
Abstract
The H19-IGF2 imprinted gene region could be implicated in the risk of developing impaired renal function (IRF). Our aim was to determine the association of several common H19-IGF2 variants and IRF in a cohort of elderly healthy individuals. The study involved 675 individuals >65 years of age, 184 with type 2 diabetes mellitus (T2DM), and 105 with IRF (estimated glomerular filtration rate [eGFR] <60). They were genotyped for two common H19 single nucleotide polymorphisms (SNPs) (rs2839698 and rs10732516), one H19-IGF2 intergenic indel (rs201858505), and one indel in the 3′UTR of the IGF2. For the H19 SNPs, we also determined the allele present in the methylated chromosome through genotyping the DNA digested with a methylation-sensitive endonuclease. None of the four H19-IGF2 variants was associated with IRF in our cohort. We found a significantly higher frequency of the 3′UTR IGF2 deletion (D) in the eGFR <60 group (p = 0.01; odds ratio = 1.16, 95% confidence interval = 1.10–2.51). This association was independent of age and T2DM, two strong predictors of IRF. In conclusion, a common indel variant in the 3′UTR of the IGF2 gene was associated with the risk of IRF. This association could be explained by the role of IGF2 in podocyte survival, through regulation of IGF2 expression by differential binding of miRNAs to the indel sequences. Functional studies should be necessary to clarify this issue.
Introduction
T
The insulin-like growth factor-2 (IGF2) is a potent survival and growth factor that binds to the IGF-receptor and promotes cell survival mechanisms (Livingstone and Borai, 2014). The IGF2 levels are controlled by differential methylation and are important for the normal growth and development of the fetus (Harris and Westwood, 2012). Recently, IGF2 has also been implicated in renal function. In an aged Igf2 transgenic mice, a reduction in the IGF-signaling caused podocyte cell death in vitro and glomerular disease in vivo (Hale et al., 2013). Podocyte insulin signaling is important for podocyte and normal glomerular function (Coward et al., 2005; Welsh et al., 2010). Because podocytes are crucial for maintaining the renal filtration capacity, the impairment of the IGF signal might result in an impaired kidney function and a reduction of the eGFR.
Podocyte loss has been associated with progressive renal damage in diabetic nephropathy (DN) and with aging renal decline (Floege et al., 1997; Pagtalunan et al., 1997). The podocyte Igf2-pathway could thus be implicated in the etiopathophysiology of DN. However, renal biopsies from T2DM patients would exhibit increased expression of IGF2 compared with normal samples (Priccit et al., 1996; Sireesha et al., 2009). This suggested that the deregulation of other IGF pathway components in kidneys exposed to high glucose levels is necessary to overcome the podocyte prosurvival effect of IGF2 (Sireesha et al., 2009).
The IGF pathway regulates adult beta-cell mass and function, and could play a role in the development of T2DM (Kulkarni et al., 1999; Modi et al., 2015). IGF2 is highly expressed and secreted by adult mouse and human beta cells and functions as an autocrine activator of the Igf1-receptor signal pathways in beta cell (Cornu et al., 2009; Nica et al., 2013). In addition, beta cell mass expansion in response to acute induction of insulin resistance was higher in normal mice compared with Igf2-null mice (Modi et al., 2015). Thus, murine Igf2 might regulate adult beta cell function to preserve glucose-stimulated insulin secretion in aging, and to adapt beta cell mass in response to metabolic stress.
The human IGF2 gene is in the short arm of chromosome 11 (11p15.5) in a region subjected to imprinting by methylation of citosine-guanine (CG) sites in an imprinting center region (ICR) (Fig. 1). IGF2 is closely linked to the H19 gene that encodes a long noncoding RNA (lncRNA) that regulates the expression of several neighbor genes (Gabory et al., 2010; Monnier et al., 2013; Lorenzen and Thum, 2016). The methylation of CG sites in the H19 region blocks its expression, whereas the methylation of the ICR blocks the binding of a CTCF insulator (a zinc-finger protein CCCTC-binding factor that blocks the interaction between enhancers and promoters) and permits the expression of IGF2. In this way, in the nonmethylated chromosome, H19 and IGF2 are expressed and nonexpressed, respectively (Nordin et al., 2014; Soellner et al., 2017). Conversely, when these sites are unmethylated, the H19 lncRNA is expressed but the IGF2 transcription is blocked (Chao et al., 2008; Bergman et al., 2013). The H19-IGF2 genome region is imprinted, and the paternal chromosome is methylated. Consequently, only the maternal H19 and the paternal IGF2 alleles are expressed (Ratajczak et al., 2013; Hur et al., 2016). The loss of the methylation/imprinting of this region leads to the Silver–Russell syndrome (SRS), a rare condition associated with prenatal and postnatal growth retardation (Azzi et al., 2014). Interestingly, some patients with SRS develop insulin resistance with elevation in fasting levels of glucose (Wakeling et al., 2017).
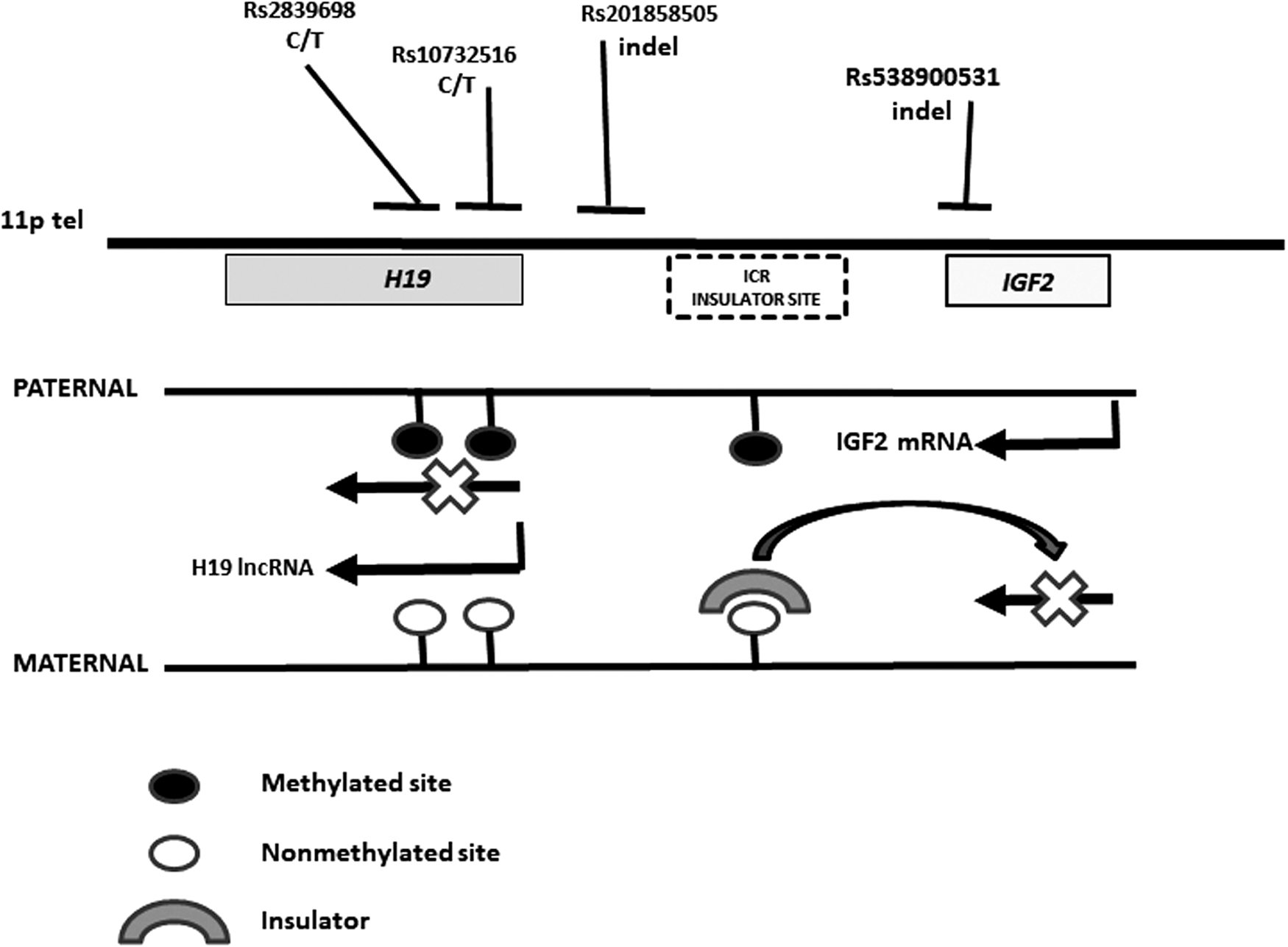
Map of the 11p region showing the location of the studied variants relative to the H19 and IGF2 genes. ICR, imprinting center region, methylated in the paternal chromosome. The methylation of this region blocks the expression of H19 and the binding of a CTCF (zinc-finger protein CCCTC-binding factor) insulator (a sequence that blocks the interaction between enhancers and promoters) to its target sites, and permits the expression of IGF2. In this way, in the maternal nonmethylated chromosome H19 and IGF2 are expressed and nonexpressed, respectively. IGF, insulin-like growth factor. ICR, imprinting center region.
Common H19-IGF2 gene variants have been associated with the risk of developing several diseases. Among others, a single nucleotide polymorphism (SNP) was correlated with IGF2 expression and associated with the risk of infantile hemangioma (Schultz et al., 2015). A common SNP (rs2839698) was also associated with higher serum H19 mRNA levels and the risk for gastric cancer in the Chinese Han population (Yang et al., 2015). The genetic variation in this imprinted region was strongly associated with cerebellum weight in a parental origin-specific manner, with maternally-inherited alleles associated with a significant increase in cerebellum weight compared with paternally inherited alleles (Pidsley et al., 2012). SNPs in the H19-IGF2 imprinted region were also related to the degree of CpG-methylation, and might thus contribute to disease susceptibility (Bell et al., 2011; Pidsley et al., 2012; Zhou et al., 2015). These and other findings raised the hypothesis of allele-specific methylation (i.e., the idea that genotype can influence epigenetics) as a putative mechanism to explain the functional effect of these variants in methylated regions (Meaburn et al., 2010).
Based on the above-referenced evidences, we hypothesized that the genetic variation of the H19-IGF genes could contribute to the risk of developing an IRF among elderly individuals. To test this hypothesis, we studied several common variants in the Spanish RENASTUR cohort.
Methods
Study subjects
A total of 675 individuals from the RENASTUR cohort were genotyped. All of them were Caucasian, 65–85 years of age (45% male) and from the region of Asturias (Northern Spain, total population one million). They were recruited through Primary Healthcare Centers to evaluate the main metabolic values and renal function in healthy elderly people, and signed an informed consent to participate in the study (Coto et al., 2014; Riobello et al., 2016). While dealing with human subjects, this study was evaluated and approved by the Hospital Universitario Central de Asturias (HUCA) Institutional Review Board (HUCA Ethics Committee protocol PI15/00542).
Individuals with a history of chronic renal disease were not eligible for the study. The main characteristics of the studied cohort are summarized in Table 1. Individuals with a documented history of hypertension or who were receiving antihypertensive drugs were considered as hypertensives, and those with a history of T2DM or who were receiving antidiabetic drugs were considered as diabetics. The biochemical profile of all the participants were obtained from fasting blood samples collected by venipuncture. Body mass index (BMI) was calculated by weight and height measured at the time of inclusion in the study.
p-Values: logistic regression eGFR <60 versus ≥60.
T2DM, eGFR <60 versus ≥60, p = 0.005, OR = 1.96, 95% CI = 1.23–3.11.
BMI, body mass index; eFGR, estimated glomerular filtration rate; HDL, high-density lipoprotein; LDL, low-density lipoprotein; OR, odds ratio; T2DM, type 2 diabetes mellitus.
The eGFR was calculated with the Modification of Diet in Renal Disease (MDRD) formulae (Levey et al., 1999):
eGFR (mL/min/1.73 m2) = 186 × [plasma creatinine (mg/dL)] −1.154 × (age) −0.203 × (0.742 if female).
H19-IGF2 SNPs genotyping
The DNA from all the participants was obtained from the leukocytes in 5 mL of blood following a salting-out method (Miller et al., 1988). We genotyped the patients and controls for the rs2839698 T/C and rs10732516 T/C SNPs in the H19 gene following a PCR–restriction fragment length polymorphism (PCR-RFLP) approach.
rs2839698: A 680 bp fragment was amplified with primers 5′CCTGCTCTGATTGGCCGGCA and 5′CCCCGAGAAGATGTCACCTTTGC. After digestion with the restriction enzyme HhaI and electrophoresis on 3% agarose gels the two alleles were visualized as fragments of 680 bp (allele T) and 310 + 370 bp (allele C) (Fig. 2).
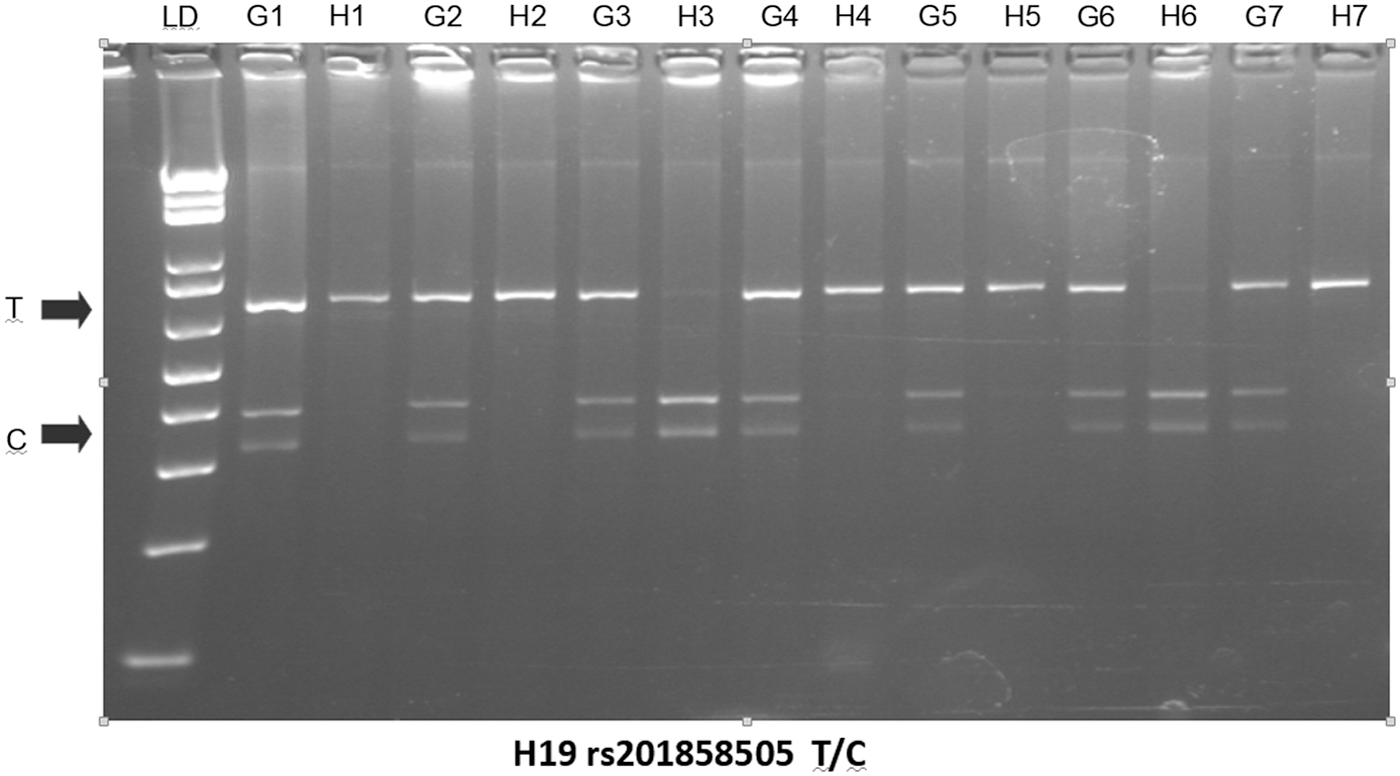
H19 rs2839698 genotypes from pairs of genomic/HpaII-digested DNAs. Genomic DNA from leukocytes was digested with HpaII. The nondigested (G lines) and HpaII-digested (H lines) DNA from each individual were amplified with primers 5′CCTGCTCTGATTGGCCGGCA and 5′CCCCGAGAAGATGTCACCTTTGC, and the 680 bp PCR fragments were digested with HhaI followed by electrophoresis on 3% agarose gels. The two alleles were visualized as fragments of 680 bp (allele T) and 310 + 370 bp (allele C). We show seven individuals who were heterozygous (the two alleles are observed in the PCR-RFLP from genomic DNA, G1–G7), but only one of the two alleles was seen in the genotyping of the HpaII digested DNA (H1–H7). This was a consequence of the full methylation of one of the two chromosomes. LD = 1 kb ladder (DNA size marker). LD, linkage disequilibrium; RFLP, restriction fragment length polymorphism.
rs10732516: A 540 bp fragment was amplified with primers 5′GAGGTATAGGAC ACTCATGGGAGC and 5′ATCACATAAGTAGGCGTGACTTGAGTC. After digestion with the restriction enzyme HhaI and electrophoresis on 3% agarose gels the two alleles were visualized as fragments of 540 bp (allele T) and 305 + 235 bp (allele C).
H19-IGF2 indel genotyping
We genotyped two insertion/deletion polymorphisms in the H19-IGF2 region by PCR amplification followed by allele-size fractioning through electrophoresis in 4% agarose gels.
rs201858505 (28 bp indel): the DNA was amplified with primers 5′CCATGCTCCCCAAACCCCAGG 5′GCTGTGAGTCTGGCTGGGGAG, and the two alleles visualized as fragments of 206 and 176 bp (Supplementary Fig. S1; Supplementary Data are available online at
We designated a protocol for genotyping the reported variant rs538900531 in the 3′UTR of IGF2. This indel was amplified with primers 5′GACCCTCACCCCCAAATCTTACATC and 5′TGCGTTTGTGTGTGTGCTGTGTGC, and the normal sequence would render a 203 bp fragment of 203 bp (Fig. 3). The IGF2-3′UTR is a highly repetitive sequence and the primer design was limited by this fact. To reduce the risk of nonspecific amplification, we checked that the two primer sequences recognized unique target sites, and added formamide (1 μL) to the PCR (25 μL final volume; annealing temperature 64°C). Under these conditions, two fragments of 203 and/or 185 bp were observed (Fig. 3 and Supplementary Fig. S2).
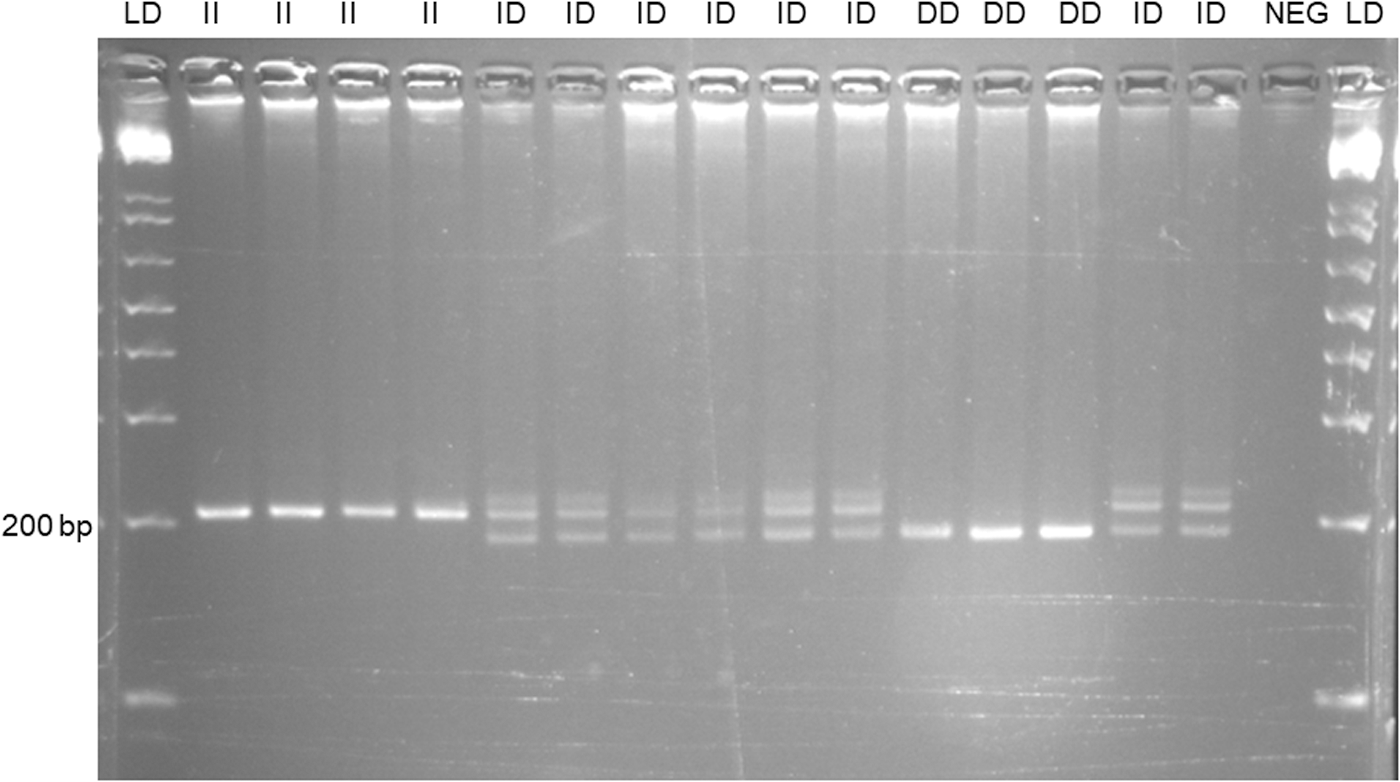
Genotypes for the indel in the 3′UTR of IGF2. A: PCR fragments were electrophoresed on 4% agarose. To reduce the risk of nonspecific amplification we added formamide (1 μL) to the PCR (25 μL final volume; annealing temperature 64°C). Under these conditions, only fragments of the predicted size were observed (203 and 185 bp). An extra band characteristic of heteroduplexes was observed in the heterozygotes. (I) and deletion (D). NEG, negative control (no DNA in the PCR tube). LD = 1 kb ladder (DNA size marker).
To characterize this indel, we amplified and Sanger sequenced several individuals representative of the three genotypes with two primers specific for the IGF2-3′UTR: 5′CAGTCACAGGGGTGGCCTGT (forward) and 5′GTGTGCTGTGTTTGTGTGTGTG (reverse) (annealing temperature at 67°C). A fragment of ∼1550 bp was amplified and Sanger sequenced, showing that the insertion was a GAGCATACAGCACACACAC sequence (Supplementary Fig. S3) and differed from the reported in the Ensembl genome server (
H19 methylation study
The H19 gene is subjected to imprinting by methylation of the paternal chromosome. There are several CCGG sites in the amplified fragments containing the rs2839698 and rs10732516 SNPs. To determine the allele methylation status we followed a PCR-RFLP of the two SNPs from HpaII-digested DNAs. Briefly, ∼200 ng of genomic DNA obtained from leukocytes were digested with the restriction enzyme HpaII that cuts unmethylated CCGG sites. The digested DNAs were further amplified with the above-referenced primers followed by digestion with HhaI to determine rs2839698 and rs10732516 genotypes (Fig. 2 and Supplementary Fig. S4).
Statistical analysis
All the participant's anthropometric, analytical, and genetic data were stored in an Excel file. The statistical analysis was performed with R-software (
Results
H19 variants in T2DM and reduced eGFR
The RENASTUR cohort was genotyped for two common H19 polymorphisms (rs2839698 and rs10732516). The observed genotype frequencies did not differ from the expected under the Hardy–Weinberg equilibrium (Table 2). The two H19 SNPs were in complete linkage disequilibrium (LD) in our population. We did not observe significant allele or genotype frequencies for the two SNPs between individuals with eGFR <60 or ≥60, and there were no differences in the allele/genotype frequencies between diabetics and nondiabetics (Supplementary Table S1). The main analytical and anthropometric values did not differ between the H19 genotypes (Table 3).
Numbers in parentheses indicate %.
H-W, Hardy–Weinberg equilibrium expected values. The difference (chi-square) between the observed and expected values was nonsignificant (p > 0.05). IGF2 3′UTR indel, eGFR <60 versus ≥60, ID+DD versus II: p = 0.01; OR = 1.16, 95% CI = 1.10–2.51.
IGF, insulin-like growth factor; MAF, minor allele frequencies.
p-Values according to logistic regression.
ICR, imprinting center region.
The rs201858505 indel variant (intergenic, between the H19 and IGF2 sequences) was not related with the risk of IRF or T2DM (Table 2), and there were no significant differences for the main analytical values between the rs201858505 genotypes (Table 3).
IGF2 3′UTR indel and IRF
The main finding of our study was a significant association between a common indel in the 3′UTR of IGF2 and the IRF (Table 2). We designated a protocol for genotyping the variant rs538900531 and observed the three indel genotypes in our cohort (Fig. 3). However, sequencing of several individuals showed that this indel differed from the reported (
The deletion (D) allele was significantly more frequent in the eGFR <60 group (0.36 vs. 0.26), with a significant difference in the recessive model (DD+ID vs. II; p = 0.01; OR = 1.16, 95% CI = 1.10–2.51). We did not observe significant differences between diabetics and nondiabetics (Table 2). In reference to the main analytical and anthropometric values, there were no significant differences between the IGF2 genotypes (Table 3).
Because the eGFR was strongly associated with age and T2DM (Table 1), we performed a multivariate analysis to determine the independent effect of these variables on the eGFR <60. Age, T2DM, and the IGF2 genotype remained significantly associated with eGFR (age, p = 0.002; T2DM, p = 0.04; IGF2 indel, p = 0.01).
H19 methylation/imprinting
We investigated the methylation status of the H19 rs2839698 T/C SNP in DNA from leukocytes. All the heterozygous patients showed a single allele in the PCR-RFLP of HpaII-digested DNA (Fig. 2). This was in accordance with the complete silencing through methylation of one of the two H19 copies. A total of 44 individuals in the eGFR <60 were rs2839698 heterozygotes and 53% (n = 23) and 47% (n = 21) showed the T and C alleles in the HpaII-digested DNA, compared with the 1:1 expected ratio (p = 0.5). We also determined the genotype from 100 heterozygotes from the eGFR ≥60 group, and found 48% and 52% were T and C, respectively (p = 0.44). Thus, the common rs2839698 SNP was not significantly associated with the methylation of one of the H19 copies in the two eGFR groups.
Discussion
Common H19-IGF2 gene variants have been associated with the risk of developing several diseases. Among others, a SNP was correlated with IGF2 expression and associated with the risk of infantile hemangioma (Schultz et al., 2015). A common SNP (rs2839698) was also associated with higher serum H19 mRNA levels and the risk for gastric cancer in the Chinese Han population (Yang et al., 2015). The genetic variation in this imprinted region was strongly associated with cerebellum weight in a parental origin-specific manner, with maternally inherited alleles associated with a significant increase in cerebellum weight compared with paternally inherited alleles (Pidsley et al., 2012).
SNPs in the H19-IGF2 imprinted region were also related with the degree of CpG-site methylation, and might thus contribute to disease susceptibility (Bell et al., 2011; Pidsley et al., 2012; Zhou et al., 2015). An interplay between polymorphisms and methylation in the H19/IGF2 region might contribute to the risk of children obesity (Hernández-Valero et al., 2013). We did not find a significant association between two common SNPs in the H19 imprinted center and IRF and type 2 diabetes. We also determined the frequency of the two rs2839698/rs10732516 linked to the methylated chromosome, and found no significant bias toward the methylation of the chromosomes with one of the two alleles. Although the allele-specific methylation assay was performed on DNA from leukocytes, this would not affect the results because the chromosome imprinting is maintained in all the tissues.
The main finding of our study was the association between the common indel in the 3′UTR of IGF2 and IRF. In the RENASTUR cohort we observed a significant higher frequency of carriers of the deletion allele in the eGFR <60 group. To our knowledge, this is the first report describing the association between IGF2 variants and IRF. Moreover, the association was independent of age and T2DM, two strong predictors of IRF in the studied cohort. An association between IGF2 SNPs and adults BMI has been reported (Gaunt et al., 2001). At least one GWAs identified SNPs close to IGF2 associated with the risk for T2DM among African Americans (Ng et al., 2014). SNPs in the IGF2 region were also associated with Igf2 concentration and weight change in T2DM (Narayanan et al., 2013). In our cohort, we did not observe a significant effect of the IGF2 indel on BMI. This variant was not associated with the main analytical values or the risk for T2DM.
The association between IGF2 gene variants and IRF might be explained by the role of the igf-2 protein on podocyte survival. Using conditionally immortalized human and ex vivo adult mouse cells of the glomerulus, Hale et al. demonstrated that podocytes are the major glomerular source and target of igf-2. In an aged transgenic mouse that produces ∼60% of the igf-2 protein, the reduction in the IGF signaling resulted in podocyte cell death in vitro and glomerular disease in vivo (Hale et al., 2013). We did not provide a functional explanation for the association between the IGF2 3′UTR indel and a reduction in the eGFR, although two lines could be proposed. This indel could be in LD with some other variant linked to differences in igf-2 expression and function. To verify this, a sequencing of the IGF2 gene in individuals with normal and IRF should be necessary to identify all the variants that could explain the association. It is also possible that the IGF2 deletion introduced a target sequence for some miRNA that regulates the stability of the mRNA and the amount of the igf-2 protein. The IGF2 expression is controlled by several miRNAs, and the loss of this regulation mechanism could enhance tumorigenesis and other pathological processes (Ma et al., 2011; Liu et al., 2013). An in silico search
Finally, although the association between IRF and the IGF2 3′UTR indel was plausible in view of the renal expression of this gene and its role in podocyte survival, we are well aware that our study was based on a limited number of individuals. Thus, our result should be confirmed on larger cohorts and from different populations. The RENASTUR cohort consists of healthy individuals >65 years of age, and we cannot exclude different effects of the H19-IGF2 variants on IRF or T2DM in other cohorts with different inclusion criteria.
Footnotes
Authors' Contributions
All the authors contributed to this work by recruiting the cohort or performing the genetic and statistical analysis.
Acknowledgments
This work was supported by a grant from the Spanish Plan Nacional de I+D+I Ministerio de Economía y Competitividad and the European FEDER, grants ISCIII-Red de Investigación Renal-REDINREN RD16/0009/0005 (principal investigator E.C.) and RD16/0009/0021 (principal investigator C.D.C.), and grant ISCIII-PI17/00648 (principal investigator J.G.).
Disclosure Statement
The authors declare no conflicts of interest related to this work.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.