
Abstract
A new hypothesis for the mechanism of Huntington's disease (HD) is driven by a small molecule lead that may connect age-associated reactive oxygen stress, oxidative DNA damage, and mitochondrial dysfunction. These pathways have also recently been defined in genome-wide association studies of cytosine-adenine-guanine-expansion polyglutamine neurodegenerative diseases, including HD and the spinocerebellar ataxias. We discuss how N6-furfuryladenine (N6FFA) nucleotide salvage and role as a kinase neosubstrate may have important mechanistic implications for both HD and familial Parkinson's disease. N6FFA highlights a mechanism of how energy dysregulation and protein misfolding in neurodegeneration may be the effect of age-associated reactive oxygen species damage to DNA and part of a feedback loop augmenting with aging.
Introduction
O
In a project screening natural products for drug leads, one compound with the most promising effect on the huntingtin protein, and a profound effect on HD mouse model symptoms, was the natural byproduct of the DNA repair process (Bowie et al., 2018). The appearance of a DNA repair product as the main hit from an unbiased screen for compounds affecting the huntingtin protein strongly reinforced the relevance of DNA repair processes to HD.
These findings took place in the context of the larger research landscape in HD, which has defined redox control, mitochondrial function, and DNA repair pathways as genetic modifiers of age of disease onset (Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium, 2015; Bettencourt et al., 2016; Moss et al., 2017). Genome-wide association study (GWAS) results were unexpected in the HD research community, which was strongly focused on proteostasis pathways and an amyloid-like hypothesis of disease mechanism in mouse and biochemical models. In 25 years of research in HD model systems, with a few notable exceptions (Kovalenko et al., 2012), DNA damage repair was not a focus of research intensity in the field. As research moves forward from a new perspective and a new hypothesis of disease mechanism, we highlight the importance of comparing multiple age-associated neurodegenerative diseases and discourage the continued siloing of HD research.
A Direct Role for Huntingtin in the DNA Damage Response
The genetic modifier pathways identified by large human studies, namely redox control, mitochondrial function, and DNA repair, may not have been such a surprise. The brain is a region of tremendously high energy consumption, with neurons burning a substantial portion of the body's glucose in their bid to generate ATP (Mergenthaler et al., 2013). An unavoidable consequence of aerobic glucose metabolism is the production of ROS, which cause oxidative stress and DNA damage (Liemburg-Apers et al., 2015). During the natural process of aging, neurons experience increased amounts of oxidative stress and perturbed energy homeostasis. Age-associated neurological disease can occur when cells are not equipped to handle this increase because of deficiencies in the function of redox, mitochondrial, or DNA repair proteins (Mattson and Magnus, 2006).
The nature of the deficiencies associated with the disease-causing mutation of the huntingtin protein, a polyglutamine expansion in the amino terminus, is not entirely understood. However, the first 17 amino acids of huntingtin, immediately upstream of the polyglutamine expansion, form a disease-modifying regulatory region called the N17 domain. Two serine residues within N17 (S13 and S16) are hypophosphorylated in HD (Atwal et al., 2011; Hung et al., 2018), and restoration of this phosphorylation is protective in a number of HD models (X. Gu et al., 2009; Atwal et al., 2011; Di Pardo et al., 2012; Bowie et al., 2018). N17 phosphorylation is, therefore, a promising target for intervention in HD, but a pharmacological challenge as most therapeutic perturbants of kinase signaling are designed to inhibit specific kinases, not augment activity.
A central methionine residue within N17 (M8) acts as an ROS sensor, sulfoxidation of which leads to disengagement of huntingtin from the endoplasmic reticulum, phosphorylation of S13 and S16, and translocation of huntingtin to the nucleus (DiGiovanni et al., 2016). This links an age-associated stress to the disease-modifying phosphorylation of huntingtin N17 domain. Although N17 is hypophosphorylated in HD cells, polyglutamine-expanded huntingtin is still capable of translocating to sites of DNA damage (Maiuri et al., 2017). Once there, its function as an ATM kinase-dependent scaffold for DNA repair factors may be impaired. This deficiency would explain the higher levels of DNA damage, and longitudinally increasing damage, seen by us and others in human HD patient cells (Bogdanov et al., 2001; Kovtun et al., 2007; Maiuri et al., 2017; Askeland et al., 2018).
A Product of DNA Damage Repair Acts as a Signal Restoring Huntingtin Phosphorylation
The relevance of DNA repair processes to HD was further reinforced when we conducted a robotic, unbiased, high-content analysis screen to identify natural compounds that could correct the hypophosphorylation of N17 in mutant huntingtin. N6-furfuryladenine (N6FFA, or kinetin), a natural human metabolite that is the DNA repair product of ROS-damaged adenosine (Barciszewski et al., 1997), was protective in HD model neurons and corrected HD phenotypes and cortical mutant huntingtin protein inclusions in an HD mouse model (Bowie et al., 2018). The mechanism by which N6FFA/kinetin achieves these effects may link DNA repair pathways with a number of long-standing observations in HD, including inverted ATP/ADP ratios, mitochondrial dysfunction, and protein aggregation.
This new hypothesis begins with the normal production of N6FFA/kinetin upon excision of damaged adenosine bases (Barciszewski et al., 1997). This compound can be locally salvaged by nucleotide salvagers and phosphorylated to produce kinetin triphosphate (KTP) (Hertz et al., 2013). KTP resembles ATP, and is thus a “neo-substrate” that can only be used as a phosphate donor by kinases with large accommodating enzymatic pockets (Hertz et al., 2013). The kinase for N17, casein kinase 2 (CK2) (Atwal et al., 2011), bears such an accommodating pocket, and can use guanosine triphosphate and KTP to phosphorylate its substrates (Niefind et al., 1999; Bowie et al., 2018). It does so under conditions of oxidative stress and DNA damage, which are associated with dangerously low levels of ATP (Sims et al., 1983; Brace et al., 2016).
Thus, in times of ATP depletion, a product of DNA damage repair acts as an alternative phosphate donor for CK2 to activate DNA repair machinery, with all factors in proximity to DNA damage (Bowie et al., 2018). The result is a positive feedback mechanism that is naturally dampened as DNA is repaired (Fig. 1). In the context of polyglutamine expansion, huntingtin is deficient in its function in DNA repair, stifling excision of N6FFA, thus KTP signaling. As a result, CK2 phosphorylation of DNA repair factors, including huntingtin N17, stagnates and DNA damage goes unrepaired. This drives ATP levels even lower, triggering a negative cascade. Providing N6FFA in trans restores CK2 activity and N17 phosphorylation, activating the positive feedback loop, augmenting DNA repair function, and alleviating the drain on intracellular ATP levels in a positive trophic cascade.
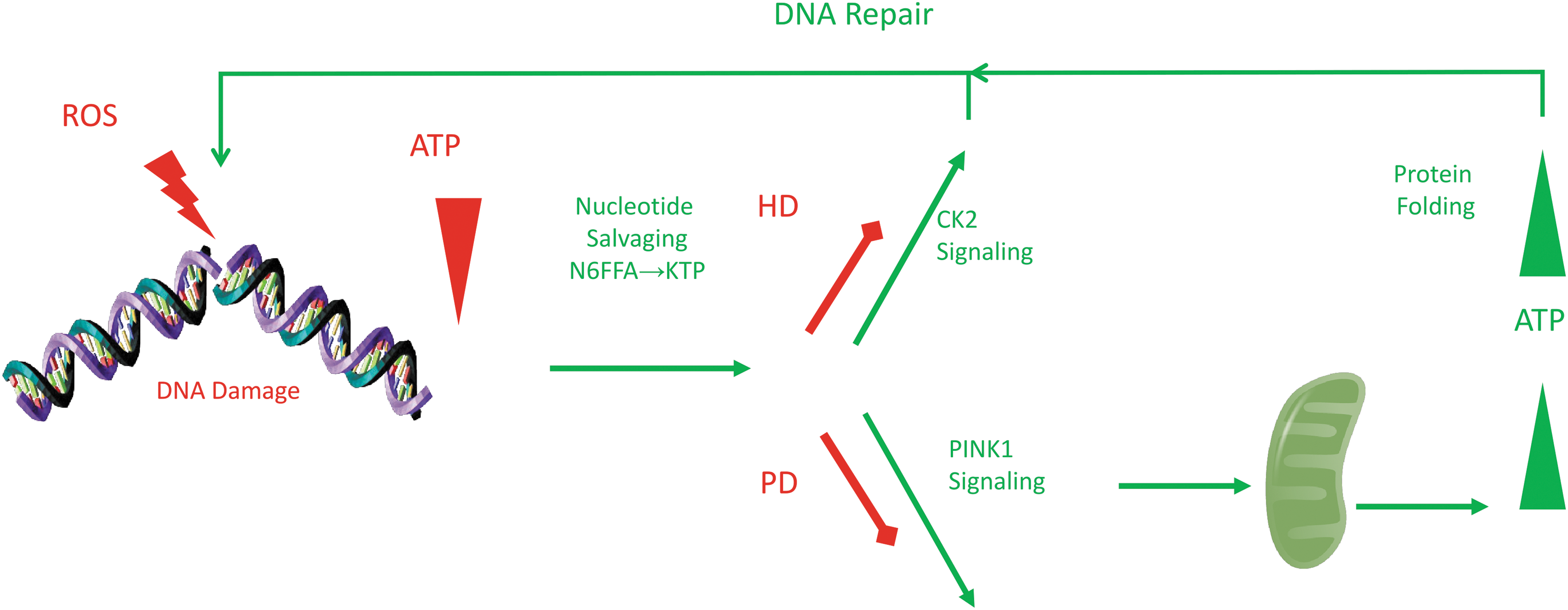
Oxidative DNA damage repair signals augmented repair and energy enhancement through CK2 and PINK1 signaling. Base excision repair of ROS lesions on adenosines nets N6FFA, which is salvaged to KTP as a neosubstrate for CK2 and PINK1 kinases. The signal augments both DNA repair and ATP production at mitochondria. Defects in this signaling by poor DNA repair, hence low KTP, would result in energy deficits in Huntington's disease or Parkinson's disease. CK2, casein kinase 2; KTP, kinetin triphosphate; N6FFA, N6-furfuryladenine; ROS, reactive oxygen species.
Suboptimal Repair of Nuclear DNA May Affect Energy Homeostasis and Protein Aggregation
One of the most striking effects of N6FFA in the HD mouse model was the reduction of mutant huntingtin protein inclusions. Protein aggregation has often been hypothesized of as a cause of neurodegenerative disease (Kumar et al., 2016; Makin, 2018). However, aggregation is likely a downstream consequence of ROS-mediated energy depletion, as ATP-dependent chaperones struggle to maintain protein quality under conditions of oxidative stress (Squier, 2001; Voth et al., 2014; Dahl et al., 2015; Weids et al., 2016).
Indeed, the concept of accumulation of misfolded huntingtin due to polyglutamine expansion has not reconciled with the biochemistry of neuronal protein half-lives (Mathieson et al., 2018). This was a disconnect that was never resolved in the polyglutamine misfolding hypothesis in HD (Truant et al., 2008). As such, strategies targeting aggregation may be addressing the correlative effect of disease, rather than the cause. In contrast, restoration of DNA repair by KTP-mediated CK2 activity would ease the ATP exhaustion, improving chaperone function and the normal folding and turnover of aggregate-prone proteins. This can explain the reduction of protein inclusions we observed in HD mice (Bowie et al., 2018).
Suboptimal repair of nuclear DNA damage can explain additional long-observed HD phenotypes. Upon detection of DNA damage, poly ADP-ribose polymerases (PARPs) tap into cellular NAD+ stores to generate poly ADP-ribose (PAR) chains, which act as recruitment scaffolds for DNA repair factors (Bürkle and Virág, 2013). Unrelented PAR production causes ATP depletion, mitochondrial failure, and energy crisis through multiple mechanisms (Berger et al., 1983; Alano et al., 2010). For example, parthanatos is a PARP1-dependent form of programmed cell death, distinct from necrosis or apoptosis, in which PAR polymers cause mitochondrial dysfunction and subsequent cell death (Fatokun et al., 2014).
Phenotypes related to energy homeostasis make up a large portion of the literature on HD. ATP/ADP ratios are abnormal in postmortem HD patient brains (Mochel et al., 2012b), transgenic HD mouse brains (Mochel et al., 2012a; Lopes et al., 2014), and striatal-derived cells (Trettel et al., 2000), as well as HD patient-derived lymphoblasts (Seong et al., 2005) and fibroblasts (Hung et al., 2018; Jędrak et al., 2018).
Mitochondrial dysfunction is well described in HD (Carmo et al., 2018) and other age-associated neurodegenerative diseases (Chaturvedi and Flint Beal, 2013). Mutant huntingtin has been observed to affect mitochondrial morphology and function in human HD lymphoblasts, postmortem HD patient brain specimens, and HD mouse model neurons (Gu et al., 1996; Panov et al., 2002; Choo et al., 2004; Squitieri et al., 2006; Song et al., 2011; Napoli et al., 2013; Yano et al., 2014), leading to hypotheses for direct interaction and effect of mutant huntingtin on the organelle.
Given the importance of DNA repair genes to HD disease onset (Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium, 2015; Bettencourt et al., 2016; Moss et al., 2017) and the number of mechanisms through which nuclear DNA damage can lead to mitochondrial failure (Berger et al., 1983; Formentini et al., 2009; Alano et al., 2010; Andrabi et al., 2014; Fouquerel et al., 2014), we propose an alternative hypothesis: that these long-standing observations can be explained by suboptimal mutant huntingtin function in the repair of nuclear DNA, leading to hyper-PARylation in the high-ROS-load neurons of the striatum. This places nuclear DNA damage upstream and causal to the mitochondrial deficits that have long been thought to be the first step in pathogenesis (Mochel and Haller, 2011; Fang et al., 2016).
This concept is supported by a recent clinical longitudinal study of HD peripheral blood mononuclear cells, in which DNA damage was seen to precede mitochondrial dysfunction (Askeland et al., 2018).
A Common Pathogenic Mechanism of Late-Onset Neurodegenerative Disease
Decades of technological advances in genetics and molecular biology have established the connection between inherited DNA repair defects and progressive neurodegenerative diseases such as ataxia telangiectasia, xeroderma pigmentosum, and Cockayne syndrome, among many others (Jeppesen et al., 2011; Madabhushi et al., 2014). Exomic sequencing in a rare age-associated ataxia oculomotor apraxia (AOA) identified mutations in the DNA repair scaffold gene XRCC1 (Hoch et al., 2017). In AOA-XRCC1, the loss of XRCC1 results in hyper-PARylation, and genetic ablation of PARP1 prevents disease onset in an AOA-XRCC1 mouse model.
Genome wide association studies in inherited progressive spinocerebellar ataxias (SCAs) have also implicated DNA repair pathways in age of disease onset (Bettencourt et al., 2016; Moss et al., 2017). It is now recognized that like huntingtin, many of the mutant proteins causal to SCAs also have direct roles in the DNA damage response (Massey and Jones, 2018). For example, in SCA3/Machado-Joseph disease, the ataxin-3 deubiquitinating enzyme regulates the double-strand break response (Pfeiffer et al., 2017). Together, these observations argue for a common pathogenic mechanism in which age-related increases in ROS overburden the neuronal DNA repair machinery because of deficits in distinct genes, leading to the death of distinct neuronal populations and culminating in related diseases with overlapping symptoms.
N6FFA Commonality in HD and Familial Parkinson's Disease: PINK1 and Cellular Bioenergetics
In addition to the action of N6FFA in DNA damage repair through CK2, KTP salvaged from N6FFA can also be used by PTEN-induced kinase 1 (PINK1) (Hertz et al., 2013). PINK1 is a mitochondrial quality control kinase that promotes mitophagy through recruitment of the E3 ubiquitin ligase parkin and mitochondrial biogenesis by promoting degradation of parkin interacting substrate (PARIS) (Lee et al., 2017).
Mutations in proteins involved in mitochondrial turnover pathways mediated by PINK1 occur in three variants of familial Parkinson's disease (PD) and both sporadic and familial PD patients show elevated PARIS levels in the brain (Shin et al., 2011; Klein and Westenberger, 2012). Similar to the way N6FFA increases the activity of CK2 on huntingtin in the context of HD, promoting DNA damage repair, N6FFA may have a parallel effect on PINK1 substrates in PD, promoting mitochondrial health.
The ability of both CK2 and PINK1 to utilize this DNA damage repair product suggests that this molecule may have a critical role in signaling DNA repair and mitochondrial turnover and points to a potential connection between the pathology of the age-associated diseases such as HD and PD (Fig. 1).
Conclusions and Perspectives
The discovery of N6FFA efficacy in HD and PD models suggests that there is a critical signaling pathway between DNA damage and mitochondria, where disruption of different branches of this pathway can lead to different diseases in brain, with the commonality of late age onset. Thus, although mitochondrial dysfunction is a classic common hallmark of neurodegeneration, recent unbiased data in human genetics and small molecule screening suggest a potent connection to DNA damage repair. One apparent node is the ATM complex, which is known to affect disease severity in HD models (Lu et al., 2014). Another is PARP activity and the resulting effect on neuronal viability (Narne et al., 2017). Each step in unraveling these mechanisms brings opportunity and hope for new therapeutic strategies.
Footnotes
Disclosure Statement
R.T. is on Scientific Advisory Board and shareholder, Mitokinin LLC. For all other authors, no competing financial interests exist.