not only further expanded the mutation spectrum of GJA3 especially in the first extracellular loop (E1) and confirmed that GJA3 is important in the maintenance of lens transmittance, but also suggested that the exome sequencing should be the effective method for hunting the mutation for the single-gene diseases. As exome sequencing is less costly, we believe that more and more researchers and doctors would like to take the exome sequencing as a primary science research process and diagnosis measure.
Footnotes
Acknowledgments
The authors greatly thank all the patients who participated in this study. This work was supported by the National Basic Research Program of China (2012CB517902), National Natural Science Foundation of China (81330027), and Natural Science Foundation of Hunan Province, China (2015JJ2160).
Disclosure Statement
No competing financial interests exist.