
Abstract
DNA hydroxymethylation is one of the major epigenetic mechanisms mediating the development of several human cancers. This study aimed to identify key hydroxymethylated genes and transcription factors (TFs) associated with alpha-fetoprotein (AFP)-negative hepatocellular carcinoma (HCC) using whole-genome DNA hydroxymethylation profiling. A total of 615 differentially hydroxymethylated regions (DHMRs) were identified from AFP-negative HCC tissues compared to adjacent normal tissues. DHMR-associated genes were significantly enriched in gene ontology functions associated with actin binding, cell leading edge, and blood vessel morphogenesis and pathways such as MAPK signaling pathway, neuroactive ligand-receptor interaction, and axon guidance. Moreover, protein–protein interaction (PPI) network analysis showed that PH domain and leucine-rich repeat protein phosphatase 1 (PHLPP1) and SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 2 (SMARCA2) had higher degrees and were hub nodes. Furthermore, TF prediction analysis showed that TFs, such as nuclear factor I C (NFIC) and GATA binding protein 3 (GATA3), regulated many DHMR-associated genes. Our findings reveal that key hydroxymethylated genes such as PHLPP1 and SMARCA2, as well as TFs such as NFIC and GATA, may be involved in the development of AFP-negative HCC. These molecules may be potential biomarkers for AFP-negative HCC.
Introduction
Hepatocellular carcinoma (HCC) is the third-ranking cause of cancer-related deaths (Di et al., 2016; Pascual et al., 2016). Most of HCC patients are diagnosed at the advanced stage and are confirmed to be refractory to chemotherapy and radiotherapy (Lo et al., 2002; Ma et al., 2010). Moreover, the 5-year survival rate of advanced HCC is <10% (Shen et al., 2016; Zaazaa et al., 2018). Therefore, early detection and early treatment are important for improving the clinical outcome in patients with HCC.
Serum alpha-fetoprotein (AFP) is the most commonly used tumor marker for HCC surveillance (Aghoram et al., 2012). Moreover, AFP may have a valuable role in postoperatively predicting the recurrence of HCC (Silva et al., 2017). Although serum AFP measurement is no longer recommended for HCC surveillance by the American Association for the Study of Liver Diseases (AASLD) (Bruix and Sherman, 2011) and European Association for the Study of the Liver (EASL) (Liver, 2012), because of lack of specificity and sensitivity, measuring AFP can remarkably improve ultrasound sensitivity for HCC detection in clinical practice (Tzartzeva et al., 2018). However, AFP-negative HCC patients account for ∼30% of the total number of HCC patients (Baig et al., 2009), and there are currently no efficient and reliable methods for diagnosing AFP-negative HCC (Zhang et al., 2016) or predicting its recurrence after curative resection (Gan et al., 2018). Therefore, it is imperative to further identify new diagnostic and prognostic biomarkers for AFP-negative HCC.
DNA methylation is an extensively characterized epigenetic mechanism for regulating gene expression (Lai and Wade, 2011). Methylation of cytosine to 5-methylcytosine (5-mC) is a well-known epigenetic feature that has been involved in both the development and progression of many cancers (Lai and Wade, 2011; Network, 2011; Paska and Hudler, 2014). In addition, by the ten-eleven translocation (TET) family of enzymes, 5-mC can be converted to 5-hydroxymethylcytosine (5-hmC) (Kriaucionis and Heintz, 2009). Emerging evidence has shown that 5-hmC alternations play a key role in the epigenetic regulation of a variety of cancers (Haffner et al., 2011; Ma et al., 2017). Furthermore, several 5-hydroxymethylated loci have been identified as promising markers in liver cancer (Muffadal et al., 2015). However, the hydroxymethylated genes that are involved in the development of AFP-negative HCC are largely unknown.
The aim of this study was to provide a landscape of DNA hydroxymethylation status in AFP-negative HCC and further to identify key hydroxymethylation associated genes. Recently, generation of the whole-genome DNA hydroxymethylation profiling by hydroxymethylated DNA immunoprecipitation sequencing (hMeDIP-Seq) is an effective tool for identifying (hydroxy)methylated genes associated with disease development (Ye et al., 2016). In this study, we investigated the 5-hmC changes in five paired AFP-negative HCC tissues and adjacent normal tissues using hMeDIP-Seq, followed by identifying differentially hydroxymethylated regions (DHMRs) and functional enrichment analysis for DHMR-associated genes. Moreover, a protein–protein interaction (PPI) network was constructed, and transcription factor (TF) prediction analysis for DHMR-associated genes was also performed. Our findings will provide new insights for identifying potential epigenetic biomarkers for AFP-negative HCC.
Materials and Methods
Patient samples
A total of five AFP-negative (AFP <20 μg/L) HCC patients in Shanghai Tenth People's Hospital (one female and four male, average age of 64.8 ± 16.7 years) were enrolled in this study. The detailed information of the enrolled patients was supplied in Supplementary Table S1. Paired HCC tissues and adjacent normal tissues were obtained, immediately frozen in liquid nitrogen, and stored at −80°C. The study was approved and supervised by the Ethics Committee of Shanghai Tenth People's Hospital (Ethical No. SHSY-IEC-KY-4.0/17-24/28, approved in 2017), and all patients gave informed consent to use their tissues in research.
Hydroxymethylated DNA immunoprecipitation sequencing
Genomic DNA was extracted from paired HCC tissues and adjacent normal tissues and sheared using a Diagenode GenDNA module (Cat No. mc-magme-03; Diagenode, Denville, NJ). Three types of DNA (unmethylated, methylated, and hydromethylated) were applied as inner controls. hMeDIP was then performed using an hMeDIP Kit (Cat No. AF-104-0016; Diagenode) according to the manufacturer's protocol. Briefly, after binding the specific 5-hmC antibodies to hydroxymethylated DNA and bead washes, magnetic immunoprecipitation was performed, and the immunoprecipitated DNA was isolated. Subsequent sequencing analysis for hMeDIP products was conducted with the Ion Proton sequencer (Thermo Fisher Scientific, Waltham, MA). The efficiency of the immunoprecipitation was verified by real-time PCR analysis, using specific primers provided in the kit.
Sequence alignment
Genomic sequences of various organisms, as well as their annotation information, were deposited in UCSC Genome Browser database (Karolchik et al., 2013). The sequencing reads were aligned to the human reference genome (hg19) in UCSC Genome Browser database using bowtie2 (Langmead and Salzberg, 2012) with default parameters. Low mapping quality (MAPQ) was defined based on a threshold of <30. After sequence alignment was completed, the nonunique mapped reads and low-quality mappings were removed.
Identification of DHMRs
Peak calling is one of the first steps for hMeDIP-seq analysis. Peaks were identified using MACS2 version 2.0.10 (Zhang et al., 2008) with default parameters. A stringent p value of <1 × 10−3 was used as the cutoff value.
The peaks identified from all samples were merged, and the read counts of each sample in peaks were calculated using the R/Bioconductor package (Lienhard et al., 2014). The expression values of all reads of each sample were normalized using the reads per kilobase per million (RPKM) method. According to the difference of read counts in samples (the read count was not 0 in at least 1/3 of the sample), the differential 5-hmC peaks (DHMRs) between AFP-negative HCC tissues and adjacent normal tissues were identified by edgeR (Robinson et al., 2010). The cutoff values were |log fold change| > 2 and p < 0.05. If the read counts of sample 1 were higher than those of sample 2, DHMRs were considered highly hydroxymethylated. Otherwise, the peaks were lowly hydroxymethylated. Moreover, the distribution of DHMRs on chromosome of each group (each paired HCC tissue and adjacent normal tissue) was visualized by circos plots using circlize (Gu et al., 2014).
Annotation for DHMRs
The identified DHMRs were then annotated using Chipseeker (Yu et al., 2015). The promoter region of each annotated gene (DHMR-associated genes) was defined as being located from 2500 bp upstream to 500 downstream of the transcription start site.
Functional enrichment analyses
Gene ontology (GO) (Ashburner et al., 2000) is a tool for unifying the biology of large-scale gene sets, mainly including cellular components, molecular function, and biological process terms. Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) pathway analysis is a tool for annotating the molecular interaction networks of large-scale gene sets. In this study, GO and KEGG pathway enrichment analyses for DHMR-associated genes were conducted using clusterProfiler v3.2.1 (Yu et al., 2012). A p < 0.05 from the hypergeometric test was defined as the cutoff value.
Construction of PPI network
PPI relationships between DHMR-associated genes were obtained based on the information of STRING version 10.0 database (Szklarczyk et al., 2015), and required confidence (combined score) >0.4 was set as the threshold value. The PPI network was constructed and visualized using Cytoscape (version: 3.2.0) (Shannon et al., 2003). Degree centrality of each node was analyzed, and the higher node degrees indicated a greater significance of the nodes in the network.
TF prediction analysis
According to the information of TFs from JASPAR_CORE and JASPAR_2014 data in the MotifDB package (Shannon and Richards, 2012), the TFs whose binding sequences were located in the promoter regions of DHMR-associated genes were predicted by the Biostrings package (Pages et al., 2014). Subsequently, the regulatory network of TFs and DHMR-associated genes in promoter regions was constructed and visualized by Cytoscape software.
Results
hMeDIP-Seq analysis
In this study, hMeDIP analysis was performed by incubation with the specific 5-hmC antibodies. Results showed the specificity and efficiency of the enrichment of hydromethylated DNA compared with unmethylated (71.12% vs 0.14%) or methylated (71.12% vs 0.13%) controls (Fig. 1), and the products could be used for subsequent sequencing analysis.
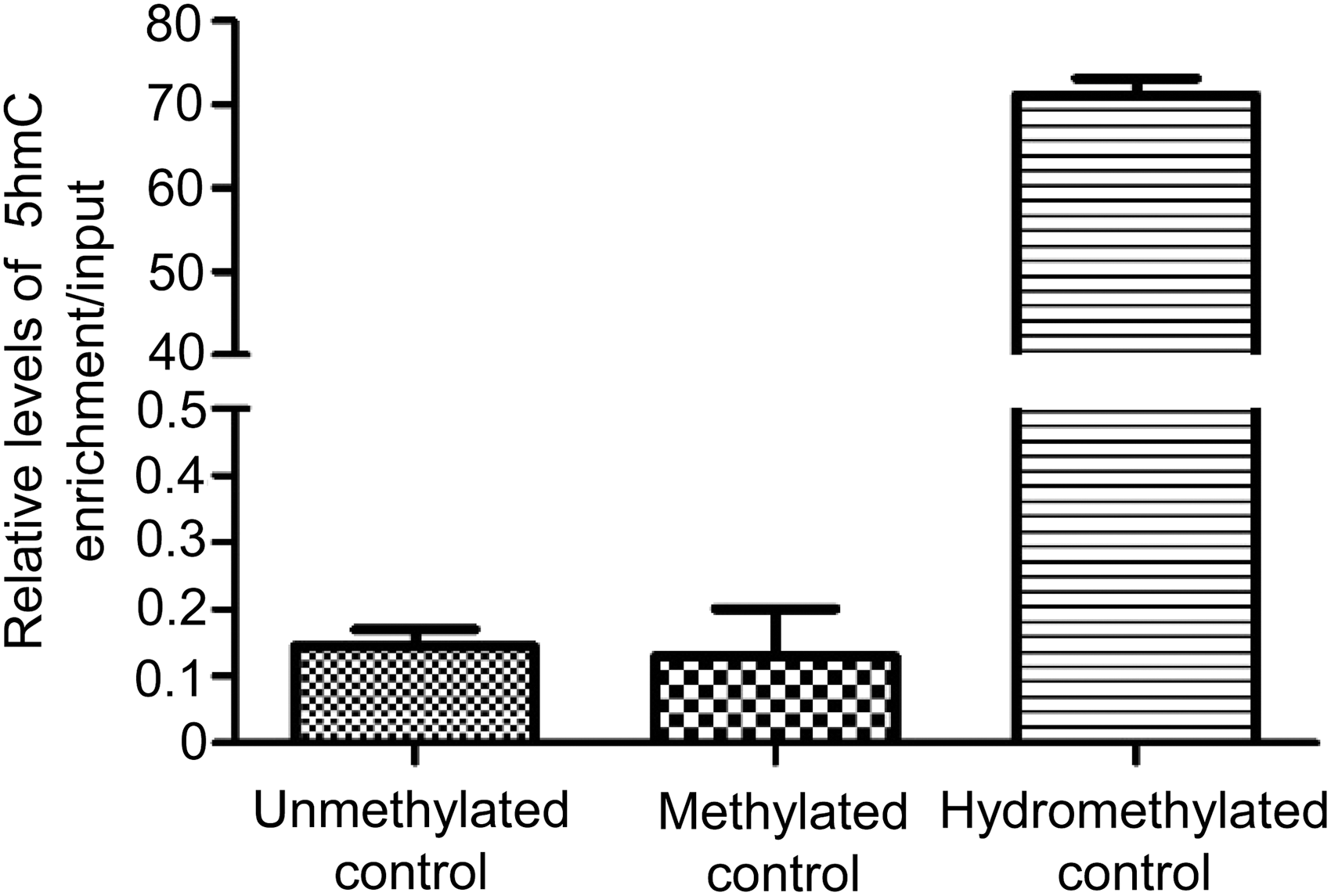
Efficiency of hydroxymethylated DNA immunoprecipitation product enrichment by incubation with the specific 5-hydroxymethylcytosine antibodies.
Sequence alignment
After sequence alignment to the hg19 human reference genome, the mapped rates of all samples were up to 90.16%, and unique mapped rates were higher than 69.25% (Table 1).
Number of Reads Generated by Hydroxymethylated DNA Immunoprecipitation Sequencing for Each Sample
Reads maps onto exactly one genome location.
The proportion of the unique mapped reads in total reads.
C, alpha-fetoprotein-negative hepatocellular carcinoma samples; N, adjacent normal tissue samples.
DHMR analysis
According to the difference of read counts in peaks, 615 DHMRs were identified from AFP-negative HCC tissues compared to adjacent normal tissues. The heatmap for DHMRs is presented in Figure 2, indicating that DHMRs could be used for distinguishing AFP-negative HCC tissues and normal tissue samples. In addition, circos plots displayed the distribution of DHMRs on chromosome of each group (each paired HCC tissue and adjacent normal tissue) (Fig. 3), indicating that the overall differential hydroxymethylation level of AFP-negative HCC tissues was lower than normal tissues, which suggested for less dynamic preprogramming rate for specific genomic region (Li et al., 2016). Furthermore, after peak annotation, the number distribution of DHMR-associated genes in functional elements is displayed in Figure 4. Most DHMRs (95.45%) were distributed in intergenic (downstream, distal intergenic) and gene body (5′UTR, 3′UTR, 1st exon, other exon, 1st intron, other intron), while others occurred at promoter regions (≤3 kb).

The heatmap for the DHMRs. A gradient map represents the hydroxymethylated status from lower to higher (blue to red). Cancer: AFP-negative HCC tissue samples; normal: adjacent normal tissue samples. AFP, alpha-fetoprotein; DHMR, differentially hydroxymethylated region; HCC, hepatocellular carcinoma.

Circos plots displayed the distribution of DHMRs on chromosome of each group (each paired HCC tissue and adjacent normal tissue). From the outside to the inside, each circle represents the name of the chromosome, chr1-22+x+y; the stripe structure of chromosome; the line graph for displaying the expression level of each normal tissue sample (CPM value); the line graph for displaying the expression level of each AFP-negative HCC tissue sample (CPM value). CPM, counts per million.
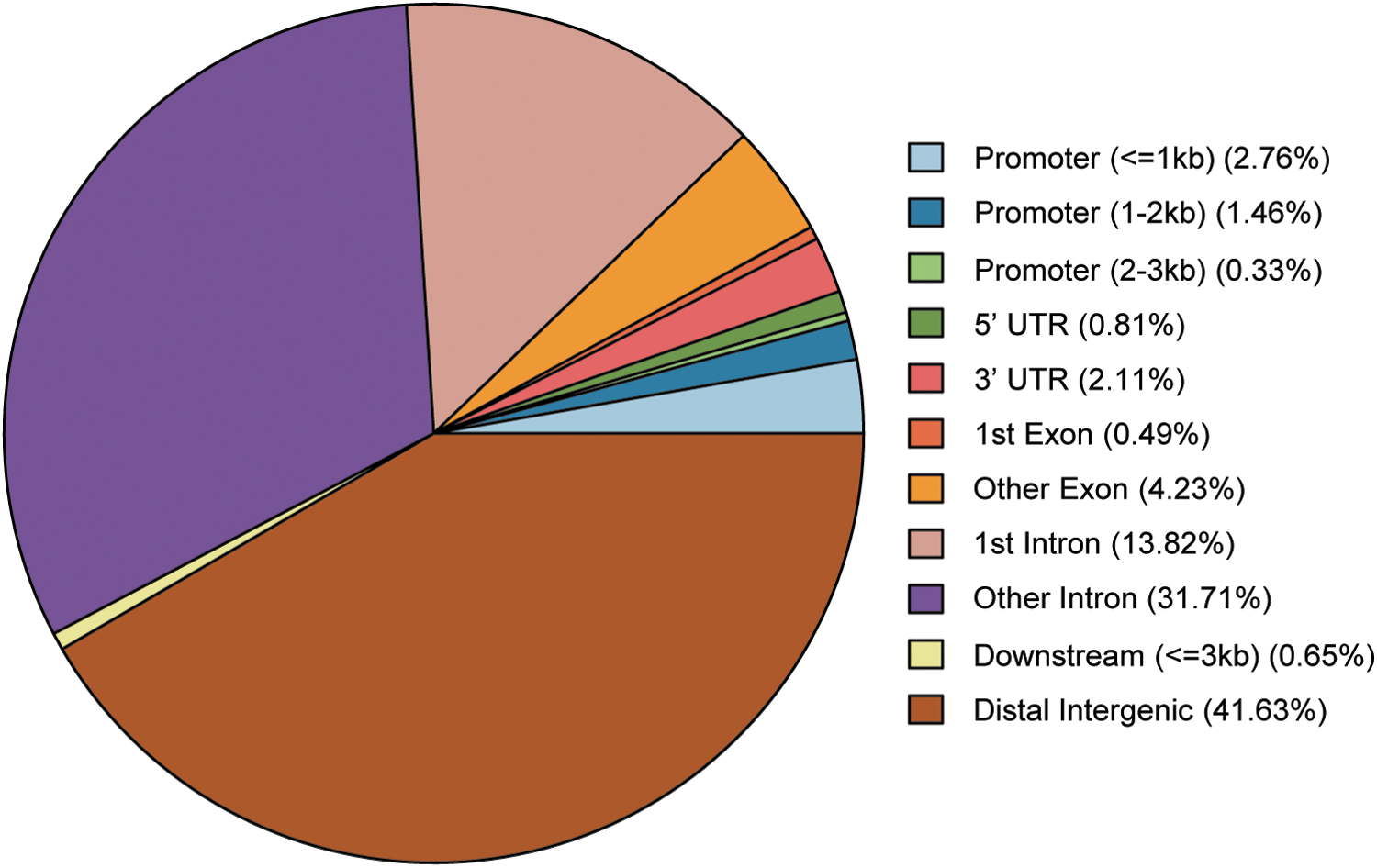
The number distribution of DHMR-associated genes in functional elements. DHMRs locate most frequently at distal intergenic, other intron, and 1st intron.
Functional enrichment analyses for DHMR associated genes
According to the peak annotation results, GO and KEGG pathway functional enrichment analyses for DHMR-associated genes, except distal intergenic regions, were performed. These DHMR-associated genes were found to be significantly enriched in GO functions associated with actin binding, cell leading edge, and blood vessel morphogenesis (Fig. 5A). KEGG pathway enrichment analysis showed that the DHMR-associated genes were remarkably enriched in the MAPK signaling pathway, axon guidance, and neuroactive ligand-receptor interaction (Fig. 5B).
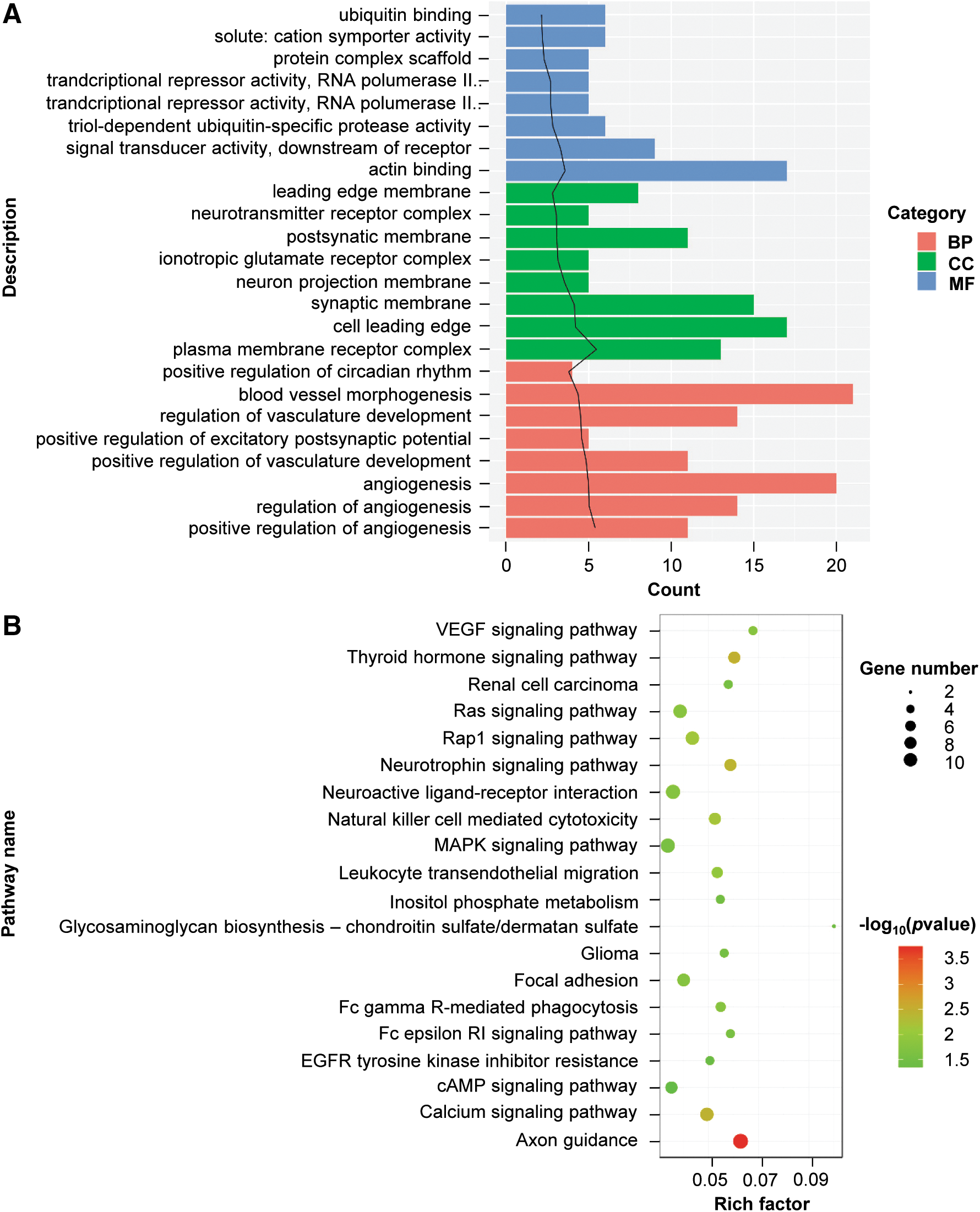
Gene ontology
PPI network analysis
According to the information from STRING, PPI network analysis for DHMR-associated genes was performed. As presented in Figure 6, 176 nodes and 254 interactions were included in the PPI network. By analyzing degree centrality, the hub nodes with the top 10 degrees were PH domain and leucine-rich repeat protein phosphatase 1 (PHLPP1, degree = 25); SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 2 (SMARCA2, degree = 14), discs large MAGUK scaffold protein 2 (DLG2, degree = 12); glutamate metabotropic receptor 1 (GRM1, degree = 11); protein kinase C alpha (PRKCA, degree = 10); P21 (RAC1) activated kinase 1 (PAK1, degree = 9); SMAD family member 2 (SMAD2, degree = 9); phospholipase C gamma 1 (PLCG1, degree = 9); protein kinase D1 (PRKD1, degree = 8); RAP1B, member of RAS oncogene family (RAP1B, degree = 8); and BRCA1, DNA repair associated (BRCA1, degree = 8).
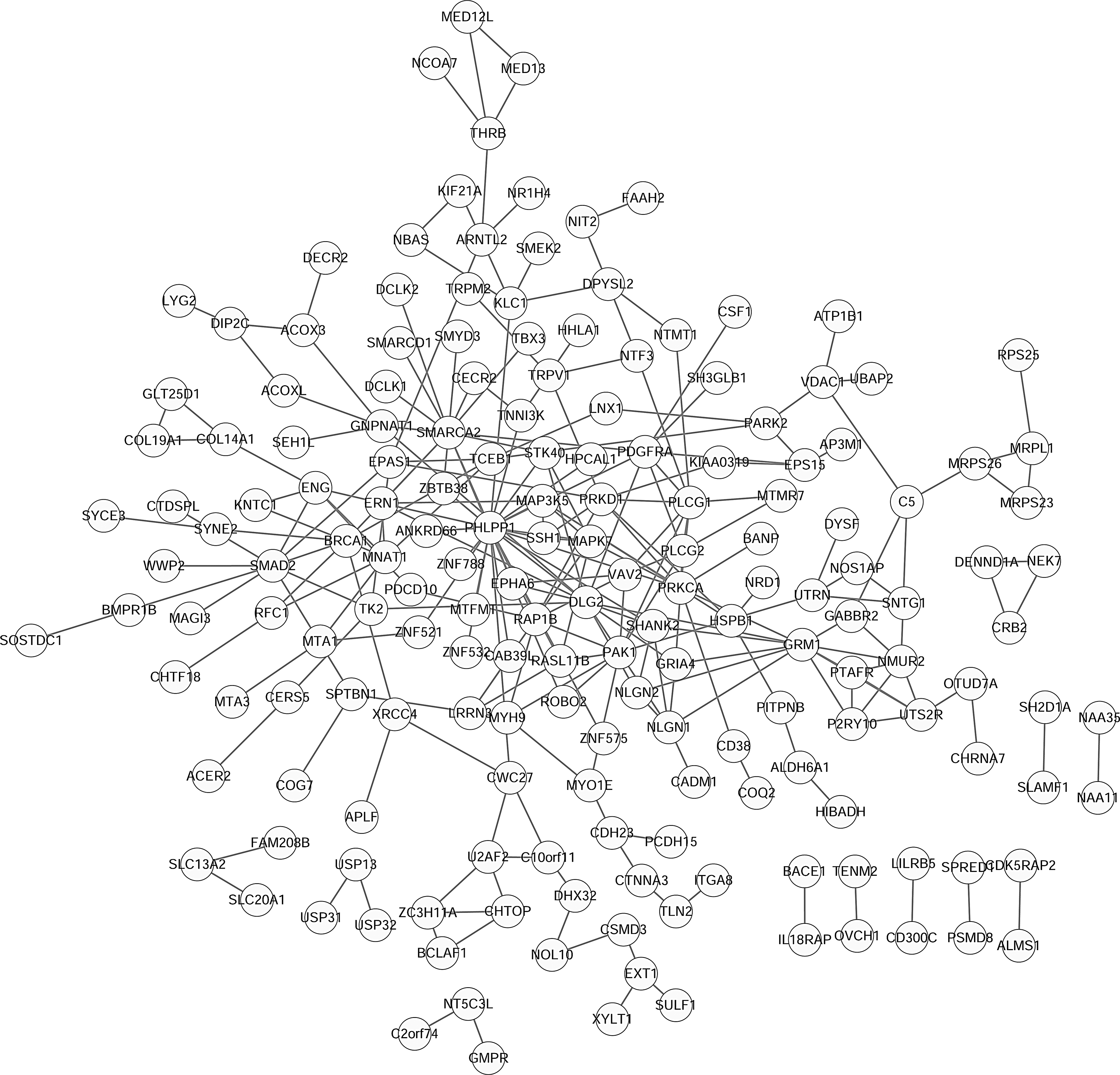
Protein–protein interaction networks for DHMR-associated genes. The circle nodes are gene/proteins, and the lines represent interactions.
TF prediction analysis
By prediction, 13 TFs were identified whose binding sequences with DHMR-associated genes were also located in the promoter region. Among them, nuclear factor I C (NFIC), GATA binding protein 3 (GATA3), Spi-1 proto-oncogene (SPI1), myeloid zinc finger 1 (MZF1), GATA Binding Protein 2 (GATA2), and ETS proto-oncogene 1 (ETS1) regulated no less than three DHMR-associated genes, as nodes of the DHMR-TF regulatory network (Fig. 7). Among the 23 relative DHMR-associated genes, replication factor C subunit 1 (RFC1), 2,4-dienoyl-CoA reductase 2 (DECR2), solute carrier family 15 member 2 (SLC15A2), and MCM3AP antisense RNA 1 (MCM3AP-AS1) were interconnected by multi TFs. These TFs and relative gene products might play pivotal roles in AFP-negative HCC progression.
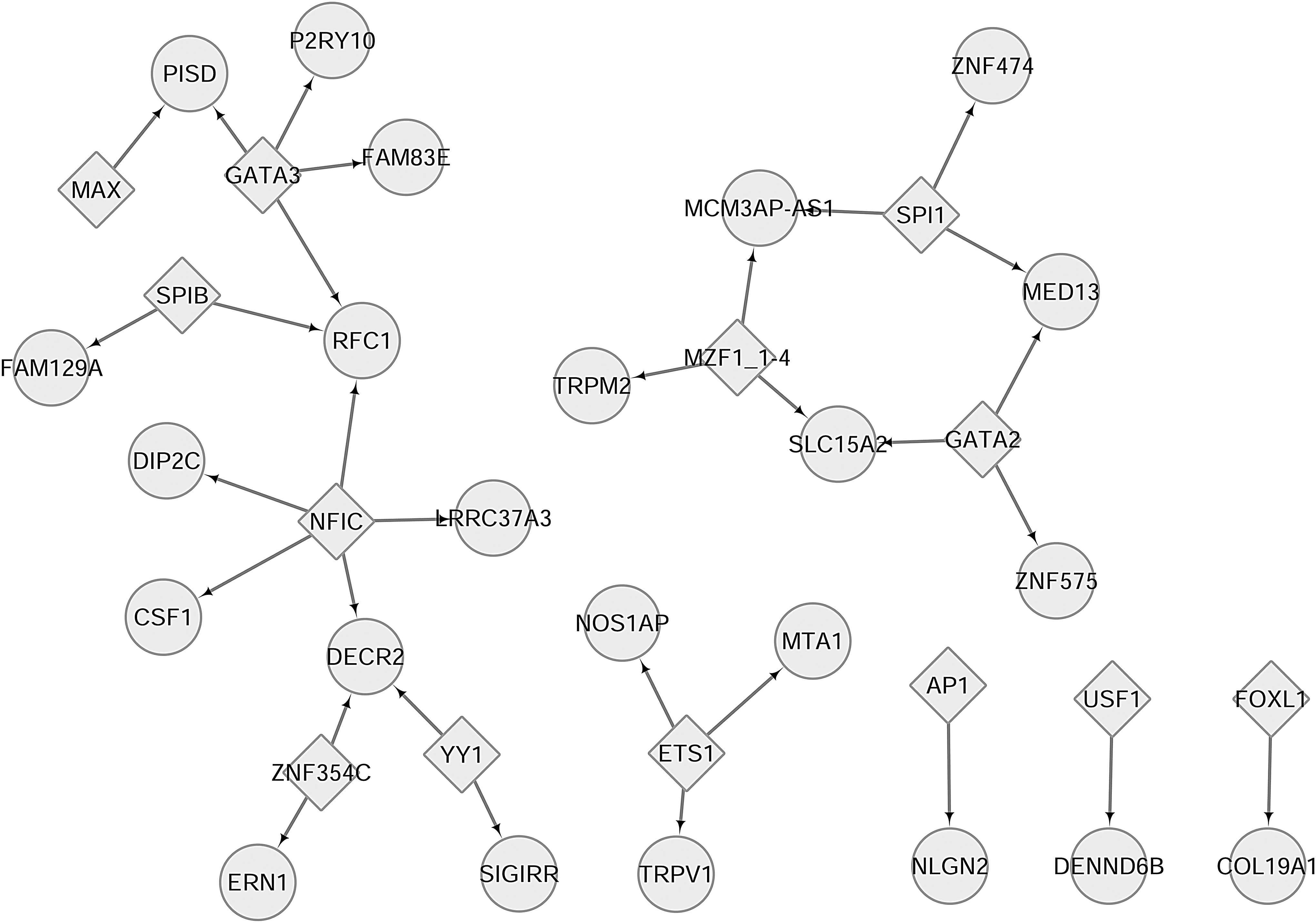
DHMR-associated gene-TF regulatory network. The rhombic nodes are TFs, and the circular nodes are DHMR-associated genes. TF, transcription factor.
Discussion
Methylation in promoter regions is widely involved in cancer development through regulating gene expression (Jones and Baylin, 2002). Identification of methylated genes will facilitate the discovery of new mechanisms underlying tumor initiation and progression. In the present study, 615 DHMRs were screened by comparing AFP-negative HCC tissues to adjacent normal tissues. Key DHMR-associated genes such as PHLPP1 and SMARCA2 had higher degrees in the PPT network and might be hub nodes. Furthermore, TF prediction analysis showed that TFs, such as NFIC and GATA3, could regulate more DHMR-associated genes. These key hydroxymethylated genes and TFs may be involved in the development of AFP-negative HCC and merit further discussion.
PHLPP1 is a protein-Ser/Thr phosphatase that is a confirmed negative regulator of the PI3-kinase/Akt pathway in cancer development (O'Neill et al., 2013). Accumulating evidence has revealed that PHLPP1 can exert tumor suppressor functions in several cancers (Nitsche et al., 2012; Yu et al., 2018). Lower expression of PHLPP1 can predict poor prognosis of gastric cancer (Hou et al., 2015). Epigenetic alterations, including DNA methylation and microRNAs, are involved in the downregulation of PHLPP1 expression (Liao et al., 2013; Dong et al., 2014). However, there have been limited reports about the role of PHLPP1 in HCC, let alone in AFP-negative HCC.
In this study, we only annotated the DHMR-associated genes in the promoter region. PHLPP1 was identified as having the highest degree in PPI network, which pointed to a potential role of dysregulated PHLPP1 by DNA hydroxymethylation in the development of AFP-negative HCC. Given the key roles of PHLPP1 in other cancers, our data suggest that DNA hydroxymethylation of PHLPP1 may be a novel epigenetic regulation mechanism of this gene and a potential biomarker of AFP-negative HCC.
SMARCA2 is also identified a hub node in PPI network with higher degrees. Hoffman et al. (2014) reported that BRM/SMARCA2 is a synthetic lethal target for BRG1-deficient cancers. Moreover, high expression of SMARCA2 is correlated with good prognosis in diverse types of tumors, including liver hepatocellular carcinoma (Guerreromartínez and Reyes, 2018). Based on our results, we speculate that DNA hydromethylation-mediated dysregulation of SMARCA2 may contribute to the development of AFP-negative HCC and predict a poor prognosis.
Furthermore, TFs are trans-regulatory factors that facilitate or repress the transcription of their target genes by binding to specific DNA sequences located within promoters (Vaquerizas et al., 2009). Therefore, identification of key TFs that can bind to DHMR-associated genes will facilitate the elucidation of the key mechanism involved in the pathogenesis of AFP-negative HCC. In this study, key TFs, such as NFIC and GATA3, regulated most DHMR-associated genes.
NFIC belongs to the NFI family that can impact developmental functions in the transcriptional modulation of many cell types (Gronostajski, 2000). Lee et al. (2015) demonstrated that NFIC could regulate Krüppel-like factor 4 and E-cadherin, thus suppressing mesenchymal–epithelial transition in breast cancer. High NFIC expression is also correlated with better prognosis in breast cancer patients (Nilsson et al., 2010). GATA3 is shown to function as a transcriptional activator in the luminal epithelial cells in the breast (Mehra et al., 2005). It has been confirmed as a prognostic marker of breast cancer (Mehra et al., 2005; Yoon et al., 2010). Nevertheless, the roles of NFIC and GATA3 in the pathogenesis of AFP-negative HCC are largely unknown. Despite these findings in breast cancer, based on our results, we speculate that NFIC and GATA3 may play a key role in AFP-negative HCC through binding to specific DNA sequences of DHMR-associated genes located within promoters. Further research is needed to confirm our speculations.
However, there are some limitations in this study. First, the sample size was small, which may be attributed to the high cost of hMeDIP-seq. Second, the expression of key hydroxymethylated genes and TFs was not detected by experiments such as quantitative PCR. Future studies with complete biochemical approaches using larger sample sizes are required to confirm our observation.
Conclusion
In conclusion, we performed a hMeDIP analysis to study the whole-genome DNA hydroxymethylation status of AFP-negative HCC. Our findings reveal that 615 DHMRs exist in AFP-negative HCC tissues. DHMR-associated genes are significantly enriched in GO functions associated with actin binding, cell leading edge, and blood vessel morphogenesis and pathways of MAPK signaling, axon guidance, and neuroactive ligand-receptor interaction. The key hydroxymethylated genes, such as PHLPP1 and SMARCA2, as well as TFs like NFIC and GATA, may be involved in the development of AFP-negative HCC. These molecules may be promising biomarkers for the diagnosis and prognosis of AFP-negative HCC.
Prospective studies with a larger sample size will elucidate the role of differential epigenetically modified target genes in AFP-negative HCC pathogenesis. Further experimental studies in vitro and in vivo will clarify the function of these genes in AFP-negative HCC carcinogenesis.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
The analysis was sponsored by National Natural Science Foundation of China (81502059, 81472582, and 81472551), Shanghai Rising-Star Program (16QB1402900), and Shanghai New Interdisciplinary Subject Funding Program of TCM: Molecular Hepatology of TCM (2017–2020).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.