
Abstract
Pierisin-5 protein (pie-5) belongs to a family of proteins possessing DNA-dependent ADP-ribosyltransferase activity, which can induce apoptotic cell death. The baculovirus-mediated expression vector system (BEVS) has been commonly used for in vitro expression of heterologous protein subunits for basic scientific research, in addition to the development and production of diagnostics and vaccines. In this study, a new method for the in vitro expression of the cytotoxic protein was established using the baculovirus expression system. The antiproliferative and apoptotic effect of the novel recombinant pierisin-5 protein (rpie-5) was investigated in different human cancer cell lines, such as HeLa, HepG2, and AGS. Cloning, in vitro overexpression, and purification of the rpie-5 protein were performed by using BEVS in Sf21 (Spodoptera frugiperda) insect cell line. The rpie-5 protein exhibits cytotoxicity in all the cell lines, but HeLa (IC50 0.6 μg/mL) was more sensitive when compared with HepG2 (IC50 1.9 μg/mL) and AGS (IC50 3.7 μg/mL) cell lines. The cytotoxic effects of rpie-5 lead to apoptotic cell death in cancer cells and resulted in nuclear fragmentation, enlargement of the nucleus, loss of mitochondrial membrane potential, and finally release of lactose dehydrogenase (LDH) enzyme from the cell membrane. This study reports the molecular mechanism of apoptotic cell death through the upregulation of Bax (Bcl-2 family activating protein-X), Bad, APAF-1 (apoptotic protease activating factor-1), Cyt-c, and caspase-3/9 and the downregulation of Bcl-2 (B-cell lymphoma 2) in rpie-5-treated cancer cells. The study concludes that rpie-5 has p53-independent apoptosis in HepG2 cells and p53-dependent apoptosis in HeLa and AGS cell lines. In the future, this study helps to understand the molecular mechanism of rpie-5 to induction of apoptosis and cell death.
Introduction
Commercially available anticancer drugs lead to adverse side effects, and hence, potent biological agents of insect origin are gaining importance. Insects produce a wide range of bioactive peptides and have been characterized as antimicrobial, antiparasitic, antiviral, and antitumor agents (Gaspar et al., 2013). An antimicrobial peptide, cecropin, was purified from the larvae of Hyalophora cecropia and demonstrated to be a potent cytotoxic agent against M14K and MDA-MB-2 cell lines (Anghel et al., 2014). The protein Alloferons that were isolated from Calliphora vicina act as immune modulatory compound against human leukemia (Majewska et al., 2016). The cytotoxic analgesic–antitumor peptide (AGAP) isolated from Buthus martensii induces cell cycle arrest in lymphoma, leukemia, human malignant glioma, and human colon cancer cell lines (Mulder et al., 2013).
We previously reported that the different column chromatography purified pierisin-5 and pierisin-6 proteins from Pieris canidia and Pieris napi butterflies larvae induced a cytotoxic effect in various human cancer cell lines (Subbarayan et al., 2016; Sarathbabu et al., 2018). This partially purified protein could not be validated for their activities due to the lack of purity and insufficient quantity.
One of the most commonly used eukaryotic expression systems is the baculovirus-mediated expression vector system (BEVS) for heterologous proteins of large molecular size (above 100 kDa) and for proper protein folding for functional studies, vaccine preparations, and diagnostics (Van Beek and Davis, 2016). In this study, we used the entire coding region (∼100 kDa) of pierisin-5 as a single polypeptide for in vitro expression in Sf21 insect cells. Baculovirus has typical properties such as early and very late gene expression for high-level gene expression and limited host range (namely for insects), which makes them highly appropriate as vectors for expression of foreign genes (van Oers et al., 2015). These advantages make it possible to express pierisin-5 proteins that are soluble and functionally similar to their natural counterparts. Thus, BEVS has been mostly used for the improvement and production of vaccines, and the significance of this study is that BEVS has been used for the production of a recombinant cytotoxic protein.
The purification of pierisin proteins from the hemolymph of developmental stage-specific butterfly larvae is very challenging in terms of purity, homogeneity, and yield. Expression of pierisin-5 in Escherichia coli-based expression is impossible because pierisin-5 protein exhibits toxicity against E. coli cells. Additionally, E. coli expression system also requires chaperones, specific transfer RNAs (tRNAs), and post-translational modifications. To overcome these drawbacks, we performed in vitro expression and purification of pierisin-5 protein in Sf21 cell lines using BEVS. Sf21 cells are derived from the ovaries of fall armyworm Spodoptera frugiperda, easily propagated as cell lines and infected by few insect lepidopteran species of Autographa californica Multiple Nucleopolyhedrovirus (Kost and Condreay, 2002).
In most of the circumstances, the post-translational modification of eukaryotic proteins expressed in Sf21 cells was more similar to the proteins, which are expressed in the mammalian system. Hence, Sf21 cells-expressed glycoproteins have comparable biological and immunological activities (Mansouri et al., 2016). Mainly, the yield of folded recombinant proteins from baculovirus expression systems was higher; costs and time duration were significantly lower than mammalian expression systems (Philipps et al., 2005). Nowadays, the baculovirus based in vitro insect culture systems is frequently engaged in the basic research in biology as well as the production of recombinant vaccines and viral pesticides (Van Beek and Davis, 2016).
This is the first study involving the cloning, in vitro expression, and purification of the recombinant pierisin-5 protein from Sf21 cell lines using BEVS. Furthermore, the purified recombinant protein was screened for their anticancer activity against HeLa (human cervical cancer), HepG2 (human hepatic cancer), and AGS (human gastric cancer) cell lines and tested for caspase-mediated apoptosis and regulation of proapoptotic and antiapoptotic genes.
Materials and Methods
Complementary DNA library preparation
Total RNA was extracted from the white cabbage butterfly P. canidia larvae using the RNeasy Mini Kit (Qiagen), and the quality and quantity were verified with Qubit® RNA HS (High Sensitivity) Assay Kit (Thermo Scientific). Dynabeads® messenger RNA (mRNA) Purification Kit (Thermo Scientific) was used to purify the mRNA from total RNA, and complementary DNA (cDNA) synthesis was carried out using Maxima First Strand cDNA Synthesis Kit (Qiagen, Venlo, Netherlands) using a mixture of oligo (dT) primer. A total reaction volume of 20 μL was used to perform reverse transcription reaction for cDNA library preparation.
Polymerase chain reaction amplification of pierisin-5 gene
Gene-specific primers were used to amplify the full-length coding region (Table 1). The product was further purified using QIAquick Gel Extraction Kit (Qiagen) to remove excess amount dNTPs, template DNA, and the polymerase enzyme, and the polymerase chain reaction (PCR) product was cloned into TA cloning vector. The pIEx/Bac-3 vector-specific sequence (VSS) was added to the 5′ end of sense and antisense gene-specific primer for ligation-independent cloning (LIC). Plasmid DNA was used as a template to amplify pierisin-5 gene with high-performance liquid chromatography purified LIC primers.
Primers Used to Study the Proapoptotic and Antiapoptotic Gene Expression in Recombinant Pierisin-5 Protein-Treated Cancer Cell Lines
VSS, vector-specific sequence.
Ligation-independent cloning
The LIC strategy was used to prepare the construction of a recombinant plasmid. The T4 DNA polymerase-treated coding region of pierisin-5 gene was cloned into pIEx/Bac-3 3C/LIC vector (Novagen; Merck) with correct orientation in the presence of dATP (Supplementary Fig. S1). The recombinant plasmid DNA contained hr5 (homologous region 5) enhancer and ie1 (immediate early 1) promoter from A. californica nuclear polyhedrosis virus (AcNPV). The amplified PCR product was cloned into pIEx/Bac-3 3C LIC linear vector (50 ng/μL) using T4 DNA polymerase.
Recombinant bacmid transformation
The ligated plasmid DNA complex was transformed into E. coli host (NovaBlue Giga Singles™ Competent Cells, K-12 strain; Novagen; Merck), and the recombinant plasmid were purified and verified by sequencing. Plasmid DNA was purified by affinity column chromatography and quantified using ultraviolet spectrophotometer method. The concentration of pIEx/Bac-Pie-5 plasmid was found to be 100 μg/mL [100 μg of rpie-5 plasmid contains: 6.023 × 1023 × 100/6,055,400 × 109 = 9.94 × 109 copy/mL].
Cotransfection in Sf21 cell lines
S. frugiperda (Sf21) insect cells were grown as a monolayer in BacVector Insect Cell Medium (Merck Millipore) with 10% heat-inactivated fetal bovine serum (FBS) and incubated at 27°C. Protein expression was carried out with cotransfection of recombinant plasmid DNA (pIEx/Bac-pie5) and linearized Baculovirus DNA (BacMagic-3) into Sf21 cells. The seeded cells (1 × 106 cells per well) were grown into log phase in six-well plates at 27°C containing serum-free BacVector insect media with gentamicin (50 μg/mL). 20 μL of Gene Juice Transfection reagent (Novagen; Merck) were diluted in 1 mL of BacVector Insect Cell Medium (serum-free; Novagen; Merck). Separately, 1 μg of purified recombinant plasmid DNA (pIEx/Bac-pie5, 100 ng/μL) and 0.2 μg of BacMagic-3 DNA (20 ng/μL) were diluted in 1 mL of serum BacVector Insect Cell Medium. Culture medium containing DNA was added dropwise to the media containing transfection reagent, mixed gently to avoid precipitation, and incubated for 1 h at room temperature. The serum containing media was removed from the plate without disrupting the cells, and the entire volume of plasmid DNA-transfection reagent containing media was added slowly drop by drop into cells and incubated for 24 h at 27°C. After incubation, 1 mL of serum containing BacVector insect cell medium were added to each well and incubated for 7 days at 27°C. The signs of viral infection were detected by observing the Sf21 cell morphology, and the supernatant was collected after 7 days and considered as the first passage of the recombinant baculovirus. The increased concentration of virions for recombinant protein production was obtained from the second passage recombinant baculovirus (p1), and the virions were harvested from the culture medium by lysis of cells at 4°C in dark. The viral load was quantitatively determined by plaque assay (Janakiraman et al., 2006). Transfection may cause distinctive changes in the size of infected cells that were monitored under an inverted phase contrast microscope (Olympus). The cell size was measured to a minimum of 100 cells from 10 different zones per well, and their arithmetic means of the cell sizes were calculated and compared with Student's t-test.
Recombinant protein expression
One hundred milliliters of Sf21 (4 × 106 cells/mL) cells were cultured (BacVector insect medium, 10% v/v heat-inactivated FBS, and gentamicin; w/v, 50 μg/mL) at an appropriate cell density in log phase growth. The cells were infected with virus concentration at multiplicity of infection of 10 pfu/cell and incubated at 27°C for recombinant protein expression. The expression of the recombinant plasmid DNA was confirmed after 72 h of post-infection by real-time PCR (RT-PCR) from the cDNA of infected cells. Viral infections may lead to cytopathic effects and structural changes in the host cell. The post-infected cells were monitored at different time points (24, 48, 72, and 96 h), and the level of recombinant protein expression was evaluated by immunoblot analysis using horseradish peroxidase (HRP)-conjugated strep tag II antibodies.
The post-infected cells were harvested by low-speed centrifugation (2000 g, 10 min) and washed thrice with ice-cold phosphate buffer solution (1× PBS, pH 7.3). Cell pellet (3 × 107/mL) was resuspended by pipetting in and out with ice-cold 1× Cytobuster Protein Extraction Reagent (Novagen; Merck) containing 1% protease inhibitor cocktail and 1% Benzonase. Sonication was used to lyse the cells, and the lysate was centrifuged at 13,200 rpm for 15 min at 4°C. The supernatant having soluble fractions was moved to a new sterile tube, without disrupting the cell debris. One hundred microliters of the final supernatant (containing solubilized proteins) were stored at 4°C for protein analysis, and the remaining was used in the purification step.
Purification of the recombinant protein
Protein purification was carried out using the Bug Buster Ni-NTA HisBind Purification Kit (Novagen; Merck). Twenty-five milliliters of 1× Ni-NTA binding buffer were mixed with 5 mL of Ni-NTA HisBind resin (50%) and the slurry was allowed to settle by gravity. The supernatant was pipetted out, and the total cell lysate was added to the Ni-NTA HisBind slurry and mixed gently by shaking at 4°C for 5 min. The entire volume of lysate-Ni-NTA HisBind mixture was poured into a column with the bottom outlet capped and was incubated for 5–10 min to allow the resin to settle down by gravity. The column was placed in a new collection tube. 1× washing buffer (20 mL) was added to the column; the column washing was repeated twice and transferred to a new collecting tube followed by the addition of 2 mL of elution buffer. Five hundred microliters of each fraction were collected and concentration was estimated (Bradford, 1976). The eluted protein fractions were stored at 4°C for subsequent studies. The purified proteins were separated under denaturing condition using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Laemmli,1970), and the rpie-5 protein was confirmed by molecular weight (MW; ∼100 kDa).
Determination of cytotoxicity
Three human cancer cell lines such as HeLa (cervical adenocarcinoma), HepG2 (liver hepatocellular carcinoma), and AGS (gastric cancer) were cultured in Dulbecco's modified Eagle's medium (Gibco Laboratories, Grand Island, NY) containing penicillin (100 U/mL), streptomycin (100 μg/mL), and 10% heat-inactivated FBS (HyClone, Logan, UT). Cell cultures were maintained at 37°C with 5% CO2 and 90% ± 5% humidity. Culture medium was changed every 2–3 days. Adherent cells were harvested using 0.25% trypsin (Thermo Scientific) and enumerated. Cells were inoculated in new dishes, grown to 80% confluence, and prepared for cytotoxicity assay.
Cell viability was determined using 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Mosmann, 1983). Briefly, the cancer cells (HeLa, HepG2, and AGS) were seeded (1 × 104 cells per well) into a 96-well plate and incubated for 24 h for attachment. After the attachment, the cells were exposed to different concentrations of recombinant protein (0.001, 0.01, 0.1, 1.0, 10.0, 100.0, and 1000.0 μg/mL) for 12, 24, 48, and 72 h. The cell viability was assessed by 20 μL of MTT (5 mg/mL) per well for 4 h at 37°C in dark, and the absorbance at 570 nm was measured using a microplate reader (SpectromaxM2; Molecular Diagnostics) and results are expressed as a percentage of controls. Positive drug control doxorubicin (0.001, 0.01, 1, 10, and 100 μg/mL) and heat-killed (80°C for 20 min) rpie-5 (HKr) protein (0.001, 0.01, 1, 10, and 100 μg/mL) were also used as a control for rpie-5 protein-specific cytotoxicity. All the assays were performed in triplicate in three different experiments and a normal cell line, Chang Liver cells, was also included. IC50 values of pierisin-5 on HeLa, HepG2, and AGS cells were determined by Prism software version 5 (GraphPad Prism).
Lactose dehydrogenase enzyme assay
Lactose dehydrogenase (LDH) release assay was used to measure the membrane permeability of cells (Kumar et al., 2018). The recombinant pierisin-5-treated cell membrane permeability of HeLa, HepG2, and AGS cancer cell lines (1 × 104 per well) were determined by LDH to release assay. Briefly, the cells were seeded for overnight in 96-well plates and treated with the IC50 concentration of rpie-5 protein. LDH enzyme release was quantified in the different time intervals (12, 24, 48, and 72 h post-treatment) using Pierce LDH Cytotoxicity Assay Kit (Thermo Scientific). The LDH release was estimated using the following equation: 100% × LDHout/(LDHout + LDHin) and compared with the total LDH in the cell lysate of each respective well (Johansson et al., 2008).
Determination of mitochondrial membrane potential
Alteration in mitochondrial membrane potential (MMP) is an indication of the early stages of apoptosis measuring rhodamine 123 (Rh123) (Baracca et al., 2003). Briefly, cells (5 × 104 cells per well) were exposed to the IC50 concentration of rpie-5 protein for 24 h. After the incubation, cells were washed twice with ice-cold 1× PBS and incubated with 10 μg/mL Rh123 for 15 min at 37°C in the dark, and images were taken using fluorescence microscope (CKX 41; Olympus) at 20× magnification. After being washed with cold PBS, cells were solubilized in 500 μL of cell lysates followed by centrifugation (3000 rpm) for 15 min. The cell supernatant was collected, and the fluorescence intensity of the sample was measured at 488 nm (excitation) and 535 nm (emission) with a multifunctional microplate reader (SpectraMax M2; Molecular Devices), and the percentage mean fluorescence intensity (% MFI) of Rh123 was calculated.
Nuclear damage and apoptotic cell death studies
DAPI (4,6-diamidino-2-phenylindole dihydrochloride) staining was used for the discovery of chromosomal changes in HeLa, HepG2, and AGS cells. The cells were cultured in a six-well tissue culture grade plate (Greiner) for 24 h. After incubation with the IC50 concentration of recombinant pierisin-5 protein for 24 h, cells were fixed with 4% formaldehyde and the cells were washed with 1× PBS (pH 7.4). The cells were stained with DAPI (1 μg/mL) and incubated in dark for 30 min and imaged using a fluorescence microscope (BX51TRF; Olympus) at 40× magnification. The stained cells were observed under a fluorescence microscope with excitation at 488 nm and emission at 550 nm, and the images were quantified using the ImageJ software.
Apoptotic cell death was identified by acridine orange–ethidium bromide (1:1 ratio of AO [5 mg/mL] and Et-Br [3 mg/mL]) (Kasibhatla, 2006). HeLa, HepG2, and AGS cancer cells were cultured individually in six-well plates for 24 h in a humidified incubator at 37°C in 5% CO2 and incubated overnight for adherence. The cells were treated with the IC50 concentration of pierisin-5 for 24 h, and at the end of the incubation, cells were rinsed twice with 1× PBS (pH 7.4). Acridine orange−ethidium bromide was incubated with the cells for 10 min at room temperature in dark. Cells were visualized under a fluorescence microscope (BX51TRF; Olympus) using blue filters along with untreated cells as a control.
Annexin-V-FITC Apoptosis detection kit (Calbiochem) was used to detect apoptosis. HeLa, HepG2, and AGS cell lines (1 × 104 cells per well) were seeded in six-well plates for 24 h at 37°C and treated with the IC50 concentration of pierisin-5 protein and incubated for 48 h. After the incubation, treated and untreated cells were washed with 1× PBS buffer (pH 7.4); further cells were stained with FITC-coupled Annexin-V and propidium iodide (Thermo Scientific). Apoptotic cells were detected under an inverted fluorescent microscope (BX51TRF; Olympus).
Caspase-3/9 enzyme activities
Activation of caspase-3/9 was measured using Caspase-3/CPP32 and Caspase-9 Mch6/Apaf-3 colorimetric protease assay kits (Invitrogen), respectively. HeLa, HepG2, and AGS cells (1 × 104 cells per flask) were seeded in a T25 flask for 24 h before the experiment. Cells were exposed to IC50 concentrations of recombinant pierisin-5 protein for 12, 24, 48, and 72 h to induce apoptosis. Triplicate and control (without induction) experiments were maintained concurrently for each experiment. Cells (3–5 × 106 cells per sample) were harvested by trypsinization and centrifuged at 150 rpm at 4°C for 10 min. The resultant cell pellet was washed twice with 1× PBS (pH 7.4) and pelleted by centrifugation at 2000 rpm at 4°C for 5 min. The clean cell pellet was lysed for 10 min on ice by adding 50 μL of chilled cell lysis buffer. Total protein concentration was determined (Bradford, 1976). Supernatant was diluted for 50 μL of cell lysis buffer containing approximately 50–200 μg protein (final concentration 1–4 mg/mL). Fifty microliters of the supernatant were transferred to a fresh 96-well plate and 50 μL of 2× reaction buffer (containing 10 mM DTT to each sample). To each sample, 5 μL of the 4 mM DEVD-pNA and LEHD-pNA substrates of caspase-3 and caspase-9 (200 μM final concentration), respectively, were added. Finally, the samples were placed at 37°C for 2 h in dark, and the optical density was measured at 405 nm in a 96-well plate reader (SpectraMax; Molecular Devices). Caspase-3/9 activities of fold-increase were estimated by direct comparison to the level of untreated controls.
Proapoptotic and antiapoptotic gene expression analysis
Total RNA extraction and quantitative analysis of proapoptotic and antiapoptotic gene expression were performed in HeLa, HepG2, and AGS cells treated with the IC50 concentration of recombinant pierisin-5 at 48 h. The total RNA was isolated, and cDNA library was prepared in a similar way as stated above. APAF-1 (apoptotic protease activating factor-1), Bcl-2 (B-cell lymphoma 2), Bax (Bcl-2 family activating protein-X), p53, CytC1, and GAPDH (internal control) gene primers were used to quantify the expression of genes using SYBR green dye in quantitative RT-PCR (qRT-PCR; Stepone plus; Thermo Scientific) (Table 1). Triplicate experiments were carried out for each concentration. Relative quantification of mRNA from each sample was performed using the Pfaffl method (Pfaffl, 2001), and GAPDH was used as the internal control gene.
Statistical analysis
Statistical results are expressed as the mean ± standard deviation of three independent experiments. One-way analysis of variance was carried out to conclude whether there were any significant differences between the IC50 means of the recombinant protein and positive drug control. Differences with p < 0.01 values were considered as significant difference. Duncan's new multiple range test was also performed to determine which particular pair of extracts differed significantly from one another.
Results
Structure of rpie-5 gene and its product
In the present study, the complete coding sequence of the pierisin-5 gene (NCBI GenBank: KJ598075) was amplified with an open reading frame (ORF) of 2553 bp encoding 850 amino acid residues. The deduced amino acid sequences had ∼98 kDa MW with an isoelectric point (pI) of 5.90. We cloned the pierisin-5 gene into baculovirus-mediated pIEX/Bac-3 3C LIC vector system, which contains 10x His-Tag in N-terminal and Strep-tag II in C-terminal of the gene. The cotransfected recombinant plasmid was transferred into Sf21 cell lines for in vitro expression and purification. The recombinant pierisin-5 (rpie-5) protein encodes 891 amino acids with a calculated molecular mass of 102.225 Da. Methionine (M) is the first amino acid in rpie-5, which encodes an ATG codon, followed by 3–12 His amino acids (10x His-Tag), which are used for the purification of recombinant protein. The amino acids 15–19 encode human rhinovirus (HRV) 3C-specific protease enzyme, which is the recognizing site for the cleavage of 10x His-Tag peptide after purification. The amino acids from 83 to 253 encode ADP-ribosyltransferase domain (ARTT), which is an active site responsible for the transfer of ADP moiety from the NAD to DNA. This domain consists of NAD binding (R-S-E) motifs such as arginine (R) 84, serine (S) 138, and glutamine (E) 188 and polynucleotide-binding sites at amino acids 88,139, and 140. The N-terminal domain has been cleaved by trypsin enzyme at Arg256 and Ser257. The N- and C-terminal domains were linked through Linker peptide region (amino acids 257–269). The C-terminal peptide of rpie-5 has target binding site, which contains four ricin binding lectin domains (RBL domains I-IV RBL-I 324–432, RBL-II 477–580, RBL-III 626–730, and RBL-IV 736–873); each domain consists of three putative sugar binding motif called Q-X-W motifs. The C-terminal end of rpie-5 was fused with strep tag-II peptide (884–891) for dual purification of rpie-5. The tag was cleaved by thrombin protease enzymes, which recognize the amino acids at 878–882 residues (Supplementary Fig. S2).
Cloning into BEVS
LIC is a simple, fast, and relatively inexpensive technique to produce expression constructs of rpie-5 plasmid DNA using VSS. Identification of positive clones using vector-specific primers (IE promoter and terminator) or in combination with insert-specific primers in colony PCR was performed (Table 1). Plasmid DNA of rpie-5 was isolated from the positive clones, and ORF of the insert was verified by sequencing. The recombinant plasmid contains a 10X-Histidine tag and Strep II tag at the N- and C-terminus of the protein, which helps to achieve better purification of the recombinant fusion protein.
Recombinant expression in Sf21 cells
The rpie-5 (pIEx/Bac-Pie-5) plasmid was cotransfected with the linearized baculovirus genomic DNA using cationic lipid Insect gene Juice into Sf21 cells. Significant differences in cell size, morphology, and cytoplasm were found after 24 h of post-transfection in infected cells. The size of the post-infected cells gradually increased after 24 h of post-infection, and it reached a maximum at 96 h and lysed at 120 h (Supplementary Fig. S3). The average size of uninfected and infected cells was 24.61 and 41.02 μm, respectively. The virions transfection also causes cytotoxicity to cells. Infected cells started detaching from the bottom of the flask and floated after 72 h, whereas the uninfected cells were intact (Supplementary Fig. S3).
The protein, rpie-5, was expressed in Sf21 cells using BEVS. The advantages of Sf21cells include serum-free media, easily scale up, and free from human pathogens. The time-dependent expression of rpie-5 was analyzed for 0, 24, 36 48, 72, 96, and 120 h by immunoblot (Fig. 1a, b). The protein expression gradually increased from 36 h after post-transfection and reached a maximum at 96 h and gradually reduced till 120 h of post-infection. Presence of some extra bands >100 kDa suggests the possibility of nonspecific aggregation of overexpressed proteins (Fig. 1c).
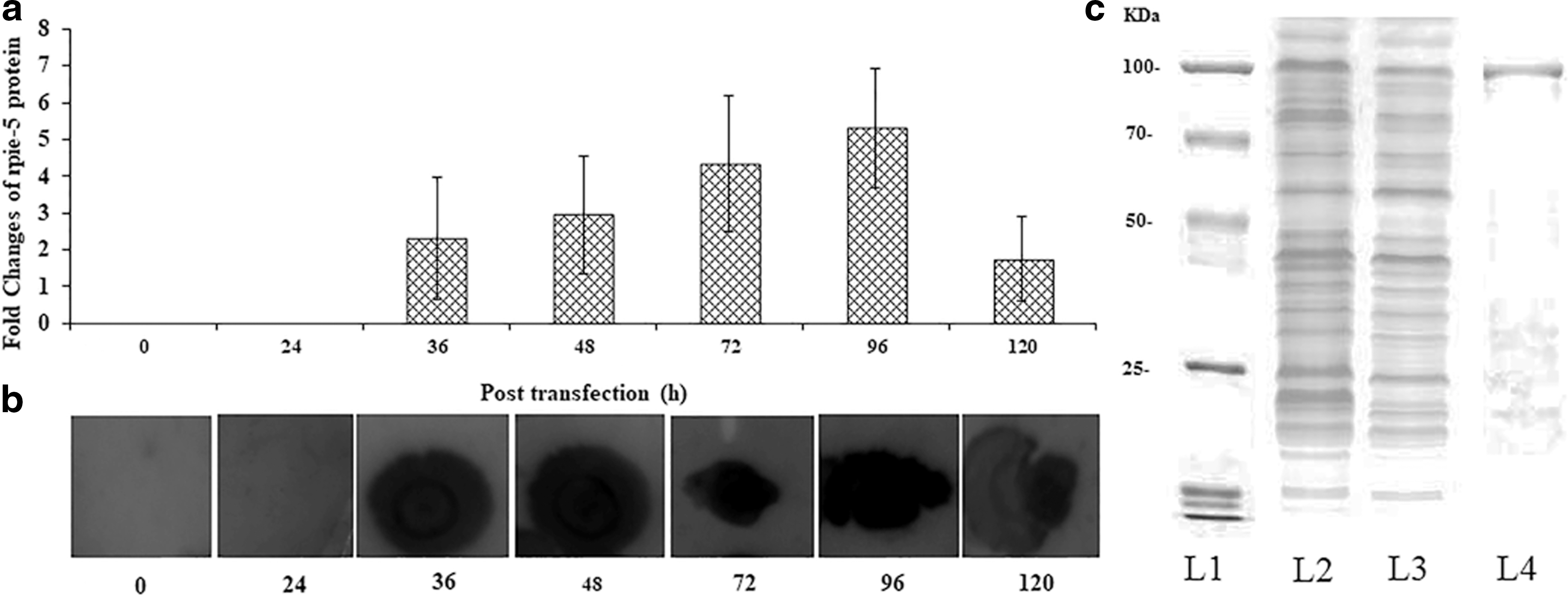
Baculovirus-mediated expression (Dot blot analyses) and separation of the rpie-5 protein from sf21 cell lines.
Altered cell morphology and viability
The rpie-5 treatment resulted in inhibition of cell proliferation at low concentration. Treatment of rpie-5 (1 ng/mL) against HeLa, HepG2, and AGS cells for 72 h resulted in a decrease in cell growth by 42%, 34% and 27%, respectively. The IC50 values were 0.6, 1.9, and 3.7 μg/mL for HeLa, HepG2, and AGS cells lines, respectively, at the end of 72 h treatment (Supplementary Table S1). Doxorubicin was used as a positive control to test the response of the cancer cell lines, and significant cytotoxicity was observed (Supplementary Fig. S4). The HKr protein showed more than 50% of the loss of cytotoxicity against the cell lines (Supplementary Fig. S5). The treated cells were morphologically distinct from untreated cells in terms of cytoplasm and nuclear condensation and cell shrinkage with the rough cell membrane.
Loss of the MMP
Quantitative data showed that the rpie-5 protein induced MMP reduction in a dose-dependent manner (p < 0.05) (Fig. 2). Untreated cells had intact mitochondrial enzyme observed by compact dense fluorescence intensity, whereas treated cells showed scattered and reduced fluorescence intensity in and around the cells indicating a significant loss of MMP (Δψm) (Fig. 2). The protein, rpie-5, was able to significantly induce loss of membrane potential in HeLa cells when compared with HepG2 and AGS cancer cells.
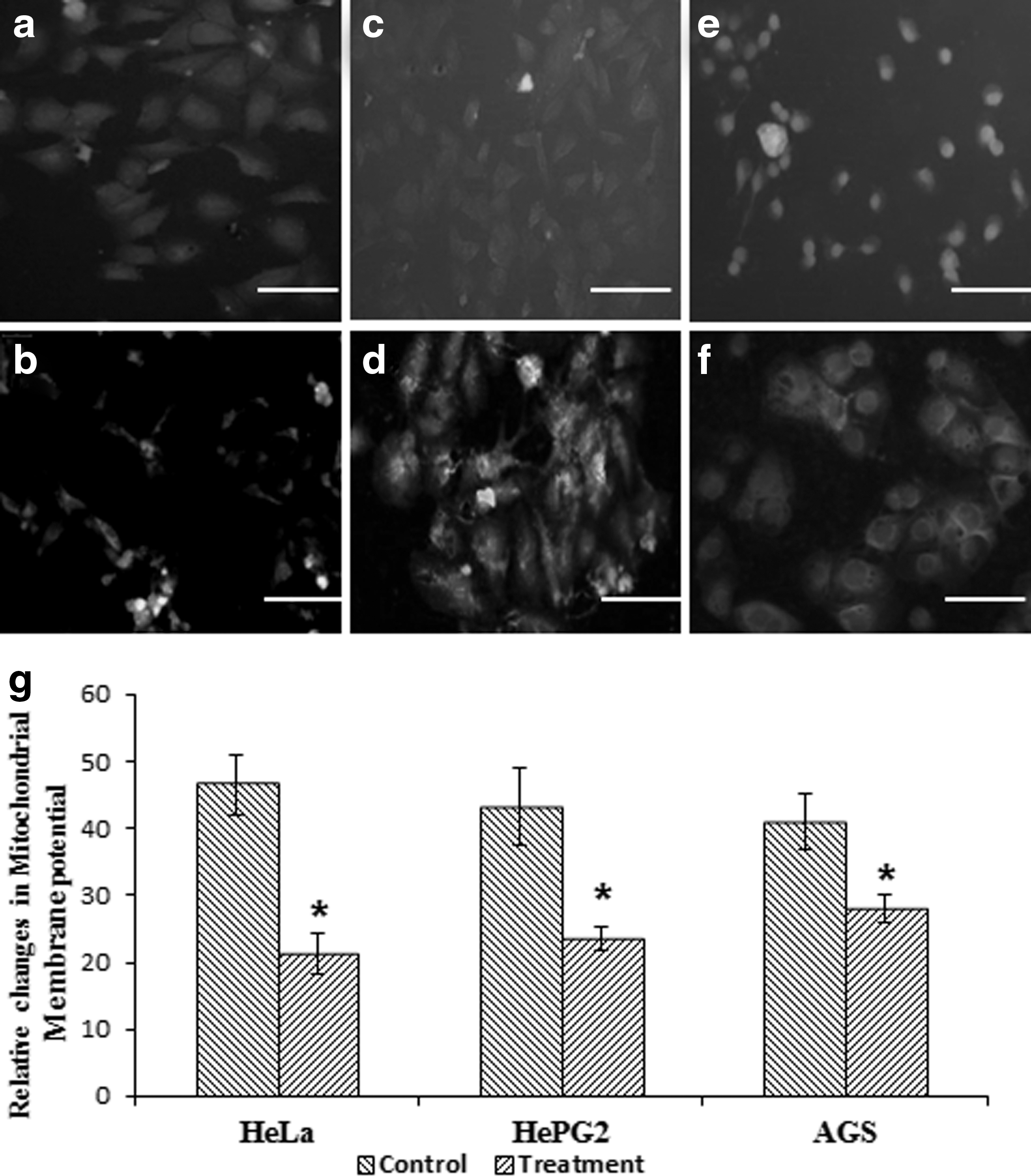
Loss of mitochondrial membrane potential (Δψm) was persuaded by rpie-5 protein.
Morphological changes in apoptosis
DAPI bound to the nucleus is used to elucidate apoptotic cell death due to the exposure of rpie-5. Under a fluorescence microscope, normal cells had compact round-shaped single nucleus, whereas exposure to rpie-5 at 1 μg/mL for 24 h altered the nuclear morphology (Fig. 3). Apoptosis resulted in cells with higher chromatin condensation and nuclear fragmentation. Correspondingly, treatment of rpie-5 resulted in a noticeable increase in the number of apoptotic cells with condensed and fragmented DNA by a more marked blue color (Fig. 3). When stained with DAPI, the untreated cells did not exhibit brighter blue fluorescence. These results suggest that rpie-5 (1 μg/mL) synergistically increases cell growth inhibition and apoptosis.
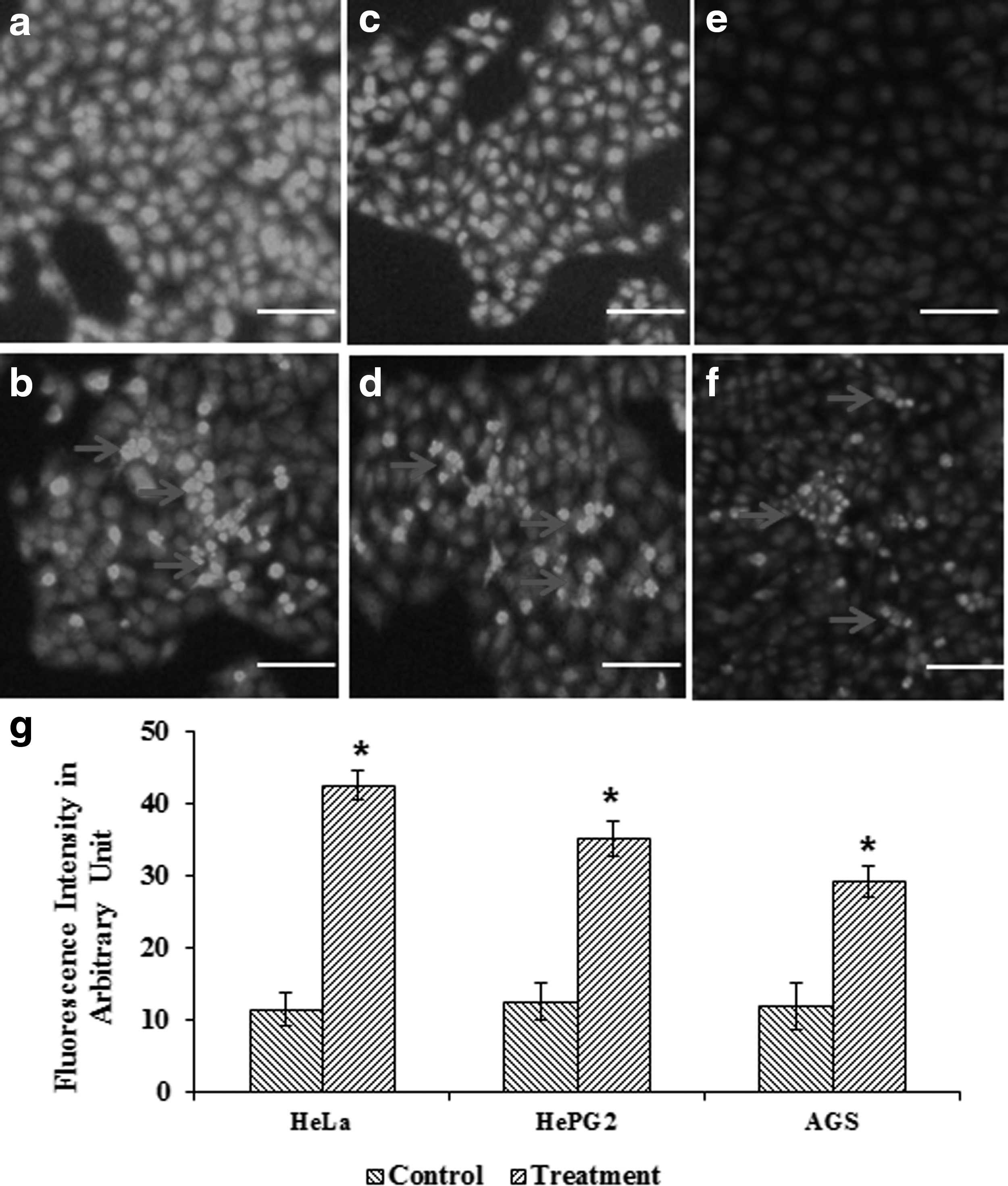
rpie-5-induced chromatin condensation was examined by 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) staining.
The untreated control cells uniformly fluoresced in green color, which indicates the features of a viable cell. When cells are treated with the rpie-5 protein, it showed orange-red fluorescence color indicating chromatin condensation and nuclear fragmentation of early apoptotic cells. In addition, cell shrinkage and formation of apoptotic bodies were also observed in some cells (Fig. 4).
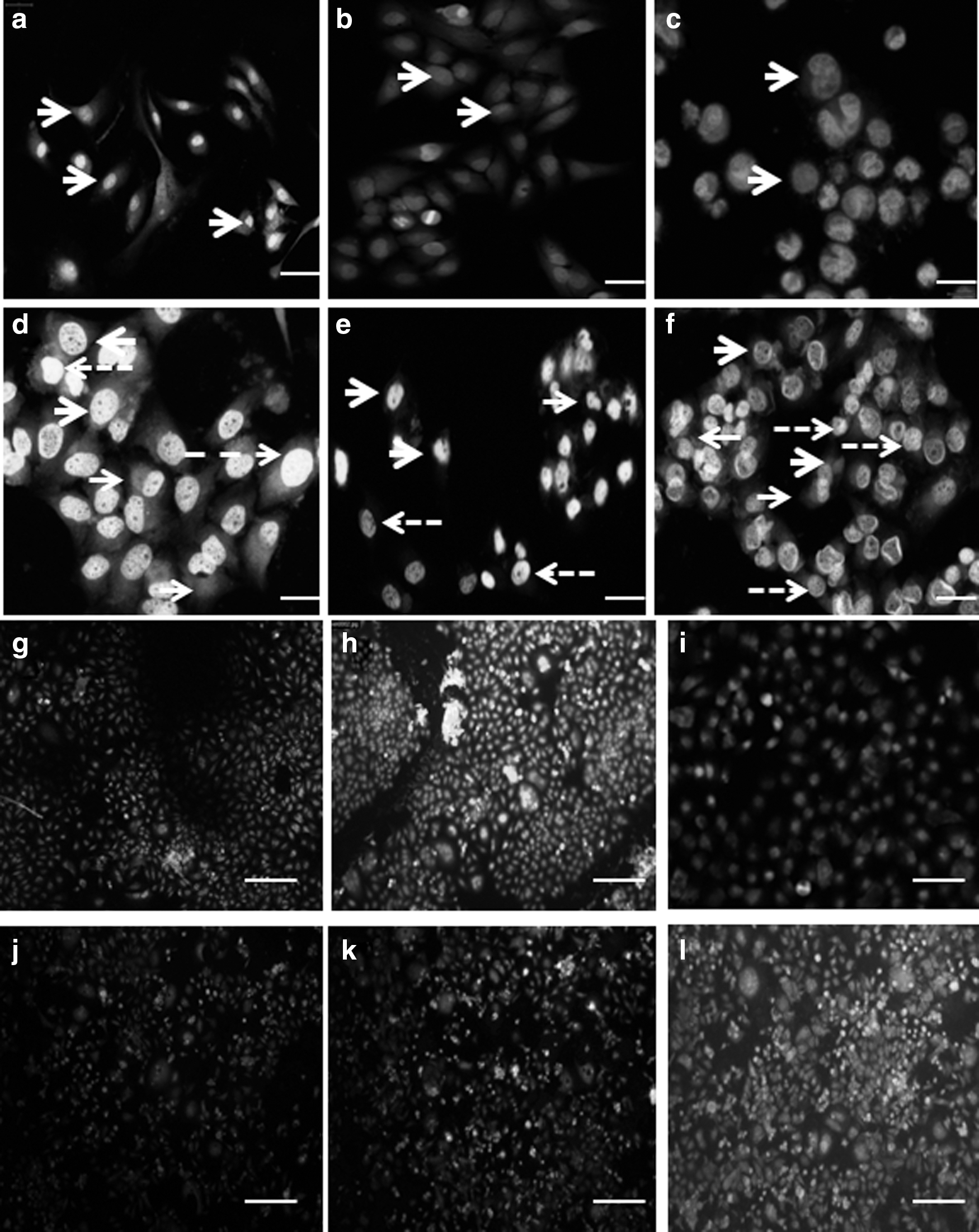
Apoptotic effect of rpie-5 protein.
The effects of rpie-5-induced apoptosis were investigated using Annexin V-FITC/PI staining combined with inverted fluorescence microscopy. Data showed that rpie-5 at IC50 initiated strong early apoptosis in HeLa cells and weak early apoptosis in HepG2 and AGS cells in 24 h while the percentage of early and late apoptotic cells was remarkably increased after exposure to 48 h. The untreated (control) cells exhibit green fluorescence, but the treated cell exhibits red fluorescence in the cytoplasm and green fluorescence in membranes, which indicates early apoptotic cell death. The cells with either green with red or only red fluorescence indicate late apoptotic and necrotic cells, respectively (Fig. 4). Thus, the study suggests that rpie-5 induced apoptosis in HeLa, HepG2, and AGS cells.
Enzymatic changes in apoptotic cell death
The release of LDH into the media was measured using LDH assay kits to assess the effects of rpie-5 on the cell membrane permeability. The rpie-5 induced cytotoxicity, and it enhances the loss of membrane permeability, which leads to the release of LDH enzyme in a time-dependent manner. HeLa cells were more susceptible to LDH release when compared with HepG2 and AGS cell lines (Fig. 5).
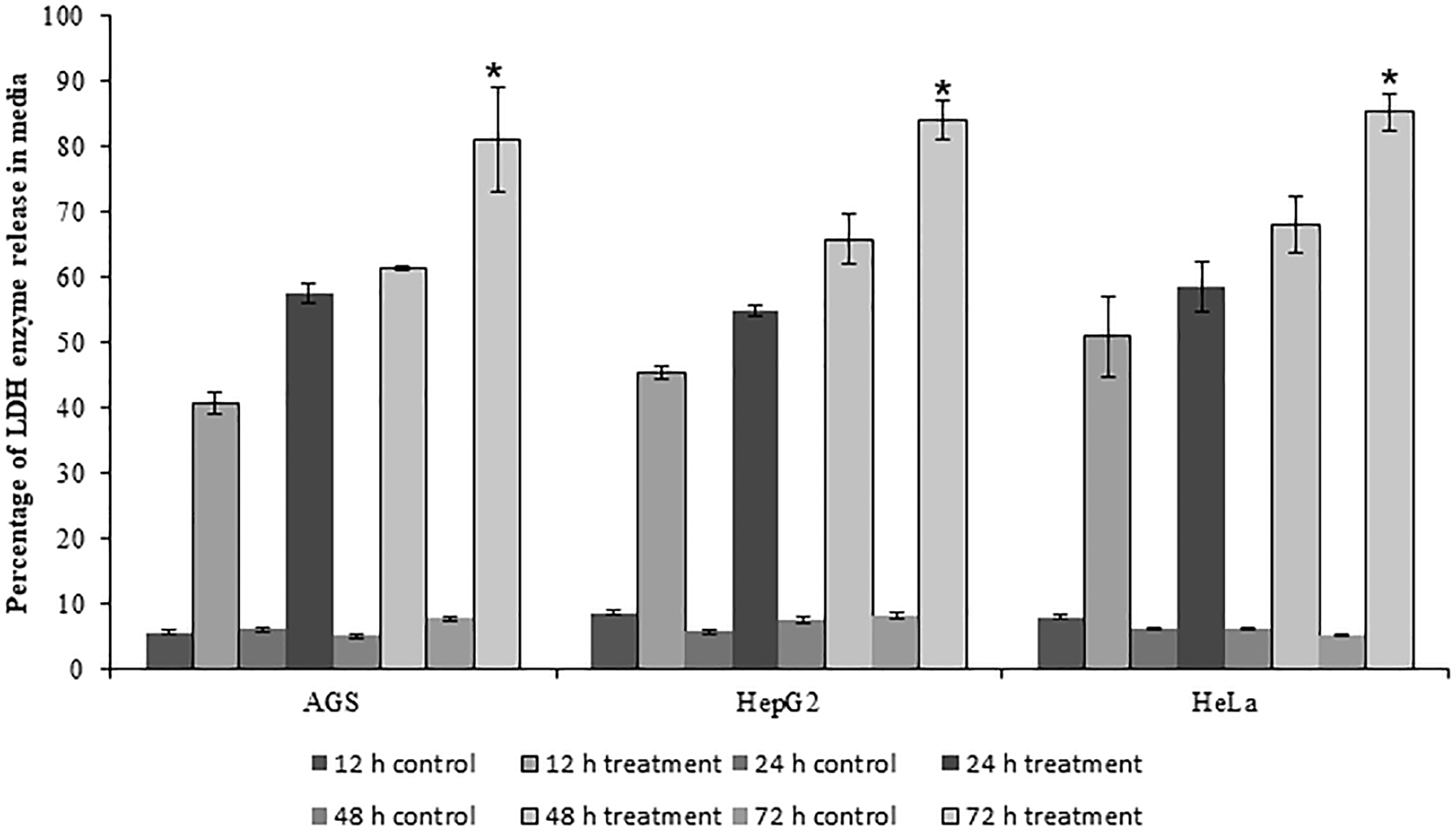
Alteration of cell membrane permeability was induced by rpie-5 protein (1–1000 μg/mL). Percentage of LDH enzyme was released in the medium at a different time intervals. Each group was tested for three times. *Indicates statistical significance (p < 0.05). LDH, lactose dehydrogenase.
To investigate the cleaved effector caspase in the apoptotic pathway, the activation of caspase-3/9 was measured by the colorimetric assay. Induction of cytotoxicity leading to the activation of caspase-3/9 by recombinant pierisin-5 protein occurs in a time-dependent manner in HeLa, HepG2, and AGS cancer cells (Fig. 6).
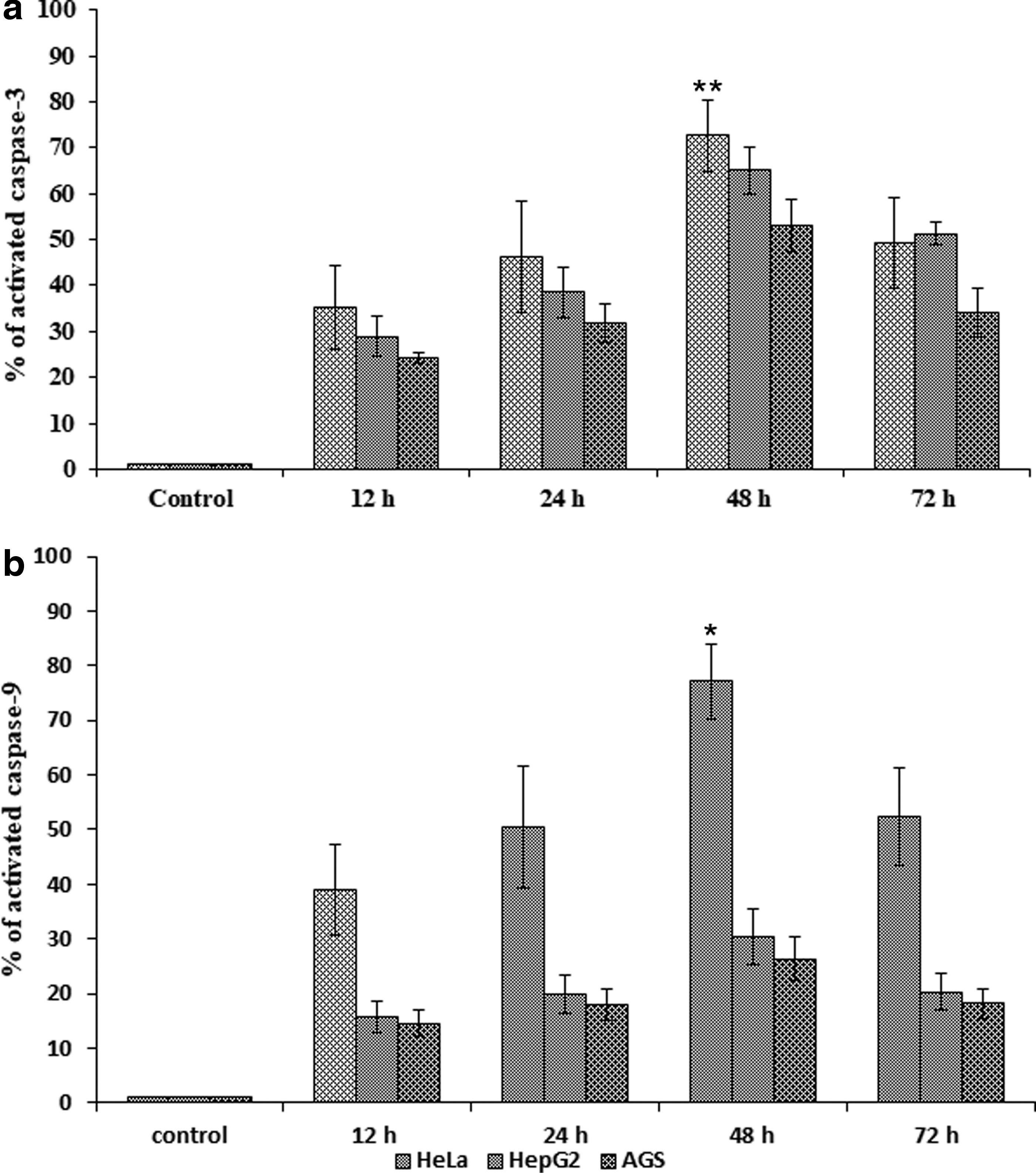
rpie-5-induced the activation of caspase-3 and caspase-9 enzymes. Quantification of rpie-5-induced activated caspase-3
The intrinsic pathway of apoptosis induced by rpie-5
To study the mechanism of rpie-5-induced cell death, we examined the expression levels of apoptotic markers by treating the cancer cell lines with the IC50 concentration of the rpie-5 for 48 h. RT-PCR results demonstrated that the rpie-5 protein upregulated the expression of proapoptotic genes (Bax) and other apoptosis genes p53, APAF1, and mitochondrial cytochrome (CytC1) genes and downregulated the expression of antiapoptotic (BCL2) in the HeLa and AGS cells (p < 0.05) compared with the control cells (Fig. 7). In HepG2 cells, pierisin-5 induced the downregulation of p53 and BCL2 genes and the upregulation of Bax, APAF1, and Cytc1 genes (Fig. 7).
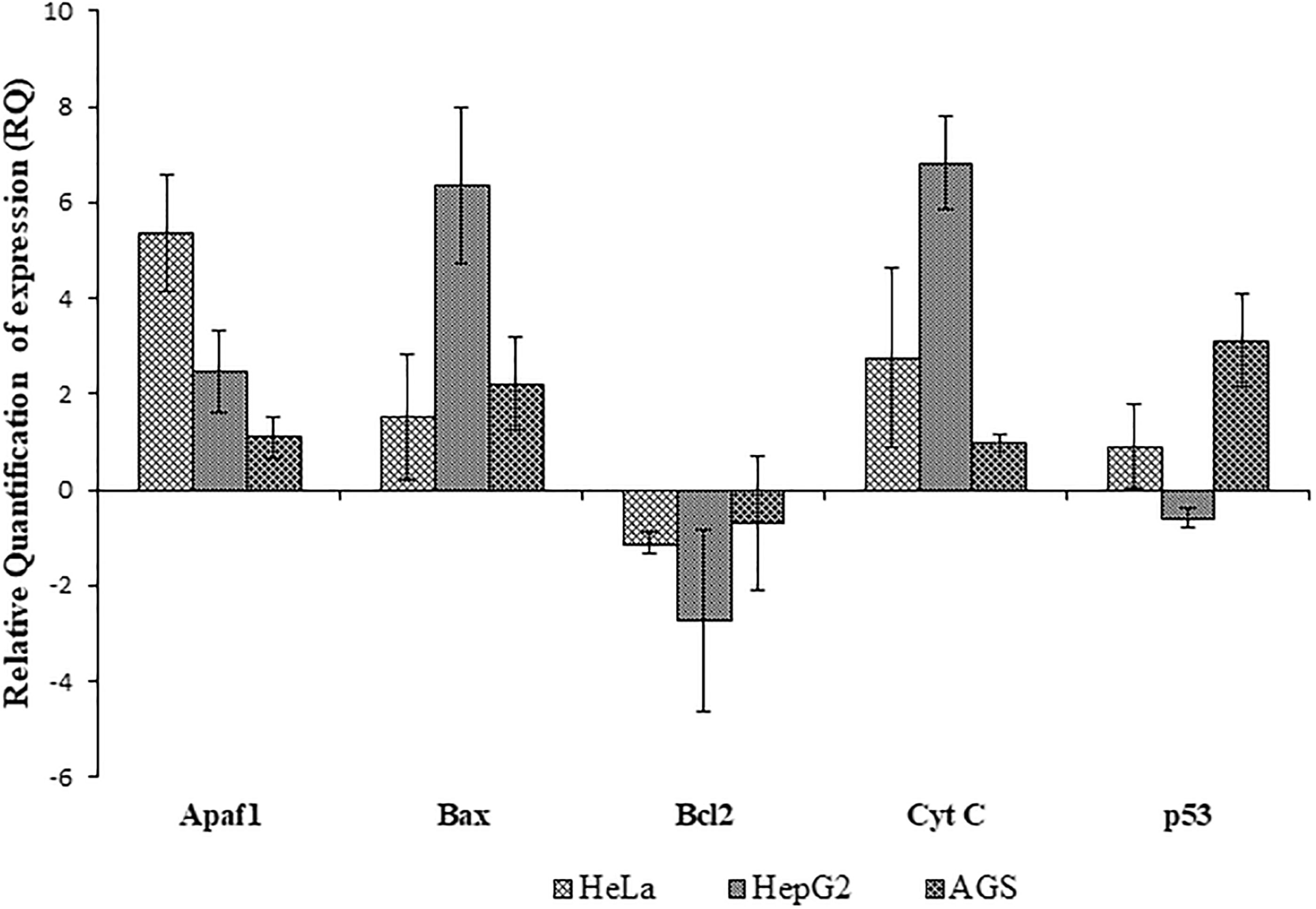
Alterations in the expression of proapoptotic and antiapoptotic genes. Proapoptotic and antiapoptotic genes (Bcl2, Bax, Apaf-1, p53, and cyt C) were relatives quantified using qRT-PCR in the presence and absence of rpie-5 protein. Representative results of three independent experiments are shown (n = 3). qRT-PCR, quantitative real-time polymerase chain reaction.
Discussion
In earlier studies, the family of pierisin-like proteins has been extensively investigated for antitumor effects against various cancers and normal cells (Watanabe et al., 1998, 1999; Subbarayan et al., 2016), including gastric, breast, cervical, and hepatic cancers (Kono et al., 1999; Orth et al., 2011). However, to the best of our knowledge, the molecular mechanism of antitumor effects of insect-derived pierisin proteins is yet to be fully investigated. We focused our study on the rpie-5 protein, which induces cytotoxic cell death via p53-related caspase-3- and caspase-9-dependent mitochondrial pathway, and this serves as the mechanism for the induction of apoptosis in HeLa, HepG2, and AGS cells. The rpie-5 protein showed high cytotoxicity and induced apoptosis at very low concentrations on HeLa, moderate effect on HepG2, and little effect on AGS cell lines. Some of the anticancer peptides and proteins (ACPs) from insects such as cecropins (Anghel et al., 2014) and defensins (Gaspar et al., 2013) induced the disruption of mitochondrial membrane and apoptosis in different human cancer cell lines.
We performed transient transfection of pIEX/Bac-3 plasmid DNA into Sf21 cell line, in which the early expression was driven by the combination of hr5/ie1 (as a weak promoter/enhancer); late and very late expression was driven by the strong p10 promoter derived from AcNPV. In this study, we used the linearized AcNPV genome that contains a deleted portion of the essential ORF 1629 for the prevention of replication of nonrecombinant virions in Sf21 cells. Similarly, linearized AcNPV genome has a deletion of nonessential chiA (chitinase), v-cath (cathespsin-like cysteine protease) genes, and three viral genes (p10, p24, and p26) to improve product quality and yield of secreted and rpie-5 protein. This will help to develop a new method for producing recombinant virus-like particles by eliminating the time-consuming plaque purification.
There were no morphological and cytoplasmic changes up to 12 h in both transduced (with recombinant VLPs) as well as untransduced Sf21 cells, and the transduced cells enlarged in size (15–18 μg) after 24 h and cytoplasm appears more granular under phase contrast microscope (Supplementary Fig. S3). The level of rpie-5 protein gradually increased from 36 h and reached a maximum at 96 h of post-transduction. After 96 h post-transduction, we observed increased cell lysis, media turbidity, nutrition depletion, pH alteration, cellular granulation, detachment, and floating of the cells in the medium. The cell viability was about 20–30%, and most of the cells obtained a secondary infection from medium containing VLPs. At this stage, the supernatant containing VLPs was infected with fresh cells containing 70–90% confluency to continue the virion production. The cytotoxic cell death in Sf21 cells was found due to either the transduction of VLPs or the production of rpie-5 proteins.
These findings improve the desirability of rpie-5 protein as a natural anticancer agent because rpie-5 protein preferentially kills HeLa cancer cells while minimizing damage to normal cells. The previous study of pierisin-5 on HeLa cells and HepG2 cells also showed the apoptotic effect (Subbarayan et al., 2016). This fact increases evidence that target-specific chemotherapeutic agents persuade cancer cell death through the mechanism of apoptosis. Moreover, cytotoxicity results showed that rpie-5 gave a higher IC50 toward normal liver cells (Chang's Liver). This finding may lead to developing a new drug as an alternative medicine to treat cancer. However, the HKr protein exhibited very low cytotoxicity against all the human cancer cells (Supplementary Fig. S4). These results suggested that HKr completely lost its activity due to heat inactivation, and rpie-5 was the only sole candidate for the induction of cytotoxicity in different cancer cell lines.
Apoptosis can be persuaded either by the intrinsic (mitochondria dependent) or by the extrinsic pathways (death receptors dependent) (Pfeffer and Singh, 2018). We showed that rpie-5 protein exhibited mitochondrial-mediated apoptotic cell death via a p53-dependent pathway in HeLa and HepG2 cells and p53-independent pathway apoptosis in AGS cell lines. Previous studies described that pierisin proteins exhibit DNA-dependent ADP-ribosylation on target cell, which leads to DNA adduct (Supplementary Fig. S6) (Takamura-Enya et al., 2001), which we believed that this might have upregulated the expression of p53 protein. Based on the result of DAPI staining, it was found that treatment with rpie-5 protein induced DNA fragmentation in all three cells in a dose-dependent manner (Fig. 3). DNA fragmentation occurring in internucleosomal was the crucial biochemical characteristic to specify an early event of apoptosis, and it signifies a point of no return from the path to cell death (Zhivotosky and Orrenius, 2001). Positive control drug, doxorubicin-treated cells (data not shown), also showed alike nuclear fluorescence, and these outcomes afford evidence that our experiments involving apoptotic pathway-mediated cell death in cancer cell lines are concluded.
The plasma membrane-bound enzyme, LDH, was mainly involved in the initiation and progression of a tumor in many tissues. Comparably, most of the tumors have high LDH enzyme depletion (Kumar et al., 2018). We showed that rpie-5 induces LDH depletion from the target cell membrane with time- and dose-dependent manner and confirmed that the loss of cellular membrane organization (Fig. 5). We further investigated the involvement of cytochrome C in rpie-5 protein-induced apoptotic pathway by measuring the MMP (Δψm) (Fig. 2). Normal mitochondrial morphology and function are maintained by MMP, which regulates mitochondrial membrane selectivity and permeability (Banerjee et al., 2016). A reduction in MMP plays a key role in triggering apoptosis, and the loss of MMP is considered an early event in the apoptosis cascade.
DAPI is a nuclear stain, which interacts with the minor groove containing adenine–thymine clusters in double-stranded DNA (Dar et al., 2016). The cytosolic cytochrome C takes part in programmed cell death by speed up the activation of a caspase cascade (Pfeffer and Singh, 2018). We believe that the upregulated cytC, Apaf-1, Bax and the downregulated Bcl-2 confirmed the involvement of cytC in rpie-5-induced apoptosis. In earlier studies, exogenous cytochrome C to cytosol can also trigger apoptotic cell death in a cell-free system (Leoni et al., 1998). AO/Etbr is a promising dye that can enter into both viable and dead cells. But EtBr only enters into dead cells due to loss of membrane integrity and distinguishes between apoptotic and necrotic cell death (Liu et al., 2015). AO/EtBr dual staining was used for qualitatively distinguishing the influence of rpie-5 on HeLa, HepG2, and AGS cells for several reasons including (1) changes in chromatin condensation, (2) fragmented nuclei, and (3) membrane blebbing (Fig. 4). Annexin-V/PI dual staining was the confirmation for apoptotic cell death used to differentiate intact cells from early apoptotic cells, late apoptotic cells, and dead (necrotic) cells (Fig. 5). Annexin, as a phosphatidylserine, which is normally present in the inner part of the plasma membrane and during the onset of apoptosis, is translocated into the outer part of the membrane and can be detected by fluorescence-tagged anti-Annexin antibodies. PI detects necrotic cells and late apoptotic cell with the permeabilized plasma membrane (Jin et al., 2017).
The caspase-cascade system acting as a significant role in the initiation, transduction, and amplification of programmed cell death (Portt et al., 2011). Quantitative analysis of the influence of rpie-5 protein on caspase cascade revealed an increase in caspase-3/9 activities in HeLa, HepG2, and AGS cells in a dose-dependent manner (Fig. 6). Similar results were observed in other anticancer protein and peptides, which lead to increased caspase-3/9 activities in different cancer cells (Slocinska et al., 2008).
Most of the human malignant cells contain TP53 gene mutations, which play a major role in transcriptional activation and inhibition of cell cycle progression and associated with other cell cycle-related genes that have been reported in different cancers (Kabel et al., 2016). The active p53 persuades the transcription of many apoptosis-related genes and recently found that Bax genes are transcriptional targets of p53 (Haupt, 2003). The upregulation of Bax gene expression might be induced by activated p53 in HeLa and AGS cell lines. The present study has observed that increased time of exposure of rpie-5 leads to the upregulation of p53, caspase-3/9, and Bax proteins and the downregulation of Bcl-2 in HeLa and AGS cell lines. In contrast, HepG2 cell has downregulation of p53 protein (Fig. 7). The Bcl-2 family of proteins is significant to regulate the apoptotic cell death, by inhibiting the mitochondrial permeability transition (PT) channel, and hence, the lowering of mitochondrial potential (Naseri et al., 2015). The level of Bax increased and concurrently Bcl-2 decreased (Fig. 7). HeLa, HepG2, and AGS cells were moderately sensitive to rpie-5, which promotes apoptosis by inhibiting the Bcl-2 protein (Kanazawa et al., 2002). The expression of Bax and Bcl-2 proteins determined the fate of each cell including both regulations of other key apoptotic proteins (Basu and Haldar, 1998; Eberle and Hossini, 2008).
In summary, we found that BEVS can be used for the high-level expression and purification of rpie-5 (∼100 kDa) cytotoxic protein at an early stage of transfection. The cytotoxic effect of rpie-5 was studied against HeLa, HepG2, and AGS cell lines, and among them, HeLa cells exhibited higher cytotoxicity. The cell death associated with apoptosis was revealed by the typical apoptotic morphology, inhibition of cell proliferation, and activation of caspase-3/9. This study for the first time reports the upregulation of Bax, Bad, APAF-1, and Cyt C and the downregulation of Bcl-2, which proves that the pierisin-induced apoptotic cell death was mediated by the intrinsic pathway. Target-specific activation of apoptosis was achieved by rpie-5 by induction of both p53-dependent (HeLa and AGS cells) and p53-independent (HepG2 cells) pathways. This area of research is promising in future investigations that identify the receptor binding mechanism and other signal transduction of rpie-5 peptide and specifically through which these receptors arbitrate its anticancer activity.
The in vitro expression of this protein allows modifications that can maximize its clinical potential, productivity, and homogeneity when compared with partially purified butterfly protein. In the future, the stable recombinant Sf21 cells can be optimized for large-scale production and purification of rpie-5 protein produced in the BEVS and used for functional studies, vaccine preparations, or therapeutically important drug against cancer.
Footnotes
Acknowledgments
The authors are thankful to the Department of Biotechnology (DBT), New Delhi, for the Twinning project on Pierisin study (Agri-94-BT/155/NE/TBP/2011) and DBT-Advanced Level State Biotech Hub, Mizoram University for the infrastructural facility. The authors are also thankful to PSG College of Technology, Coimbatore, India, for the support.
Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.