
Abstract
Propofol is a widely used intravenous agent for the induction and maintenance of anesthesia. An increasing number of studies have shown that propofol modulates autophagy, which is an evolutionarily conserved catabolic process that maintains cellular homeostasis by degrading long-lived proteins and damaged cellular proteins or organelles. Extensive studies have been performed to better understand the regulation of autophagy by propofol, the majority of which have demonstrated that the effects of propofol on autophagy are beneficial to organs and tissues. In this review, we retrospectively analyzed studies to assess the effects of propofol on autophagy in different diseases and evaluated the underlying mechanisms.
Introduction
Autophagy
Autophagy, the term coined by Christian de Duve in 1963, refers to a conserved intracellular pathway that delivers cytosolic constituents to lysosomes for degradation through double-membrane autophagosomes (Ohsumi, 2014). As a dynamic circulation system, autophagy plays an important role in maintaining cellular homeostasis (Weidberg et al., 2011). At present, the function of autophagy can be classified into two principal categories: a physiologic stress response and intracellular quality control mechanism (Mizushima and Komatsu, 2011). The former serves as a salvage mechanism to sustain core metabolic functions during pathologic stresses, such as starvation, infection, ischemia, and cancer. The second function is to play a vital quality control function in the cell by degradation and recycling the damaged and redundant organelles, long-lived proteins, and abnormal macromolecules (Murrow and Debnath, 2013).
From yeast to mammals, there are three primary types of autophagy: microautophagy, macroautophagy, and chaperone-mediated autophagy (Klionsky, 2005). Microautophagy refers to the direct uptake by lysosomes of cytoplasmic contents through invagination of the lysosomal membrane (Marzella et al., 1981). Macroautophagy involves the sequestering of cellular constituents in double-membrane autophagosomes that subsequently fuse with lysosomes to form an autolysosome that degrades the cellular debris (Yang and Klionsky, 2010). In chaperone-mediated autophagy, the cargo is selectively delivered to lysosomes upon recognition by hsc70 and binding to the LAMP2A receptor on the lysosome (Massey et al., 2004). All three types of autophagy lead to degradation of cellular contents and release of the breakdown products back into the cytosol for reuse by the cell. Among the three types of autophagy, macroautophagy has become the best characterized form of autophagy and is the focus of this review. Therefore, herein we refer to macroautophagy simply as autophagy.
The autophagy mechanism has four primary stages: induction of autophagosome formation, nucleation of the phagophore, elongation of the phagophore, autophagosome completion and fusion with the lysosome. First, the activation of autophagosome formation in macroautophagy is controlled by the ULK1 complex (Ulk1/2-Atg13-FIP200-Atg101 complex) in mammals (known as the Atg1 complex in yeast) (Hurley and Young, 2017). In the presence of nutrients, the active mammalian target of rapamycin (mTOR) combines directly with the ULK1 complex, and phosphorylates Ulk1/2 and Atg13 and results in the inhibition of Ulk1/2 kinase activity and autophagy induction. In contrast, inactive mTOR separates from ULK1 and dephosphorylates Ulk1/2 and Atg13 under the nutrient-poor conditions, thereby activating Ulk1/2 kinase activity leading to the initiation of autophagy (Hosokawa et al., 2009). Second, nucleation of the phagophore is mediated by the class III phosphatidylinositol 3-kinase (PI3KC3) complex containing ATG14L, Beclin-1, and VPS34/15 (equivalent to the Atg14/Atg6/Vps34/15 in yeast) (Itakura and Mizushima, 2010). Under nutrient-rich conditions, the antiapoptotic protein B cell lymphoma/leukemia-2 (Bcl2) combines with Beclin-1 and suppresses the association of Beclin-1 with Vps34, which results in the inhibition of autophagy. Beclin 1 is released upon phosphorylation of Bcl-2 by JNK1 (Furuya et al., 2005). In the third stage, the expansion of the phagophore is closely associated with two ubiquitin-like protein systems (Weidberg et al., 2011). The first system involves formation of the Atg12-Atg5-Atg16 complex and the second is the Atg8/LC3 system, both of them play important roles in the elongation of the phagophore (Wilson and Gelb, 2002). As for the last stage, the expanding phagophore matures and becomes a complete autophagosome, which fuses with an endosome and/or lysosome to become an autolysosome, ultimately leading to the degradation of autolysosomal contents (Monastyrska et al., 2009).
Propofol
Propofol (2,6-diisopropylphenol), commonly dubbed as “milk of anesthesia,” is an intravenous anesthetic agent widely used for induction and maintenance of anesthesia during surgical performance, and is also used in the intensive care unit for long-term sedation (Marik, 2004). Similar to other intravenous anesthetic agents like benzodiazepines and barbiturates, propofol exerts its hypnotic actions by activating the central inhibitory neurotransmitter gamma-aminobutyric acid in the central nervous system (Brohan and Goudra, 2017). Furthermore, propofol has many pharmacological advantages over other anesthetic agents such as rapid effect, short action, and fewer side effects like postoperative nausea, which has enabled it to become one of the popular intravenous anesthetic agents in modern medicine (Bang et al., 2016). Propofol still has some clinical drawbacks including microbial infection (Langevin et al., 1999), hyperlipidemia (Wolf et al., 2001), pain upon injection (Nathanson and Gajraj, 1998), and propofol infusion syndrome (Hemphill et al., 2019). Propofol can cause hypotension, arrhythmia, and respiratory depression in a dose-dependent manner (Langley and Heel, 1988; Roekaerts et al., 1993).
In addition to its anesthetic purpose, propofol exerts other effects such as affecting cardioprotection (Noh et al., 2010), neuroprotection (Cui et al., 2012), lung protection (Gu et al., 2012), and antitumor activity (Du et al., 2013), and protecting other organs and tissues in a certain context (Kim et al., 2016). Propofol has been claimed to protect animals against oxidative injury and ischemia/reperfusion (I/R), and hypoxia/reperfusion (H/R) injury, whereas other studies have shown that propofol exerts either a detrimental or beneficial effect on several different types of cells such as neural progenitor cells (NPCs) (Qiao et al., 2017), DT40 or SH-SY5Y cells (Ren et al., 2017), and C2C12 myoblast cells (Chen et al., 2018b) In various cell culture models, high-pharmacologic concentrations of propofol adversely affect cell viability by impairing autophagy flux (Qiao et al., 2017; Ren et al., 2017). Other intravenous anesthetics, such as ketamine and barbiturate, and volatile anesthetics also exert the protection of organs and tissues. Increasing evidence indicates volatile anesthetics protect the myocardium against I/R injury as shown in both animal and clinical studies (Tanaka et al., 2004; Van Allen et al., 2012). In this review, we focus on the effects of propofol on autophagy on different diseases and its underlying mechanisms (Table 1).
Summary of the Recent Studies About How Propofol Affects Autophagy
AMPK, AMP-activated protein kinase; BMP-2, bone morphogenetic protein-2; DRAM, damage-regulated autophagy modulator; ECS, electroconvulsive shock; ECT, electroconvulsive therapy; ERK, extracellular signal-regulated protein kinases; hFOB, human fetal osteoblasts; HIF-1α, hypoxia-inducible factor 1α; H/R, hypoxia/reperfusion; I/R, ischemia/reperfusion; JNK, c-Jun NH2-terminal kinase; LPS, lipopolysaccharide; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor-κB; OGD, oxygen and glucose deprivation; TGF-β1, transforming growth factor-β1.
Protective effect of propofol on myocardial damage
A growing body of research suggests that autophagy plays a crucial role in maintaining the cardiac homeostasis and function by eliminating the misfolded proteins and damaged organelles (Nakai et al., 2007; Ikeda et al., 2015). Furthermore, autophagy also reduces the cardiac damage during many pathologic stresses, such as ischemia and hypoxia. During myocardial ischemia, autophagy is motivated by activating the AMP-activated protein kinase (AMPK) pathway and PERK/ATF4 pathway (Sciarretta et al., 2013) and inhibiting the Rheb/mTORC1 pathway. Destruction of these pathways leads to the disruption of autophagy and facilitates myocardial injury (Matsui et al., 2007; Sciarretta et al., 2012, 2013). However, during reperfusion, autophagy is excessively activated and causes myocardial injury because of the massive accumulation of Beclin-1 in the heart in a ROS-dependent manner (Matsui et al., 2007). Recent studies have shown that some anesthetics can reduce myocardial injury by the regulation of autophagy, such as sevoflurane and propofol (Tanaka et al., 2004). We mainly discuss the effect of propofol on autophagy in myocardial I/R and H/R (Fig. 1).
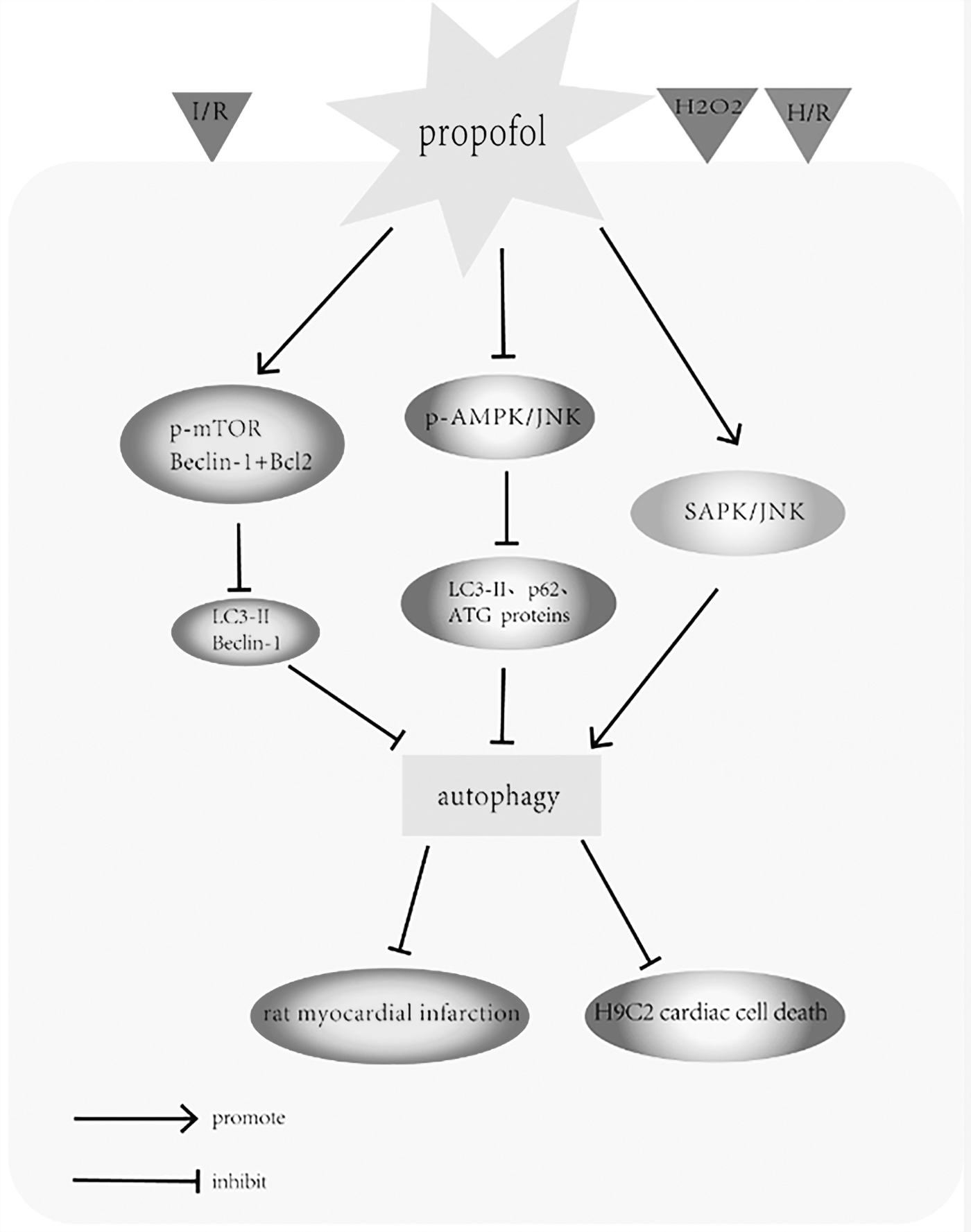
The protective effect of propofol on myocardial damage. The diagram illustrates the modulatory effects of propofol on rat myocardium and H9C2 cardiac cells. Stimuli are represented by triangles of different shades. Lines with arrowheads and dots at the end represent “promotion” and “inhibition.” Propofol preconditioning dramatically reduced the infarction size in the rat myocardium by inhibiting autophagy through promotion of phosphorylation in the mTOR pathways and by increasing Beclin-1/Bcl-2 interactions during I/R injury. Similarly, propofol preconditioning reduced H2O2-induced H9C2 cardiac cell death through mitigating autophagy by suppressing phosphorylation in the AMPK and JNK pathways. However, propofol postconditioning improved cell survival through the induction of autophagy in H9c2 cardiac cells by activating the SAPK/JNK survival signaling pathway. AMPK, AMP-activated protein kinase; I/R, ischemia/reperfusion; JNK, c-Jun NH2-terminal kinase; mTOR, mammalian target of rapamycin.
To explore the influences of propofol in the I/R-injured rat myocardium, rats were randomly divided into sham procedure, or left coronary artery occlusion with 25 min of ischemia and reperfusion with saline, propofol, or intralipid for 24 h by Noh et al. (2010). They found that the infarction size in the myocardium was increased during I/R injury, but was dramatically reduced in rats treated with propofol compared with untreated rats. By electron microscope analysis (TEM), they observed a great number of autophagic vacuoles (autophagosomes and autophagolysosomes) in the cardiomyocytes of I/R-injured rats but rarely in I/R-injured rats treated with propofol. They also found that the autophagy-related protein LC3-II was upregulated in the I/R-injured myocardium, but was significantly downregulated in the myocardium of rats treated with propofol. Furthermore, propofol promoted the phosphorylation of mTOR during I/R, increased Beclin-1/Bcl-2 interaction in cells, and inhibited the expression of Beclin-1. These data clearly suggest that propofol prevents myocardial damage after I/R by inhibiting autophagy (Noh et al., 2010).
Although Noh et al. asserted the concepts of “autophagic cell death” in their article, we should know this concept is still controversial nowadays. The term “autophagic cell death” was initially referred to a morphology that cells die with the emergence of autophagosomes (Schweichel and Merker, 1973). In many cases, it is agreed that this autophagic cell death is cell death accompanied by autophagy rather than cell death driven by autophagy (Kroemer and Levine, 2008). Furthermore, recent studies suggest that autophagy may promote other types of cell death, such as apoptosis and necrosis, rather than playing as a primary type of death. So, the cell death in the presence of autophagy could not simply be defined as autophagic cell death (Doherty and Baehrecke, 2018).
Similarly, to investigate the mechanism underlying the protective effect of propofol in oxidative stress-induced cell death in cardiomyocytes, another study by Ha et al. (2012) suggested that severe oxidative stress generally induces both apoptotic cell death and autophagy in H9C2 cells. Autophagy has been proved to accelerate apoptosis by eliminating the antiapoptotic and cell survival factors (Doherty and Baehrecke, 2018). These authors further demonstrated that propofol protected cardiomyocytes from H2O2-induced cell death by mitigating autophagy. They observed that acidic autophagic vacuoles decreased to a greater extent in the propofol-treated H2O2 group than in the untreated H2O2 group. Furthermore, many autophagy-related proteins such as LC3-II, ATG proteins, p62, AMPK, and c-Jun NH2-terminal kinase (JNK) were significantly deactivated after pretreatment with propofol. Thus, propofol blocked oxidant stress-induced autophagy in cardiomyocytes by suppressing the phosphorylation of the AMPK and JNK pathways (Ha et al., 2012). However, details of the underlying mechanisms promoting the protective effect of propofol in cardiomyocytes require further elucidation.
Of interest, Li et al. (2018) studied the effect of propofol postconditioning on H/R injury in H9C2 cardiac cells and found that it improved cell survival through the induction of autophagy in H9c2 cardiac cells through activation of the SAPK/JNK survival signaling pathway. These conflicting findings may be because of the different interventions and differing durations of propofol application in the experimental environment. Nonetheless, it can be seen that propofol exerts its protective effect on myocardial cells through different autophagy mechanisms, and this aspect deserves further study.
Protective effect of propofol on neuronal injury
It is well known that autophagy is a double-edged sword in the nervous system after traumatic brain injury and ischemic stroke. Some studies suggest that autophagy is neuroprotective and beneficial for cell survival (Carloni et al., 2010), whereas other studies deem autophagy is neurotoxic (Wen et al., 2008). The controversial findings can be explained by the variation of autophagy flux. The autophagy exerts neuroprotective effects if the autophagy flux is unimpeded, but it becomes detrimental when autophagy flux is interrupted (Lipinski et al., 2015). The dysfunction of autophagy flux is found to participate in the development of neurological disorders, such as Huntington's disease, Alzheimer's disease (AD), and Parkinson's disease (Lee et al., 2011; Tanik et al., 2013). In this part, we mainly discuss the effect of propofol on autophagy in the central nervous system. It has been shown that I/R, oxygen and glucose deprivation (OGD), and electroconvulsive therapy (ECT) result in damage in the nervous system, whereas the neuroprotective effect provided by propofol has been confirmed in recent years (Fig. 2).
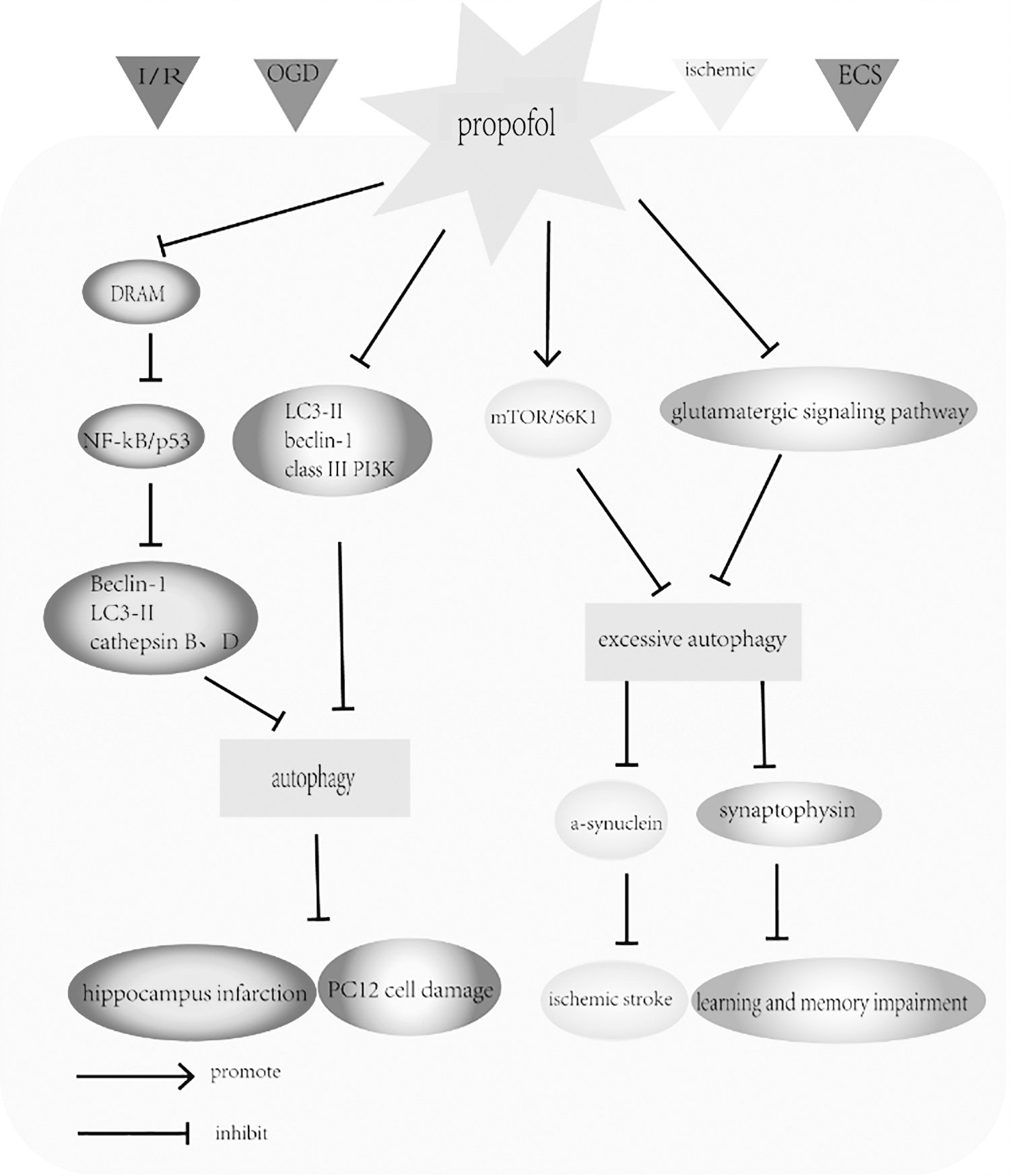
The protective effect of propofol on neuronal injury. The schematic diagram illustrates the modulatory effects of propofol on rat hippocampus and neuronal PC12 cells. The administration of propofol effectively decreased infarct size in the rat hippocampus by inhibition of autophagy by suppressing the DRAM, NF-κB and P53 signaling pathways during I/R injury. Propofol also reduced OGD-induced PC12 cell damage by inhibition of autophagy by decreasing the expression of LC3-II, Beclin-1, and class III PI3K. Furthermore, propofol administration decreases the neurotoxic aggregation of a-synuclein and reduces the excessive autophagy by increasing the mTOR and S6K1 signaling pathway in a mouse model of ischemic stroke. In addition, propofol suppressed the overexpression of autophagy by inhibiting the glutamatergic signaling pathway and then suppressed ECS-induced overactivation of synaptophysin to reduce the learning and memory impairment after ECT in depressed rats. DRAM, damage-regulated autophagy modulator; ECS, electroconvulsive shock; ECT, electroconvulsive therapy; OGD, oxygen and glucose deprivation.
Cui et al. (2012) investigated the neuroprotection mediated by propofol in neuronal PC12 cells after OGD and in rat pyramidal hippocampal neurons after I/R insults. After 6 h of OGD, PC12 cell viability was sharply decreased, but the amounts of autophagosomes and autolysosomes, autophagy-related proteins such as LC3-II, Beclin-1, and class III PI3K were increased, whereas propofol treatment significantly reversed the situation. These changes suggested that propofol in OGD-injured PC12 cells did indeed inhibit autophagy. After 10-min ischemia generated by transient two-vessel occlusion, rat hippocampal pyramidal neurons were damaged with excessive autophagy, but propofol treatment effectively decreased infarct size in the hippocampus by ameliorating autophagy (Cui et al., 2012). In later research, these same authors also observed that the nuclear factor-κB (NF-κB) and p53 signaling pathways play crucial roles in the autophagic mechanism after cerebral ischemic exposure in the rat hippocampus (Cui et al., 2013). As they observed, propofol exerted neuroprotective effects and reversed I/R-induced upregulation of damage-regulated autophagy modulator (DRAM). DRAM is regarded as a novel regulator of autophagy that links p53 to autophagy (Garufi et al., 2017). It was observed that the I/R-induced elevations of LC3-II, Beclin 1, active cathepsin D, and cathepsin B protein levels were reversed in the hippocampus after the administration of propofol. The NF-κB inhibitor SN50 and the p53-specific inhibitor pifithrin-α also suppressed the increase in DRAM, Beclin 1, LC3-II, active cathepsin D, and cathepsin B induced by I/R. These results suggest that propofol prevents autophagy activation in response to cerebral ischemia, likely through the DRAM-mediated NF-κB/p53 signaling pathway (Cui et al., 2013). In addition, Wang et al. (2019a) indicated that propofol administration decreases the neurotoxic aggregation of a-synuclein and reduces the excessive autophagy by increasing the mTOR and S6K1 (ribosomal protein S6 kinase beta-1) signaling pathway in a mouse model of ischemic stroke. Aggregates of a-synuclein, consisting of higher level oligomers and insoluble fibrils, are proved to be implicated in neurotoxicity and exacerbate neuronal damage after acute ischemia (Winner et al., 2011; Pieri et al., 2012). In their study, propofol treatment significantly decreased the infarct area, attenuated the neurological deficits, and improved the outcome of acute ischemic stroke (Wang et al., 2019a). However, we should consider that propofol may be only secondarily decreasing autophagy because of reduction of cell stress; thus, autophagy as a cytoprotective mechanism is no longer necessary in these already rescued cells.
ECT is an effective treatment for depression, but it can result in learning and memory impairment (Pagnin et al., 2004). Li et al. (2012) demonstrated that propofol can alleviate ECT-induced learning and memory impairment in depressed rats by activating hippocampal CaMKIIα (calcium/calmodulin-dependent protein kinase IIα) in the hippocampus. The authors showed that propofol could mitigate ECT-induced impairments in learning and memory function by suppressing the overexpression of autophagy. They found that propofol not only suppressed electroconvulsive shock (ECS)-induced overactivation of synaptophysin, reduced synaptic overgrowth, and restored synaptic homeostasis, but also partially inhibited ECS-induced overexpression of glutamate, by which it suppresses the excessive activation of autophagy. These findings suggest that propofol effectively relieves learning and memory impairments after ECT (Li et al., 2016). Nonetheless, the authors cannot ignore the consideration that overexpression of autophagy can be because of the futile effort of cells trying to survive and propofol is indirectly decreasing autophagy because of reduction of cell stress.
Recent studies suggested that propofol appears in dual effects of cytoprotection and cytotoxicity depending on the concentration and exposure duration (Ren et al., 2017). Using cortical-derived NPCs, Qiao et al. (2017) demonstrated that propofol affected neurodegeneration and neurogenesis by regulating intracellular calcium homeostasis. They suggested that a clinically relevant dose of propofol promoted cell viability and proliferation by adequately activating autophagy through moderate activation of ryanodine receptors (RyRs) and the inositol 1,4,5-trisphosphate receptor (InsP3R) (Qiao et al., 2017). InsP3R is situated on the membrane of the endoplasmic reticulum (ER) and type-1 InsP3Rs (InsP3R-1) in particular play vital roles in many pathological conditions among the three subtypes of InsP3R. Elevation of cytoplasmic Ca2+ concentration from the ER through InsP3Rs-1 has been shown to irritate autophagy through an mTOR-dependent pathway (Wagner and Yule, 2012; Ren et al., 2017). However, cell cytotoxicity was induced in high pharmacological concentrations and prolonged duration of propofol, which results in abnormal elevation of cytoplasmic Ca2+ concentration by overactivation of InsP3R and RyRs and impair autophagy flux by reducing ATP production, increasing lysosomal pH, and subsequently impairing lysosome and autolysosome function, eventually promote cell death or neurodegeneration (Ren et al., 2017). Impairment of autophagy function contributes to neuropathology in AD (Nixon and Yang, 2011), which may result in the accumulation of amyloid or tau pathology (Hamano et al., 2008; Yang et al., 2011) and increase neurodegeneration (Komatsu et al., 2006). To model AD cells, rat pheochromocytoma cells (PC12) were transfected with point-mutated (L286V) presenilin 1 (PS1). Yang et al. (2019) found that propofol affects cell activity in a dose-dependent manner. Propofol at clinically relevant concentrations (1–20 μ) moderately activates InsP3R and RYR, maintaining normal lysosome H+/Ca2+ homeostasis and supporting cell survival. Nevertheless, propofol at pharmacological concentrations (>50 μ) sensitized AD cells to cause the overactivation of InsP3Rs and RYRs and abnormal elevation of cytosolic Ca2+ concentration, which destroy the lysosome H+/Ca2+ homeostasis and impair the lysosome acidity and autophagy function, eventually inducing cell death (Yang et al., 2019).
Protective effect of propofol on pulmonary injury
Autophagy also plays a homeostatic role in lung epithelia and disruption of autophagy in the lung participates in the development of pulmonary diseases, such as airway hyperresponsiveness and cystic fibrosis (Inoue et al., 2011; Racanelli et al., 2018). As discussed previously, propofol provides benefits of cardioprotection and neuroprotection by regulation of autophagy. In addition, other researchers have provided new insights into lung protection afforded by propofol. Previous study found that the antioxidative sedative propofol could improve recovery from paraquat acute toxicity in A549 cells (Ariyama et al., 2000). In this study, we mainly discuss whether propofol can exert lung protection by autophagy (Fig. 3).
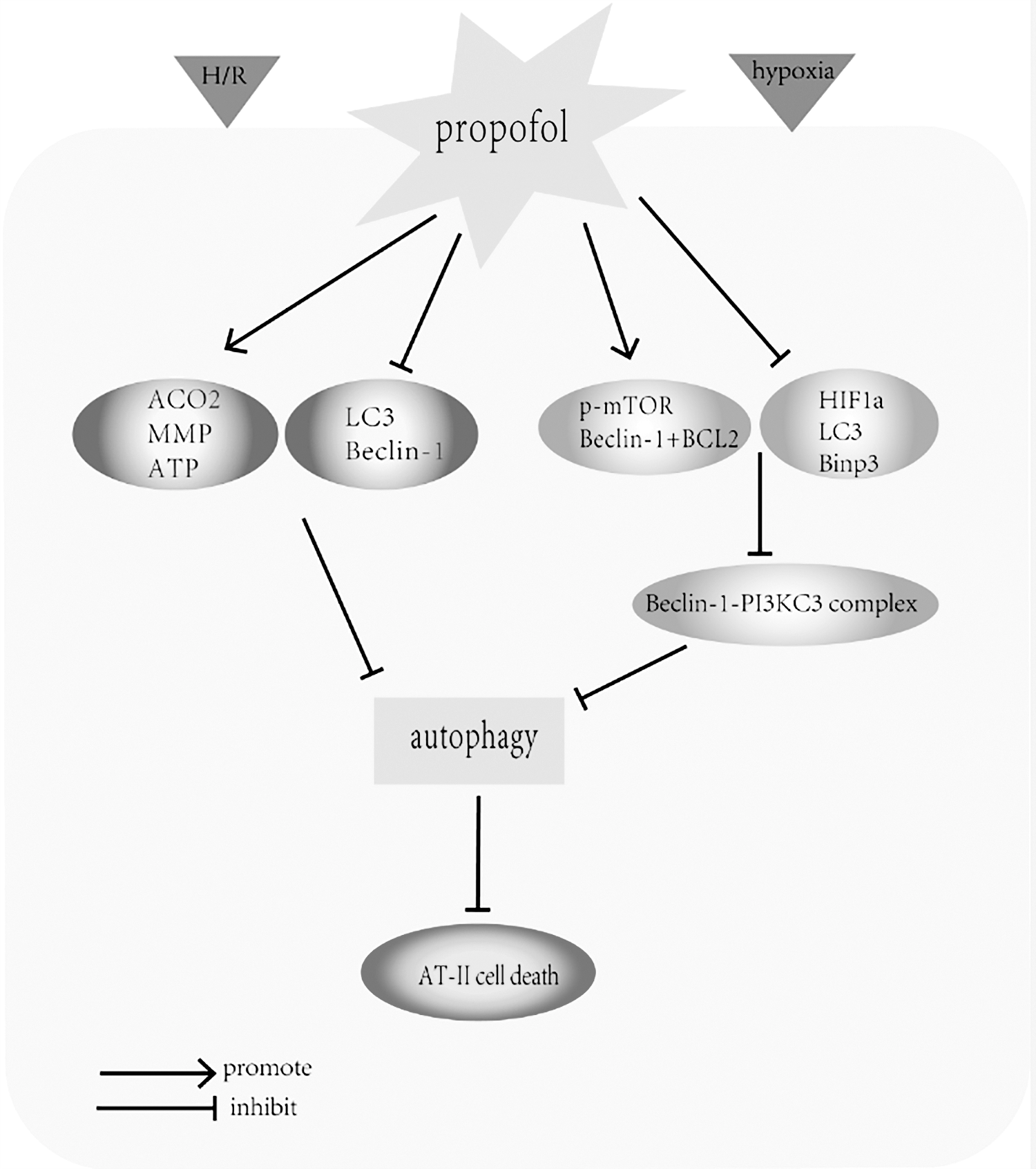
The protective effect of propofol on pulmonary injury. The diagram illustrates the modulatory effects of propofol on pulmonary epithelial cells. Propofol treatment protected AT-II cells against LPS-induced cell death by upregulating the expression of ACO2 and ATP, increasing MMP and by inhibiting the overexpression of the autophagy-associated proteins such as LC3 and Beclin-1. Similarly, propofol protected ATII cells from hypoxia-induced cell death. Pretreatment with propofol markedly decreased the accumulation of LC3-II, HIF-1α, and Bnip3, increased p-mTOR, and stimulated the interaction between Beclin-1 and Bcl-2, which prevents the formation of the Beclin-1/PI3KC3 complex and eventually inhibits autophagy. HIF-1α, hypoxia-inducible factor 1α; LPS, lipopolysaccharide.
Gu et al. (2012) found that propofol protects pulmonary epithelial cells against lipopolysaccharide (LPS)-induced cell death by blocking apoptosis and probably autophagy. LPS, as a constituent of the outer membrane of Gram-negative bacteria, contributes to the pathogenesis of sepsis-induced acute lung injury and induces direct injury of lung epithelial cells (Sun et al., 2012). In the study by Gu et al., propofol treatment inhibited apoptosis by reversing the increase of ROS production and caspase 9 activity and reduction of the mitochondrial membrane potential (MMP) and aconitase 2 (ACO2). Propofol also inhibited the overexpression of the autophagy-associated proteins such as LC3 and Beclin-1 (Gu et al., 2012). These findings suggest that the protection offered by propofol in A549 cells against LPS-induced cell death may provide a novel therapeutic strategy for acute lung injury induced by Gram-negative bacteria.
Similarly, another study indicated propofol can protect ATII cells against hypoxia-induced cell death by reducing apoptosis and autophagy (Ning et al., 2017). This study reported that autophagy was markedly increased by hypoxic treatment of ATII cells in a time-dependent manner. From western blot analysis, hypoxic ATII cells markedly increased the production of LC3-II, hypoxia-inducible factor 1α (HIF-1α), and B cell lymphoma-2 interacting protein 3 (Bnip3), and reduced the levels of phosphorylation of p-mTOR and Beclin-1 (Beclin-1 is also known as autophagy-related gene Atg6). Immunoprecipitation assays revealed that hypoxia markedly decreased the interaction between Beclin-1 and Bcl-2. However, the opposite was true after pretreatment with propofol. The levels of HIF-1α and Bnip3 were reduced by propofol but the level of p-mTOR was increased (Ning et al., 2017), and previous studies have shown that the phosphorylation of mTOR reduces autophagy by preventing the formation of the autophagosome (Jung et al., 2009; Noh et al., 2010). In addition, propofol promotes the interaction between Beclin-1 and Bcl-2, which prevents the formation of Beclin-1/PI3KC3 complex and eventually inhibits autophagy (Noh et al., 2010). Although the above authors agree that propofol reduces the autophagic cell death, we must emphasize that propofol's reversal of increased autophagy in dying cells does not directly mean that autophagy causes cell death. It can also mean that the cells are using autophagy as organ and cell-protective mechanisms, and the reduction of autophagy by propofol can only suggest secondary decrement because of the amelioration of the cellular stress. In brief, additional studies will be required to fully illuminate the mechanisms underlying the protective effects of propofol through autophagy regulation in the lung.
Antitumor effects of propofol and effects of propofol-induced autophagy on other organs and tissues
Antitumor effects of propofol in different tumors. Recent studies have suggested that propofol exerts antitumor effects against various cancers. Propofol has been shown to enhance tumor sensitivity to anticarcinogens or directly suppress tumor cell viability by modulating autophagy (Fig. 4). Du et al. suggested that propofol can not only act as an anesthetic agent, but also strengthen the antitumor effect of pancreatic cancer therapy. They demonstrated that propofol increased gemcitabine sensitivity in pancreatic cancer cells by inactivating the NF-κB signaling pathway (Du et al., 2013). Previous study has shown that autophagy and NF-κB possess common upstream signals and interact with each other through positive or negative feedback loops to maintain cellular homeostasis. The complex interaction between autophagy and NF-κB signaling pathways affects the effectiveness in tumor responses to therapy (Trocoli and Djavaheri-Mergny, 2011). Therefore, we suggest that the propofol may produce chemosensitization of pancreatic tumors to gemcitabine by regulating both the NF-κB and autophagy pathways. However, the detailed mechanism that explains how propofol strengthens the antitumor effects of gemcitabine through the interplay between autophagy and NF-κB signaling pathways requires further elucidation.
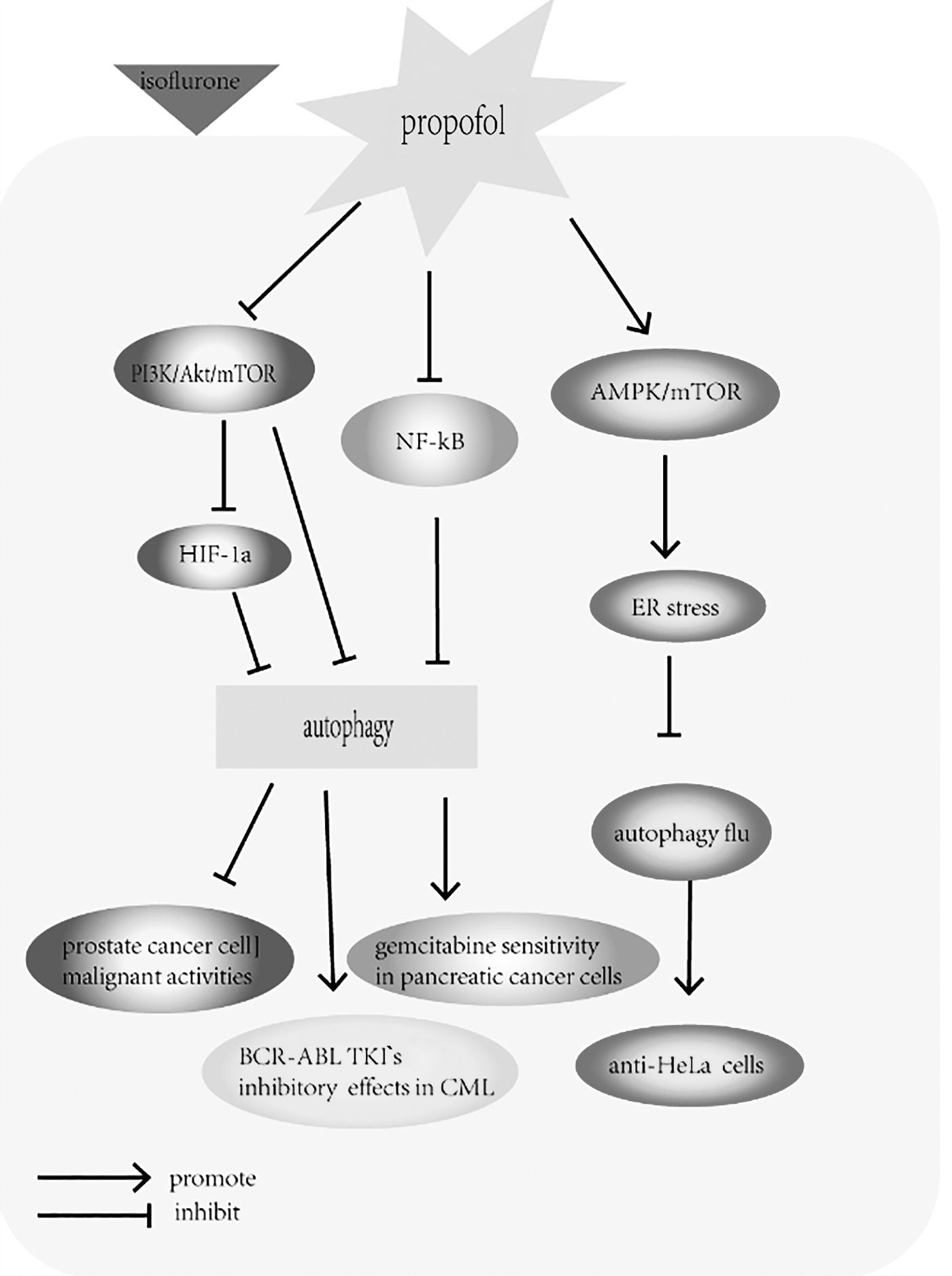
The antitumor effects of propofol in different kinds of tumor. The figure illustrates the modulatory effects of propofol on tumor cells. Propofol has been shown to inhibit the overactivation of HIF-1α induced by isoflurane and reduce prostate cancer cell malignant activities through suppression of the PI3K/Akt/mTOR pathway. Similarly, propofol augmented the inhibitory effects of BCR-ABL tyrosine kinase inhibitors in CML by suppressing the Akt/mTOR signaling pathway, and propofol increased gemcitabine sensitivity of pancreatic cancer cells by inactivation of the NF-κB signaling pathway. Furthermore, propofol decreased viability of HeLa cells by impairing autophagic flux through induction of ER stress and by activating the AMPK/mTOR signaling pathway. CML, chronic myeloid leukemia; ER, endoplasmic reticulum.
Huang et al. (2014) demonstrated that isoflurane enhanced the synthesis of HIF-1α through the PI3K/Akt/mTOR pathway in prostate cancer cells. It has also been shown that high levels of HIF-1α within a tumor promote the malignant activities of cancer cells, whereas the use of propofol to inhibit HIF-1α activity can improve the prognosis (Semenza, 2003). Therefore, they suggested that propofol inhibits the overactivation of HIF-1α induced by isoflurane and partially reduces prostate cancer cell malignant activities by suppression of the PI3K/Akt/mTOR pathway (Huang et al., 2014). Similarly, Tan et al. (2017) suggested that propofol has properties that weaken the malignant activity of cancer cells. They showed that propofol augmented the inhibitory effect of BCR-ABL tyrosine kinase inhibitors in chronic myeloid leukemia by suppressing the Akt/mTOR signaling pathway (Tan et al., 2017).
Recent accumulating evidence has shown that propofol plays a crucial role in the antitumor activity of drugs and chemicals (Cheong et al., 2012; Jung et al., 2016). Chen et al. (2018a) found that propofol suppresses cell viability and increases autophagosome generation in HeLa human cervical cancer cells . The major findings of this study were that propofol decreased the viability of HeLa cells by blocking autophagosome–lysosome fusion and promoting autophagosome accumulation, which resulted in the inhibition of autophagic flux. Impairment of autophagic flux by propofol involved induction of ER stress regulated by calcium and activation of the AMPK/mTOR signaling pathway. On the one hand, propofol impaired autophagosome clearance by interfering with autophagosome–lysosome fusion through the AMPK/mTOR signaling pathway. Propofol thus appears to be both an mTOR inhibitor and an AMPK activator, and both contribute to propofol-induced accumulation of autophagosomes. On the other hand, propofol induced ER stress and disrupted the intracellular Ca2+ balance, thereby enhancing autophagosome accumulation. This is the first study to indicate that propofol exerts antitumor effects in HeLa cells by increasing autophagosome accumulation through inhibition of autophagic flux (Chen et al., 2018a).
Among the several signaling pathways regulating autophagy, the PI3K/Akt/mTOR signaling pathway plays a pivotal role in regulating the balance between cell proliferation and autophagy in response to cellular stress induced by chemotherapeutics in cancer cells. Therefore, we propose that propofol mediates autophagy by regulating the PI3K/Akt/mTOR signaling pathway, which ultimately enhances antitumor activity. However, additional research is needed to explore and clarify whether propofol suppresses other tumor cell types through the same mechanism.
Effects of propofol on autophagy in other organs and tissues
Many studies have examined the impact of propofol on autophagy in other organs and tissues. In the vast majority of in vitro and in vivo studies, autophagy modulated by propofol is beneficial (Fig. 5). Yoon et al. carried out four studies in succession, which demonstrated the effects of propofol-induced autophagy against ROS in several types of cells, including human osteoblasts, human keratinocytes, and COS-7 cells (Park et al., 2014; Kim et al., 2016; Yoon et al., 2017). In 2016, they demonstrated the protective effect of propofol against H2O2-induced oxidative stress on human fetal osteoblast (hFOB) cells through activation of autophagy (Kim et al., 2016). They found that propofol preconditioning abated H2O2-induced oxidative stress in osteoblasts and effectively decreased hFOB cell death. In western blot analysis, pretreatment with propofol induced the expression of bone-related proteins such as collagen type I, bone morphogenetic protein-2 (BMP-2), osterix, and transforming growth factor-β1 (TGF-β1) in osteoblasts after H2O2 injury. These proteins play a crucial role in the differentiation and proliferation of osteoblasts, and contribute to increasing bone nodular mineralization (Noda and Camilliere, 1989; Ducy et al., 1996; Li and Cao, 2006).
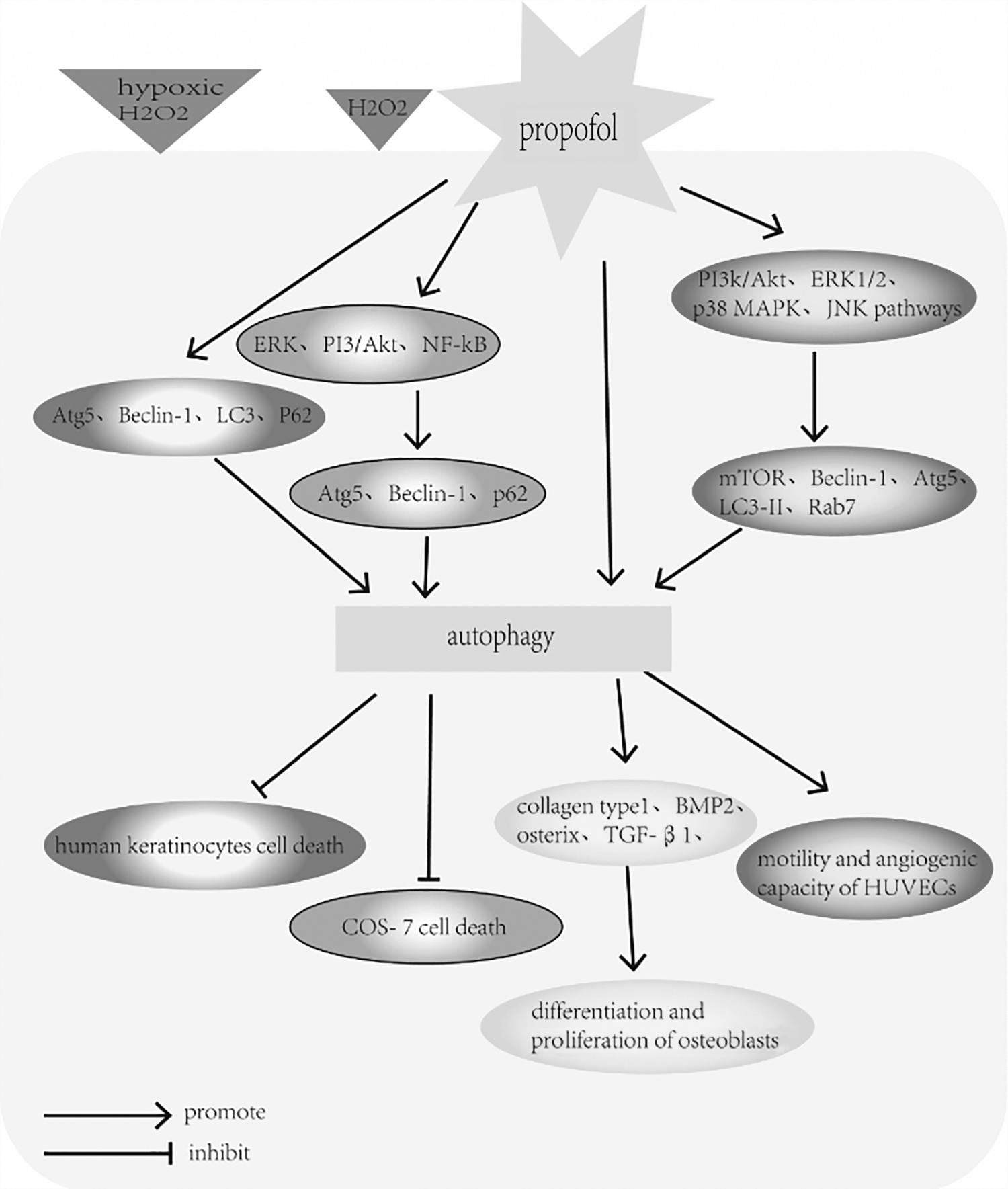
The effects of propofol on autophagy in other organs and tissues. The figure illustrates the modulatory effects of propofol in other organs and tissues. Propofol preconditioning effectively decreased both hypoxic and H2O2-induced cell death by inducing autophagy and elevating levels of Atg5, Beclin-1, LC3-II, and p62 in human keratinocytes cells. Similarly, propofol protected COS-7 cells from H2O2-induced oxidative injury by activating autophagy, which partly through activation of the ERK, PI3K/Akt and NF-κB signaling pathways. In addition, propofol preconditioning abated H2O2-induced oxidative stress in osteoblasts by activating autophagy and contributed to the differentiation and proliferation of osteoblasts. Furthermore, propofol seemed to improve the motility and angiogenic capacity of HUVECs by stimulating autophagic flux. The underlying molecular mechanisms may involve the propofol-driven signal activation of the PI3K/Akt, ERK1/2, p38 MAPK and JNK pathways. ERK, extracellular regulated protein kinases; HUVEC, human umbilical vascular endothelial cells.
Skin cells are equipped with an elaborate system that can protect the cells from oxidative stress damage by maintaining the balance between oxidative stress and antioxidant defense (Tsuchiya et al., 2001). However, excessive cellular levels of ROS can induce cell death and damage skin integrity (Wilson and Gelb, 2002). Propofol has a phenolic hydroxyl group, which is structurally similar to vitamin E, a type of endogenous antioxidant (Gulcin et al., 2005). Accordingly, propofol possesses antioxidant properties and some studies have demonstrated the antioxidant properties of propofol both in vitro and in vivo (Li Volti et al., 2008; Ha et al., 2012; Yoon et al., 2017). These studies examined the effects of propofol on hypoxic and H2O2-induced cell death in human keratinocytes. The results showed that propofol preconditioning effectively decreased both hypoxic and H2O2-induced cell death by inducing autophagy and also stimulated wound healing by increasing cell migration. As shown in western blot analysis, Atg5, Beclin-1, LC3-II, and p62 were elevated after propofol preconditioning of human keratinocytes, but were decreased when autophagy was suppressed by the autophagy pathway inhibitor 3-methyladenine (3-MA) (Park et al., 2014).
A similar event occurred in H2O2-treated COS-7 cells (Yoon et al., 2017), where fluorescence microscopy showed that propofol pretreatment dramatically increased the formation of autophagosomes, which was inhibited by 3-MA. Furthermore, western blot analysis showed that the expression of autophagy-related proteins including Atg5, Beclin-1, and p62 was higher in the propofol+H2O2 group than in the H2O2 group. The authors concluded that propofol protects COS-7 cells from H2O2-induced oxidative injury, partly through activation of the extracellular signal-regulated protein kinases (ERK), PI3K/Akt, and NF-κB signaling pathways and through the heat shock response (Yoon et al., 2017).
Chang et al. (2015) investigated the effects of propofol on cultured human umbilical vascular endothelial cells (HUVECs). Propofol seemed to stimulate autophagic flux by augmenting the expression of autophagy markers including mTOR, Beclin-1, Atg5, and LC3I/II, and the late endosomal indicator Rab7. The underlying molecular mechanisms may involve propofol-driven signal activation of the PI3K/Akt, ERK1/2, p38 MAPK, and JNK pathways. Furthermore, they also demonstrated that propofol improved the motility and angiogenic capacity of HUVECs. However, this enhancement was completely blocked in the presence of autophagy inhibitors bafilomycin A, 3-MA, and chloroquine, suggesting a proangiogenic contribution of propofol by increasing autophagy in cultured HUVECs (Chang et al., 2015). In fact, as early as 2013, Chen et al. found that propofol protected HUVECs from H/R-induced cell death and showed that differential autophagy-related gene expression regulated by miRNAs played a key role in this process.
Conclusion
In general, propofol at moderate concentration and duration can protect organs and cells from some types of damage, such as oxidative injury and I/R and H/R injury. Increasing evidence indicates that propofol exerts beneficial effects in neuroprotection, cardioprotection, and lung protection and exerts antitumor effects by regulating the expression of autophagy. Conversely, it is clear that propofol at high concentrations and prolonged duration results in overactivation of autophagy and that eventually promotes cell and organ damage, but it does not mean excessive autophagy promotes cell death. It is highly likely that autophagy increase in the excessive treatment by propofol is because of either futile effort of the cells to evade cell death by augmenting cell protection scheme of autophagy or impaired autophagy flux providing an appearance of accumulated autophagy machinery in the overtly stressed cells owing to the downstream blockade. The underlying mechanisms have not yet been fully elucidated and further research is needed to clarify the association between propofol and autophagy and the underlying mechanisms. For example, autophagy induction can ameliorate inflammatory responses in intestinal I/R (Wang et al., 2019b), which can lead to multiple organ dysfunction syndrome with a high mortality rate ranging from 30% to 90% (de Pinho-Apezzato et al., 2011; Wu et al., 2013). However, whether propofol can protect against I/R-induced intestinal injury through the autophagic pathway has not been clarified and deserves further study. A more explicit understanding of these issues will pave the way for clinical guidelines and ultimately refine perioperative management. In summary, the effects of propofol on autophagy are promising areas for future research.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (grant no. 81500387).