
Abstract
Grb10 (growth factor receptor-bound protein 10)-interacting GYF protein 1 (GIGYF1) can modulate insulin-like growth factor 1 receptor (IGF1R) signaling pathway, which plays an important role in regulating diabetes-associated cognitive impairment, by linking to Grb10 adapter. However, it remains unclear whether endogenous GIGYF1 expression is associated with the development of diabetes-related cognitive impairment. In this study, we measured the expression level of GIGYF1, Grb10, phosphorylated IGF1R/IGF1R, phosphorylated AKT serine/threonine protein kinase/protein kinase B (AKT)/AKT, and phosphorylated extracellular signal-regulated kinase (ERK)/ERK in human neuroblastoma SHSY-5Y cells. Meanwhile, we detected cell apoptosis, proliferation, and migration. Our results showed that the percentage of apoptotic cells increased along with the increasing concentrations of glucose (0–200 mM). The expression of GIGYF1 had a significant increase in the presence of 25 mM concentration of glucose in SHSY-5Y cells. In addition, high glucose augmented the expression of IGF1R and Grb10, but decreased the expression of p-IGF1R, p-AKT, and p-ERK. However, GIGYF1 knockdown reversed the decline in the expression of p-IGF1R, p-AKT, and p-ERK. In addition, knocking down GIGYF1 promoted the proliferation and migration of SHSY-5Y cells, but inhibited the apoptosis in SHSY-5Y cells. These results demonstrate that the expression of GIGYF1 can regulate IGF1R signaling pathway in high glucose-induced SHSY-5Y cells.
Introduction
N
Cerebral neuronal deficits are the main cause of brain dysfunction, induced by diabetes mellitus. In addition, aberrant signaling pathways of the insulin receptor (IR) and insulin-like growth factor 1 receptor (IGF1R) induce the apoptosis of neurons and contribute to the development of DE (Hoffman et al., 2010; Ito et al., 2012). It has been established that signaling pathways mediated by insulin/insulin-like growth factor 1 (IGF1) and its receptors play important roles in controlling the normal growth and development of organisms, and are also widely distributed in the central nervous system (Sharma et al., 2008), whereby insulin/IGF1 signaling pathway contributes to neuronal growth and differentiation as well as energy metabolism and neuroprotection (Ma et al., 2015).
There is evidence indicating that insulin and IGF1 exert direct anabolic actions through activation of their cognate receptors and then play an important role in the pathogenesis of DE (Zhang et al., 2015). Meanwhile, the IGF1/IGF1R signaling pathway plays an important role as a suppressor of cell apoptosis. IGF1 binds to cell surface tyrosine kinase receptor IR/IGF1R, making it phosphorylate, and then activates the two families of insulin receptor substrate (IRS) and Src homologous and collagen protein (Shc) (Hakuno and Takahashi, 2018).
In addition, through two major signaling pathways of phosphoinositide 3-kinase (PI3K)/AKT serine/threonine protein kinase/protein kinase B (AKT) and extracellular signal-regulated kinase (Ras/ERK), IGF1 regulates cell metabolism. The former mainly regulates the acute metabolism of insulin by serine phosphorylation and the latter prevailingly the proliferation and differentiation of cells (Siddle, 2012).
Growth factor receptor-bound protein 10 (Grb10) is an adaptor protein and belongs to the Grb7/10/14 family, which possesses an SH2 domain at or near the C-terminus, a central PH domain and a proline-rich region near the N-terminus, and a BPS (between PH and SH2) domain (Depetris et al., 2009). Grb10/14 associates with the activation loop of insulin/IGF1 receptor through the IR/IGF1R SH2 domains and the IR/IGF1R unique BPS domain. Association with multiple proteins allows Grb10 to regulate receptor-mediated downstream signaling pathways differentially (Kabir and Kazi, 2014). Grb10 modulates insulin/IGF1 signaling pathway involved in the regulation of metabolism, growth, cell survival, and several tissue-specific effects by interacting with their downstream signaling components (Holt and Siddle, 2005; Desbuquois et al., 2013).
In addition, Grb10 plays an important role in tissue-specific regulation and function during growth and neuronal commitment (Yamamoto et al., 2008; Plasschaert and Bartolomei, 2015). Other studies have shown that overexpression of full length of Grb10 automatically inhibits insulin-stimulated receptor phosphorylation and glucose intake, while the truncated N-terminal of Grb10 (only contains BPS-SH2 area) lost such a role, suggesting that Grb10 N-terminal fragment is essential for biological functions of Grb10. Meanwhile, it has been demonstrated that Grb10-interacting GYF protein (GIGYF) interacts and specifically binds with Grb10 with high affinity (Mori et al., 2005).
Grb10-interacting GYF protein 1 (GIGYF1) and Grb10-interacting GYF protein 2 (GIGYF2) are two novel proteins, which modulate IGF1R signaling pathway by binding with Grb10 adapter. GIGYF1 and GIGYF2 have a 17-amino acid sequence, which is homologous to GYF domain and bind to tandem proline-rich region at the N-terminus of Grb10 (Giovannone et al., 2003). Previous reports have demonstrated that GIGYF2 may play an important role in the development of diabetes-associated cognitive impairment through modulating the phosphorylation of IGF1R and its downstream ERK1/2 signaling pathway, but not AKT signaling pathway (Higashi et al., 2010; Xie et al., 2014). However, the pathophysiological function of GIGYF1, especially its role in the aberrant IGF1R signaling pathway in diabetes, is still an enigma.
To further explore the function of GIGYF1 in regulating the IGF1R pathway, we used human neuroblastoma SHSY-5Y cells, a well-established model for the study of neuronal development and differentiation (Pahlman et al., 1990), to explore the effects of GIGYF1 on cell migration and cell apoptosis regulated by IGF1R-mediated signaling pathway under high-glucose conditions.
Materials and Methods
Cell culture
SHSY-5Y human neuroblastoma cells purchased from the American Type Culture Collection (ATCC, Manassas, VA) were grown in a 1:1 mixture of Dulbecco's modified Eagle's medium (DMEM) and F12 Ham (Hyclone, UT) with 10% fetal bovine serum (FBS; PANSera, ES, South America) and 1% penicillin/streptomycin at 37°C in a humidified incubator with 5% CO2. Subcultures were digested with 0.25% trypsin when the cells grew to 90%. The cells were inoculated to six-well plates according to the density of (1.5–2.0) × 105 cells for each hole. Cells were serum-starved for 1 day before treatment with high glucose.
ShRNA-expressing plasmid DNA
Three different targeted sequences designed to be homologous to GIGYF1 were designed, using the lentiviral expression vector (pHB-U6-MCS-CMV-ZsGreen-PGK-PURO). Target sites in human genes encoding GIGYF1 were as follows: shGIGYF1_1 sense strand, 5′-GACTACCGTTATGGGCGAGAGGAAA-3′, shGIGYF1_1 antisense strand, 5′-TTTCCTCTCGCCCATAACGGTAGTC-3′; shGIGYF1_2 sense strand, 5′-GAGAGGCGGTTTGAGAAGTCAGCAA-3′, shGIGYF1_2 antisense strand, 5′-TTGCTGACTTCTCAAACCGCCTCTC-3′; and shGIGYF1_3 sense strand, 5′-GGACACGCTGGAAGCCAAAGAAT-3′, shGIGYF1_3 antisense strand, 5′-ATTCTTTGGCTTCCAGCGTGTCC-3′.
These plasmid DNAs transcribed shRNA with loop sequences of 5′-TTCAAGAGA-3′. A negative control sequence was also designed by the same process, which had no homology with human proteins (shScramble sense strand, 5′-TTCTCCGAACGTGTCACGTAA-3′ and shScramble antisense strand, 5′-TTACGTGACACGTTCGGAGAA-3′). Each DNA was used to transform the Escherichia coli strain DH5α and purified with a Plasmid Mini/Maxprep Kit (Beijing ComWin Biotech). The ligation product was confirmed by PCR and sequencing.
Lentivirus construction and establishment of the SHSY-5Y cell line stably expressing ShRNA
293T cells were seeded at a density of 3 × 106 cells on 10-cm culture plates. After 24 hours, lentiviral vector with shGIGYF1 or shacking was transfected into 293T cells with the lentiviral helper plasmids to generate respective lentiviruses, and Lipofiter™ reagent was used. After 48 and 72 h, the supernatant was collected. The supernatant was centrifuged at 2000 g for 10 min at 4°C and then purified using ultracentrifugation at 82,700 g for 120 min at 4°C.
Virus titer was determined by fluorescence-activated cell sorting analysis of GFP-positive 293T cells and was ∼1 × 108 transducing units/mL medium. When the SHSY-5Y cell grew to 60% confluent in six-well plates, lentivirus-mediated cell transfection was performed. SHSY-5Y cells were transduced by the lentiviral particles at a multiplicity of infection of 20 followed by puromycin (5 μg/mL) selection for 3 generations. Transduction efficiency of SHSY-5Y cells with GFP signals was determined by fluorescence microscope. The knockdown of GIGYF1 was evaluated by real-time quantitative PCR and Western blot analysis.
Real-time quantitative RT-PCR
SHSY-5Y cells were divided into four groups: control (CON), control high glucose (HG), empty vector+high glucose (sh0+HG), and GIGYF1 shRNA+high glucose (GIGYF1-shRNA+HG). After high glucose (25mM) induced for 48 h, the total RNA of cells was extracted with a total RNA isolation Kit (Promega, CHN). Reverse transcription (RT) was performed using a PrimeScript™RT reagent kit (TaKaRa, Japan). It was amplified according to instructions for the real-time PCR kit. HS-RPS18 was used as an endogenous control. Quantitative polymerase chain reaction (qPCR) was performed in triplicate using SYBR® Premix Ex Taq™ II (TaKaRa, Japan).
The qPCR cycling conditions were as follows: predegenerate at 95°C for 30 s, followed by 40 cycles of denaturation at 95°C for 5 s, annealing at 60°C for 30 s, and Melt Curve, using the CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA). Values were normalized to the expression of the HS-RPS18 gene using the 2−ΔΔCt method. GIGYF1, forward: 5′-TCCTTGTCGGTGCCAGATTC-3′; reverse: 5′-AGAATTGGACCCTGAGTCGAAG-3′; GRB10, forward: 5′-CCATGGCCAGTGAGAGTAAAT-3′; reverse: 5′-CTGGCACCAAGTAACCATCT-3′; and IGF1R, forward: 5′-TGGCCGACGAGTGGAGAAAT-3′; reverse: 5′-TGGAGGTAGCCCTCGATCAC-3′.
Western blotting
SHSY-5Y cells that were transfected or high glucose induced for 48 h in six-well plates were washed with phosphate-buffered saline (PBS), pH 7.4 thrice. Then, the cells were lysed with 150 μL of 1% sodium dodecyl sulfate (SDS) lysis buffer. The lysed complex was collected and boiled at 95°C in a water bath for 10 min, followed by centrifuging at 12,000 rpm for 10 min to gather the proteins in supernatant. The concentration of proteins was measured using the Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA) based on the manufacturer's instructions. A total of 30 μg of protein were loaded per lane in the 10% gel and separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred to polyvinylidene difluoride membranes (Millipore Corporation, Billerica, MA).
After blocking with 5% nonfat milk at room temperature for 2 h, the membranes were incubated in 5% bovine serum albumin (BSA), 1× tris-buffered saline (TBS), and 0.1% Tween-20 at 4°C overnight with the following antibodies: a rabbit polyclonal antibody for GIGYF1 (1:2500; Abcam, Cambridge, MA), a rabbit antibody for Grb10 (1:1000; Proteintech, Chicago, IL), IGF1 Receptor β (D23H3) XP® Rabbit mAb (1:1000; Cell Signaling Technology, Tokyo, Japan), Phospho-IGFI Receptor β (Tyr1135/1136)/Insulin Receptor β (Tyr1150/1151) (19H7) Rabbit mAb (1:1000; Cell Signaling Technology), AKT (pan) (C67E7) Rabbit mAb (1:1000; Cell Signaling Technology), Phospho-AKT (Ser473) (D9E) XP Rabbit mAb (1:2000; Cell Signaling Technology), ERK1/2 (1:1000; Proteintech), and Phospho-ERK1-T202/Y204+ERK2-T185/Y187pAb (1:1000; ABclona, Wuhan, China). Rabbit Anti-β-Actin (Loading Control) antibody (1:5000; Bioss, Beijing, China) and Anti-GAPDH rabbit polyclonal antibody (1:5000; Sangon Biotech, Shanghai, China) were used as an internal control.
After washing thrice with TBST, they were cultured with horseradish peroxidase labeled goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (1:5000, SA00001-2; Proteintech) for 1 h at room temperature, and then membrane washing was performed thrice. The semiquantitative analysis of the bands was performed using Quantity One software version 4.6.2 (Bio-Rad). Levels of phosphorylated proteins were determined as a ratio of total protein: p-IR/IGF1R relative to IR/IGF1R, p-AKT relative to AKT, and p-ERK1/2 relative to ERK1/2. Densitometric analysis was performed using ImageJ software.
Annexin V/PI staining assay
The number of apoptotic cells in the different groups was determined using an Annexin V-FITC apoptosis detection kit according to the manufacturer's protocol (MultiSciences Biotech, Hangzhou, China). After washing twice with PBS, cells were resuspended in the binding buffer followed by incubation with 5 mL of Annexin V-FITC and 10 mL of PI in the dark for 5 min. At least 10,000 cells were counted per sample. This test discriminates between intact cells (Annexin V−/PI−), early apoptotic cells (Annexin V+/PI−), late apoptotic cells (Annexin V+/PI+), and dead cells (Annexin V−/PI+).
Wound healing assay
GIGYF1-shRNA SHSY-5Y and empty virus (sh0) SHSY-5Ycells were plated in six-well plates in DMEM and F12 Ham supplemented with 10% FBS. When the cells grew to 95–100%, the monolayer cells were generated scratch wounds using a pipette tip, then washed twice with PBS, pH 7.4 to wipe off cell debris. Cells were incubated in complete DMEM and F12 Ham at 37°C, 100% humidity, and 5% CO2. The images of wound width were taken at different time points (0, 6, 12, and 24 h) using a fluorescence microscope (ECLIPSE Ti-s; Nikon, Tokyo, Japan). Four images were collected from independent selected fields of each sample, and the areas of wound were calculated by ImageJ.
5-Ethynyl-2′-deoxyuridine assay
GIGYF1-shRNA and empty virus (sh0) SHSY-5Y cells were seeded onto 96-well plates (10,000 cells each well) and grew for about 24 h so that the cells were well adherent. According to the manufacturer's instructions, the proliferation of SHSY-5Y cells was detected using the EdU (5-ethynyl-2′-deoxyuridine) DNA Proliferation in Detection kit (RiboBio, Guangzhou, China).
Statistical analysis
All experiments were performed independently thrice and the results were presented as the mean ± standard errors of the mean or standard deviation. Comparisons among values for all groups were performed by one-way ANOVA (analysis of variance). Holm's t-test was used for analysis of differences between different groups. The GraphPad Prism 7 software (GraphPad, San Diego, CA) was used to evaluate the statistical results. Statistical differences were considered significant with *p < 0.05, **p < 0.01, ***p < 0.001.
Results
High glucose induces apoptosis and increases GIGYF1 expression in SHSY-5Y cells
To determine the effect of high glucose on cell viability of SHSY-5Y cells, we treated the serum-starved SHSY-5Y cells with various doses of glucose or mannitol (0–200 mM) for 48 h and detected the apoptosis rate using Annexin V-FITC assay, whereby 0 mM glucose or mannitol was set as control. The results showed that the percentage of apoptotic cells increased along with increasing concentrations of glucose or mannitol. In addition, the extent of apoptosis induced by mannitol was significantly less than that induced by high glucose, and the most significant additive effects of glucose toxicity above the hyperosmolar effect occurred at a concentration of 50 mM (Fig. 1A–C).

Effects of glucose and mannitol on apoptosis in SHSY-5Y cells.
We then detected the expression levels of GIGYF1 mRNA and protein in SHSY-5Y after treatment with the same regimen. We found that SHSY-5Y cells treated with 25 mM glucose had a significant increase in the expression level of GIGYF1 compared to other groups (Fig. 2A, B). To further investigate the function of GIGYF1, we knocked down GIGYF1 expression with three lentiviruses in SHSY-5Y cells. Results from real-time PCR and Western blot analysis indicated that the GIGYF1-shRNA3 could efficiently knock down the expression of GIGYF1 in SHSY-5Y cells (Fig. 2C, D); we then used this lentiviral-shRNA vector for the following studies:
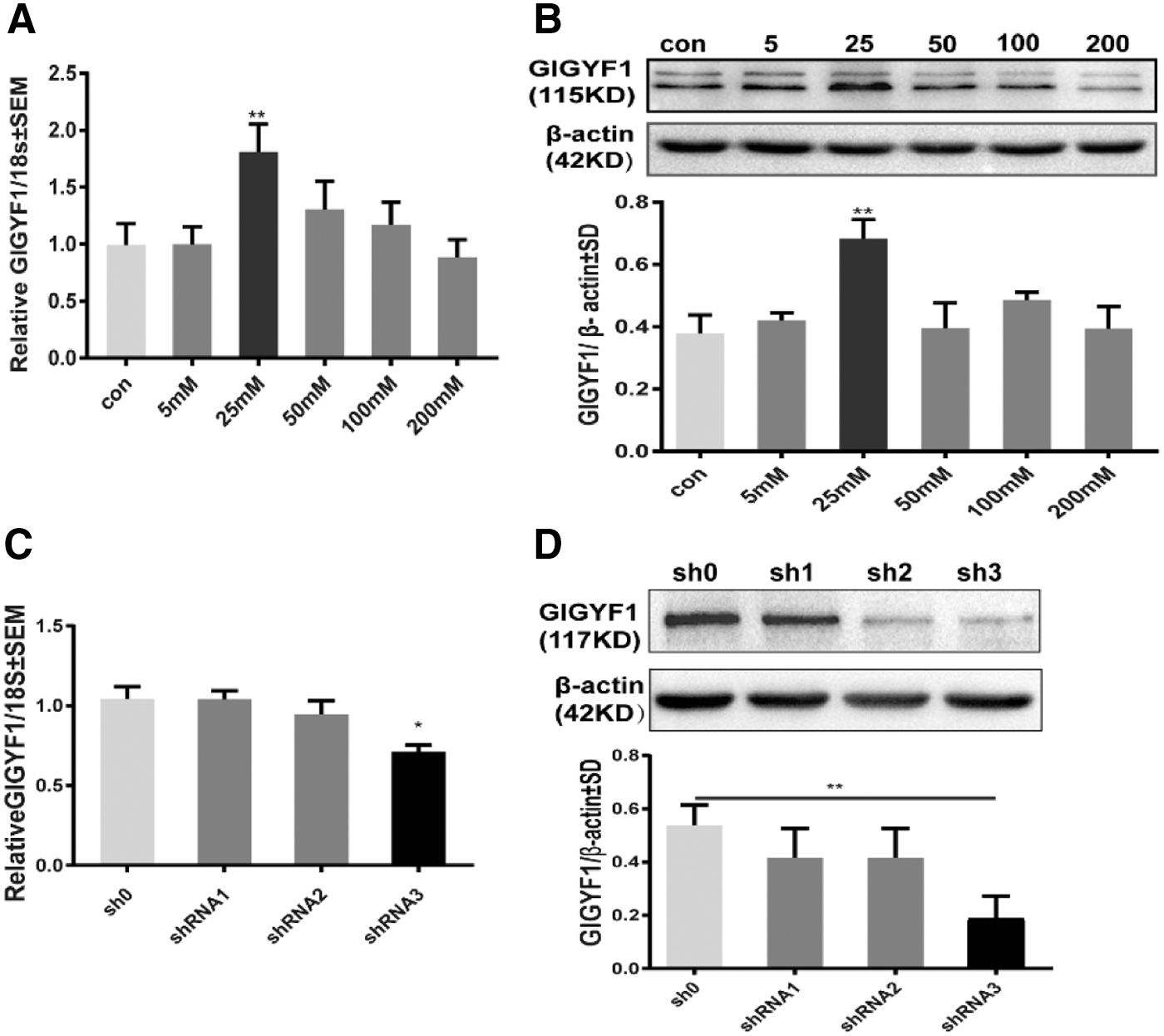
Effects of high glucose on the level of GIGYF1 expression in SHSY-5Y cells.
Lentiviral shRNA-mediated GIGYF1 knockdown and its effect on IGF1R signaling
Next, we determined the effect of GIGYF1 on IGF1R signaling in high glucose (25 mM)-stimulated SHSY-5Y cells. Our results showed that HG significantly increased the mRNA and protein expressions of GIGYF1 (Fig. 3A, D), Grb10 (Fig. 3B, E), and IGF1R (Fig. 3C, F), respectively. As expected, transduction of cells with the GIGYF1-shRNA3 robustly reduced the expression of HG-induced upregulation of GIGYF1 at both mRNA and protein levels (Fig. 3A, D), but had no obvious attenuation effects on HG-induced upregulation of Grb10 (Fig. 3B, E), and IGF1R (Fig. 3C, F). Nevertheless, HG exposure suppressed the phosphorylation of IGF1R at Tyr1135/1136, whereas such HG-mediated inhibitory effects on IGF1R phosphorylation were reversed, to certain degrees, by transduction with GIGYF1-shRNA (Fig. 3G). These results suggest that GIGYF1 is negatively involved in regulating IGF1R activation.
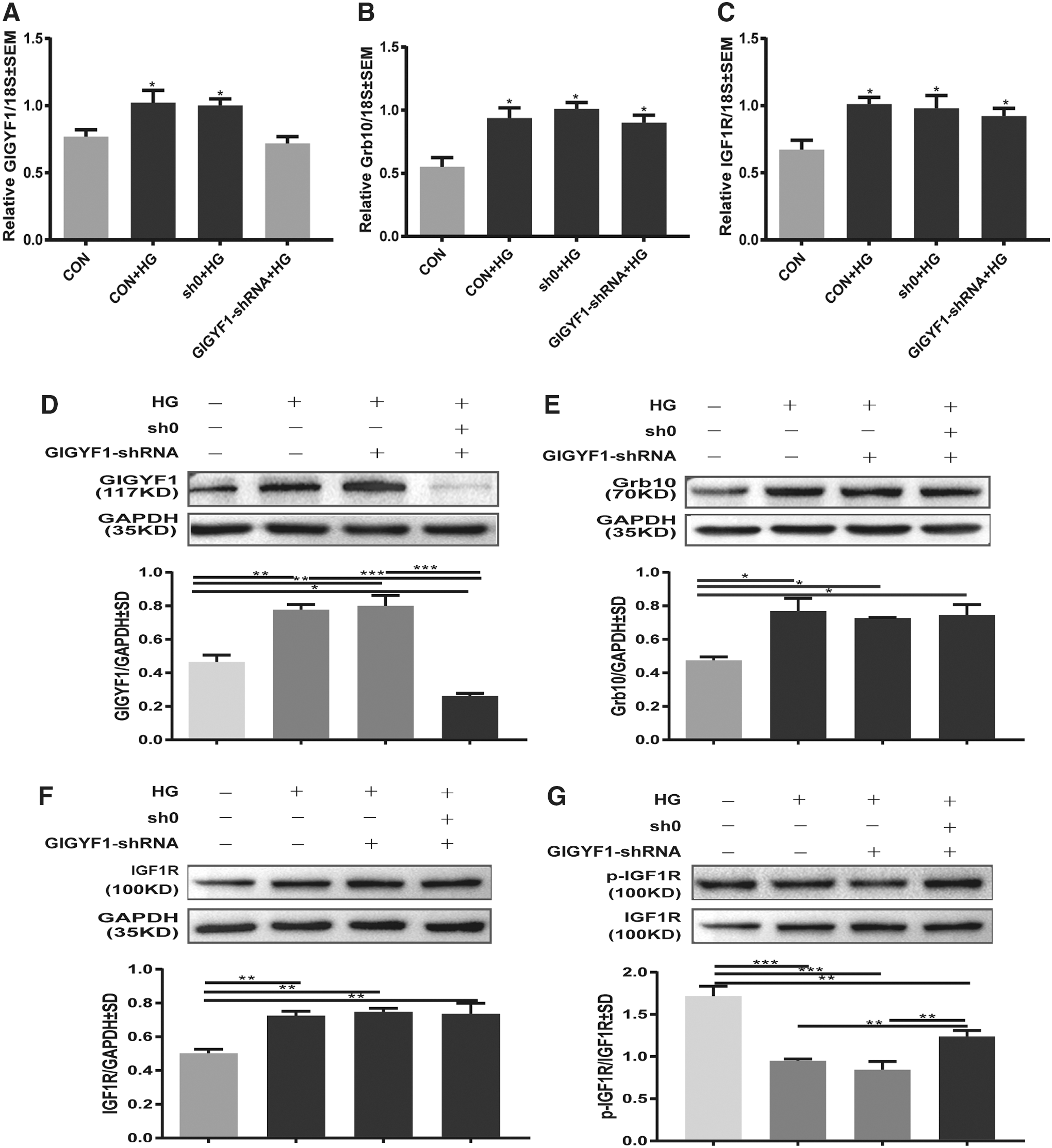
Impact of GIGYF1 to Grb10 and IGF1R in the level of mRNA and protein. Twenty-five millimolars glucose were added to serum-starved normal, sham-knocked down, and GIGYF1-knockdown SHSY-5Y cells, respectively. After inducing for 48 h, we used the real-time PCR and Western blot to detect the expression levels of target gene mRNA and protein.
To further elucidate the mechanism of GIGYF1 involved in the regulation of IGF1R-mediated downstream signaling pathways, the levels of total and phosphorylated AKT (the phosphorylation site of Ser473), phosph-ERK1/2 (the phosphorylation site of Thr202/Tyr204 Thr185/Tyr187), were examined by Western blot analysis. As shown in Figure 4A and B, exposure to HG had little effects on the total levels of either AKT or ERK1/2, but significantly decreased the phospho-AKT to total AKT and phospho-ERK1/2 to total ERK1/2 ratios in SHSY-5Y cells. Nevertheless, such HG-mediated inhibitory effects on the phosphorylation or activation of AKT and ERK1/2 were attenuated, to certain degrees, by knocking down the GIGYF1 gene expression (Fig. 4A, B). Again, these results further support the notion that GIGYF1 is negatively involved in regulating the IGF1R signaling pathway.
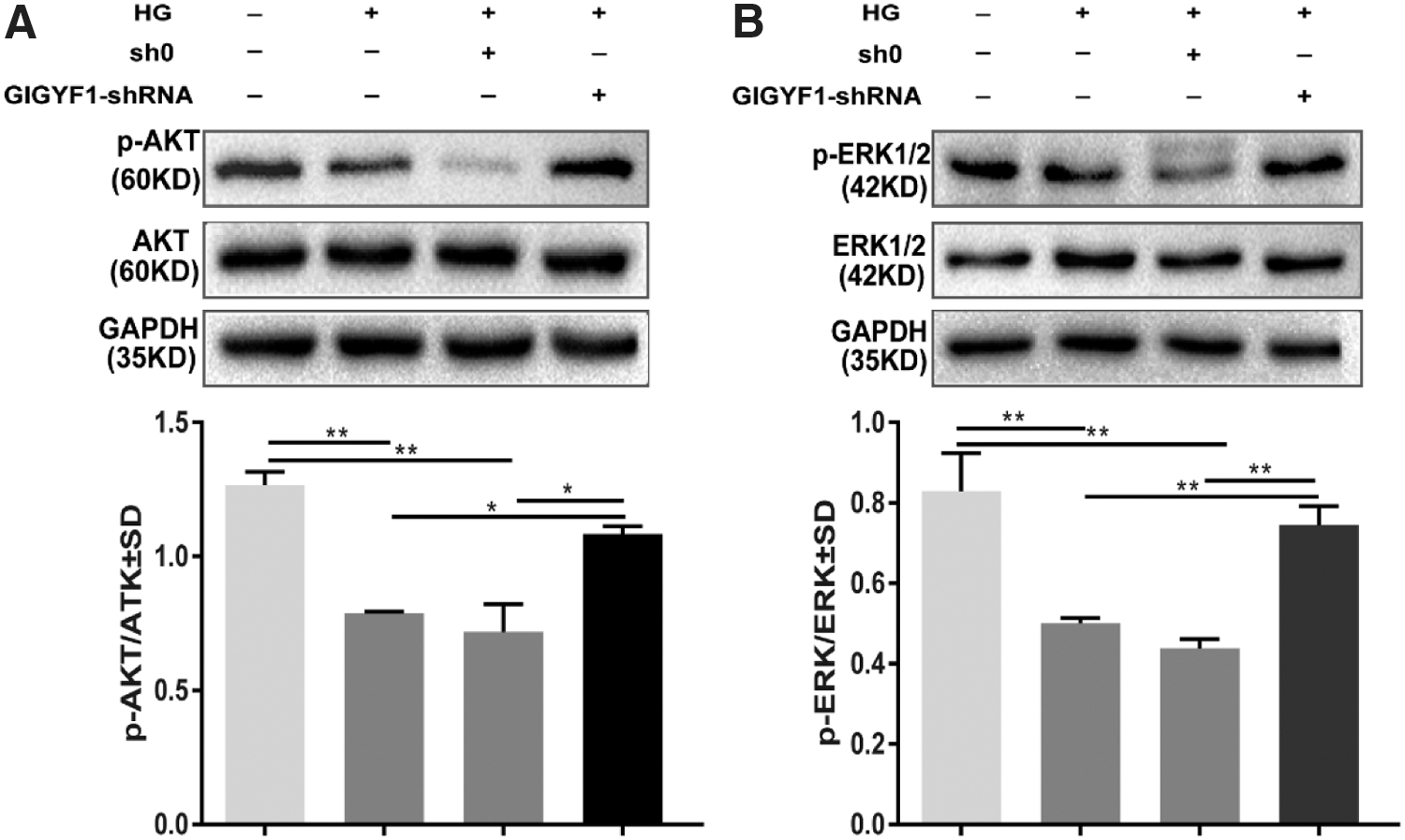
Impact of GIGYF1 to activate AKT and ERK1/2 in the level of protein. After glucose inducing for 48 h in SHSY-5Y cells, we used Western blot to detect the expression levels of target gene protein.
GIFYF1 regulates the proliferation, migration, and apoptosis of SHSY-5Y cells
Since the IGF1R signaling pathway plays an important role in regulating the proliferation and apoptosis of cells, we then determined whether GIGYF1 is involved in such IGF1R-related functions. To this end, we performed EdU assay and scratch-wound healing assay, finding that deficiency of GIGYF1 resulted in an increase in the ratio of EdU positive and wound healing rate compared with the empty virus group (Fig. 5A–D). Meanwhile, we determined cell apoptosis in SHSY-5Y cells following stimulation with 50 mM glucose for 48 h. Our results indicated that knockdown of GIGYF1 expression significantly attenuated HG-induced apoptosis in SHSY-5Y cells (Fig. 5E, F). These data suggest that endogenous GIGYF1 is functional to inhibit cell proliferation and migration, while promoting apoptosis in SHSY-5Y cells.
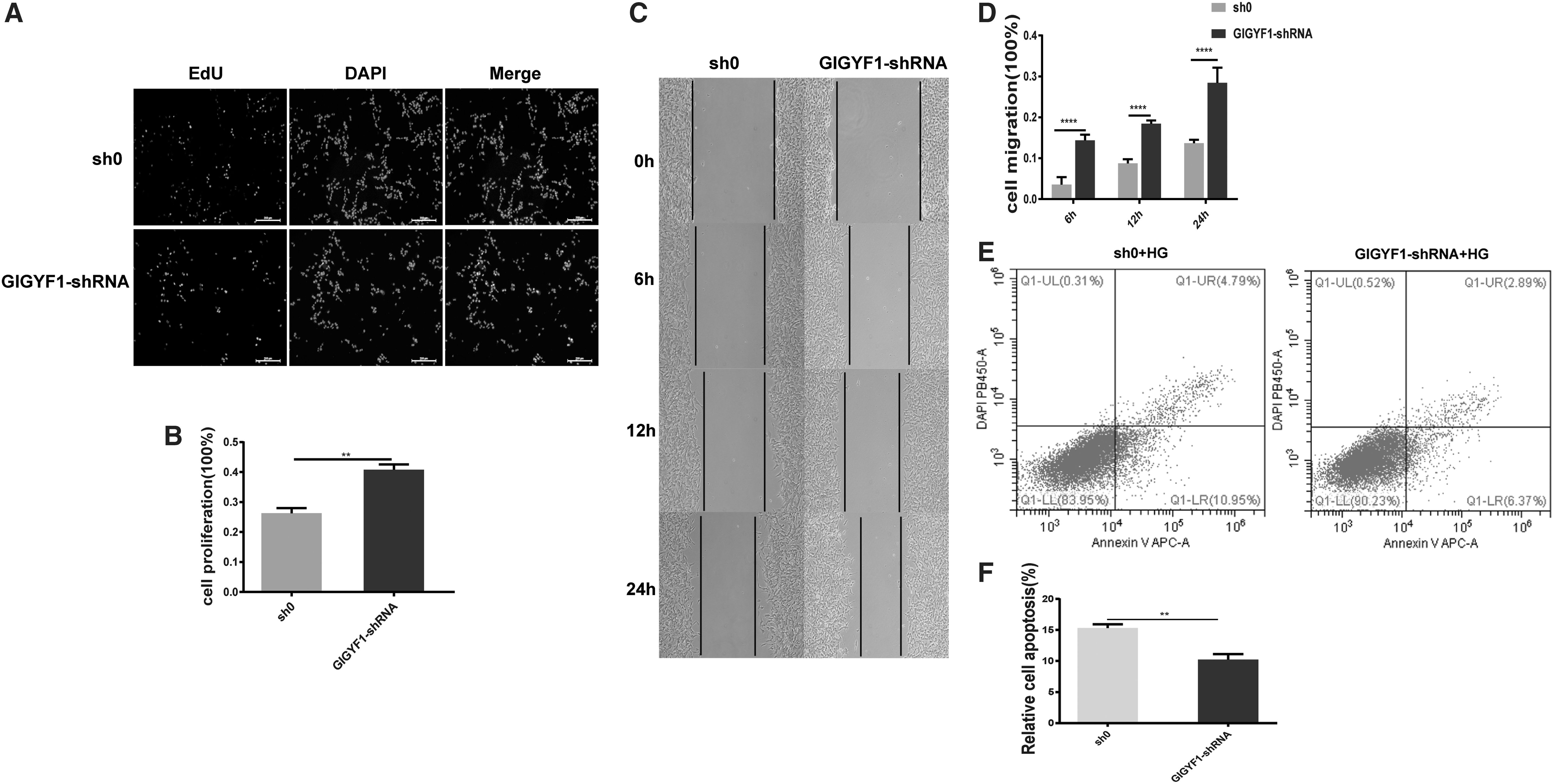
Knockdown of GIGYF1 promotes SHSY-5Y cell proliferation and migration, while inhibiting cell apoptosis with inducing 50 mM glucose 48 h later.
Discussion
In this study, we explored the effects of GIGYF1 in regulating IGF1R-mediated signaling pathway and related biological functions in HG-stimulated SHSY-5Y cells. Our results showed that exposure of SHSY-5Y cells to increasing concentrations of glucose (0–200 mM) increasingly induced apoptosis (Fig. 1B, C). Meanwhile, we found that the level of GIGYF1 expression was obviously upregulated in SHSY-5Y cells following stimulation with 25 mM glucose for 48 h (Fig. 2A, B). Moreover, to distinguish the effect of glucose toxicity from that of hyperosmolarity, the effects of the same concentrations of mannitol on SHSY-5Y cell apoptosis were evaluated in parallel (Fig. 1A). Also, it had been reported that the extent of apoptosis induced by mannitol was significantly less than that induced by high glucose, suggesting that glucose toxicity per se adds to the hyperosmolar effect (Li et al., 2003). In addition, the most significant additive effects of glucose toxicity above the hyperosmolar effect occurred at a concentration of 50 mM. Therefore, we used 25 mM glucose in the most of subsequent mechanistic studies, while 50 mM of glucose was employed for functional studies on apoptosis, migration, and proliferation.
To further explore the potential function of GIGYF1 comprehensively, we knocked down GIGYF1 expression by transducing cells with lentiviral-shRNA vectors (Fig. 2C, D). As is well known, IGF1, a single-chain polypeptide, is widely expressed in the central nervous system and acts primarily through its receptor, IGF1R, which is widely distributed in the brain (Meng et al., 2013; Zhang et al., 2015). The disruption of IGF1R-mediated downstream signaling pathways is involved in the development of some of these cognitive disorders, including DE (Saenger et al., 2011; Werner and LeRoith, 2014; Jiang et al., 2015). Meanwhile, previous reports have shown that IGF1R is increased in patients with T2DM, and IGF1R might be involved in diabetes-related tumorigenesis and tumor growth (Ding et al., 2013; Janku et al., 2013; Yan et al., 2017). Our current research confirmed that the levels of both total GIGYF1 and IGF1R were significantly increased in SHSY-5Y cells stimulated with 25 mM high glucose (Fig. 3A, D and C, F). However, high glucose exposure led to an obvious decrease in the phosphorylated IGF1R expression level in SHSY-5Y cells, while such high glucose-induced inhibitory effects on IGF1R phosphorylation were attenuated, to certain degrees, following transduction of cells with GIGYF1-shRNA (Fig. 3G). These results indicate that GIGYF1 is negatively involved in regulating phosphorylation or activation of IGF1R signaling pathway.
It is well accepted that IGF1R phosphorylation mediated by IGF1 stimulation leads to the activation of two distinct downstream signaling pathways, which are the Shc/mitogen-activated protein kinase (MAPK) pathway and the PI3K/AKT pathway (Chitnis et al., 2008). Herein, we further determined the potential role of GIGYF1 expression in the activation of AKT and ERK1/2 signaling pathways in HG-stimulated SHSY-5Y cells. Notably, we found that the levels of phosphorylated AKT and ERK1/2 were decreased in high glucose-stimulated SHSY-5Y cells, which were reversed, to certain degrees, by downregulation of GIGYF1 expression (Fig. 4A, B). These results further support the notion that GIGYF1 is negatively involved in regulating IGF1R-mediated downstream AKT and ERK1/2 signaling pathways.
Of note, recent studies have demonstrated that the activation of IGF1R signaling pathways protected cells from apoptosis, with an associated increase in IGF1-mediated DNA synthesis (Trojanek et al., 2003; Yang et al., 2013). In addition, numerous studies have reported that the PI3K/AKT pathway plays a role in cell proliferation and cell survival; however, there are some exceptions, whereby the PI3K pathway is also reported to promote cell apoptosis (Shin et al., 2009; Das et al., 2014). At the same time, there is a report showing that PI3K/AKT and the MAPK family such as ERK play an important role in the antiapoptosis signaling pathways in many types of cells (Zhu et al., 2016). Our studies showed that the knockdown of GIGYF1 expression could attenuate high glucose-induced apoptosis (Fig. 5E, F), which might be due to the restoration in the decreased levels of phosphorylated IGF1R, AKT, and ERK1/2. What is more, we found that the cells with GIGYF1 knocked down had significantly improved proliferation and migration ability than the control group (Fig. 5A–D). Together, these findings suggest that GIGYF1 may play a role in the regulation of cell proliferation, migration, and apoptosis. However, further studies are warranted to explore the detailed mechanisms.
It has been demonstrated that Grb10 adaptor modulates insulin/IGF1 action by interacting with signaling components downstream of receptors and exerts several tissue-specific effects (Desbuquois et al., 2013). Meanwhile, previous studies have also shown that GIGYF1 was associated with IGF1R and regulated its signaling pathway through Grb10 in IGFI receptor-expressing R+ fibroblasts (Giovannone et al., 2003). Our previous studies have proven that the overexpression of Grb10 was harmful to cognitive function in diabetic rats, and a regulatory axis composed of IGF1/IR and the downstream Grb10 regulator had a role in regulating biological functions of the hippocampus (Ma et al., 2013). In this study, we found that the level of Grb10 expression was significantly increased in the high glucose-stimulated SHSY-5Y cells (Fig. 3B, E). However, disruption of GIGYF1 expression had no obvious effect on Grb10 expression, indicating that GIGYF1 expression might have no obvious relationship with the Grb10 expression. Based on these findings, we can speculate that the effect of GIGYF1 on IGF1R signaling is not only mediated by cooperation with Grb10 but also by interaction with some other molecules. A recent study has shown that the GYF1/2 proteins directly contribute to the repressive activity of 4EHP, thus uncovering an unexpected facet of a mechanism that regulates mRNA expression (Peter et al., 2017). Therefore, future studies are needed to further explore the molecular mechanisms underlying GIGYF1-mediated regulation of IGF1R and its downstream signaling pathway.
In summary, our findings have indicated that the expression of GIGYF1 is negatively involved in regulating IGF1R-mediated signaling pathway, while downregulation of GIGYF1 could promote migration and inhibit apoptosis in HG-stimulated SHSY-5Y cells. Of note, since SHSY-5Y cells cannot fully represent neurons, high glucose-stimulated SHSY-5Y cells could not be a best cell model for mechanistic studies on the pathophysiology of DE.
Conclusions
Our study implies that the downregulated expression of GIGYF1 can ameliorate the aberrant IGF1R signaling pathway that's induced by high glucose in SHSY-5Y cells. Meanwhile, GIGYF1 involves in the regulation of migration, proliferation, and apoptosis in SHSY-5Y cells. It can be speculated that GIGYF1 may provide a potential novel approach to treat diabetes-associated cognitive impairment caused by aberrant IGF1R signaling pathway.
Footnotes
Acknowledgments
This research was funded by the Chongqing Postgraduate Research Innovation Project (CYS17164). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the article. We are sincerely thankful for the support by the Chongqing Health Bureau: the promoter CamKII-alpha mediated Grb10-siRNA intervention of diabetes encephalopathy (No. 2011-1-009) to Q.W. We also thank the Division of Molecular Nephrology and The Creative Training Center for Undergraduates, The Ministry of Education Key Laboratory of Clinical Diagnostics, School of Laboratory Medicine of Chongqing Medical University, for providing laboratory facilities.
Disclosure Statement
No competing financial interests exist.