
Abstract
The chemokine receptor CXCR2 is a receptor for CXC chemokines, including CXCL1 and CXCL2. CXCR2 is expressed by resident cells of the central nervous system, including neurons, microglia, oligodendrocyte progenitor cells (OPCs), and oligodendrocytes. CXCR2 signaling is important in regulating OPC biology with regard to positional migration and myelination during development. More recently, studies have argued that CXCR2 is involved in controlling events related to remyelination after experimentally induced demyelination. This review examines the concept that targeting CXCR2 may offer a novel therapeutic target for promoting remyelination.
Introduction
Multiple Sclerosis (MS) is characterized as a chronic neurodegenerative disease of the central nervous system (CNS) that preferentially affects young adults. Disease pathogenesis is thought to be driven by focal neuroinflammation, demyelination, and axonal damage culminating in a spectrum of neurological disabilities, including impaired motor skills, speech/auditory problems, and cognitive decline (Lassmann et al., 2007; Steinman, 2014). Although the cause of MS is unknown, it is considered an autoimmune disease characterized by a misguided attack by the immune system against proteins embedded within the myelin sheath (Steinman, 2014). Infiltration of inflammatory lymphocytes, for example, T cells and B cells along with activated monocytes/macrophages, and neutrophils have all been implicated in contributing to demyelination either directly or through secretion of proinflammatory cytokines/chemokines that augment the inflammatory response (Steinman, 1996, 2014; Khaibullin et al., 2017). The importance of activated lymphocytes in contributing to lesion formation is supported by preclinical animal studies as well as genome-wide association studies yet recent findings of the MS genomic map have now implicated microglia, the resident immune cell of the brain, in contributing to disease susceptibility (International Multiple Sclerosis Genetics Consortium, 2019).
Current FDA-approved disease-modifying therapies have been effective in dampening new lesion formation and disease progression, in part, by limiting access of activated lymphocytes into the CNS (Steinman, 2014; Greenfield and Hauser, 2018). Nonetheless, no therapies to date have shown any effect on promoting remyelination of demyelinated axons and this is an area of intense research (Cunniffe and Coles, 2019).
Ongoing studies have focused on both oligodendrocyte progenitor cells (OPCs) and mature oligodendrocytes to better understand the paucity in remyelination in MS patients. OPCs are present within areas of demyelination (Chang et al., 2000); however, a failure to mature into myelinating oligodendrocytes ultimately results in a failure to remyelinate axons. Therefore, strategies to promote remyelination have focused on methods to promote the maturation of OPCs to mature myelinating oligodendrocytes with the goal of reversing underlying myelin damage and limiting axonopathy. This brief review will examine how targeting the chemokine receptor CXCR2 on oligodendrocytes influences remyelination within the context of preclinical models of demyelination.
Neurobiology of the Chemokine Receptor CXCR2
Chemokine receptors are G protein-coupled receptors that bind small chemoattractant peptides called chemokines (Olson and Ley, 2002; Turner et al., 2014). Chemokines are small (8–10 kDa) proteins expressed by almost all nucleated cell types that are divided into four subfamilies based upon the number and spacing of conserved cysteine residues present within the amino terminus of the protein. Chemokines are considered critical mediators of a variety of biological processes, including development, tissue homeostasis, and coordinated immune responses after microbial infection. With this in mind, chemokine signaling is important for cell trafficking, immune cell activation, and differentiation and have been shown to be involved in numerous inflammatory disease conditions (Zarbock and Stadtmann, 2012; Griffith et al., 2014).
The CXC chemokine receptor CXCR2 was first cloned in 1991 (Murphy and Tiffany, 1991) and is an ELR-positive receptor characterized by a glutamic acid-leucine-arginine (ELR) sequence and has high binding affinity to the chemokine ligands CXCL1 and CXCL2 (Wolpe et al., 1989; Moser et al., 1990; Schumacher et al., 1992). CXCR2 is expressed in the CNS by resident cells, including microglia (Filipovic et al., 2003), neurons (Horuk et al., 1997), mature myelinating oligodendrocytes, and OPCs (Omari et al., 2005; Hosking et al., 2010; Tirotta et al., 2011) as well as in neutrophils and endothelial cells (Zhang et al., 2004; Wu et al., 2015). Chemokine ligands specific for CXCR2 are secreted by numerous different cell types, including macrophages, endothelial cells, neutrophils, and astrocytes (Rubio et al., 2006; Rubio and Sanz-Rodriguez, 2007). Ligand binding to targeted chemokine receptors induces different cellular mechanisms, including adhesion and migration associated with changes in cell morphology (Baggiolini, 1998).
CXCR2 is closely involved in the chemotaxis of neutrophils to sites of injury through high-affinity binding with a gradient of its cognate ligands. Indeed, several preclinical mouse models of MS have shown that CXCR2-mediated recruitment of neutrophils influences the severity of demyelination (Carlson et al., 2008; Simmons et al., 2014; Liu et al., 2015a; Marro et al., 2016; Grist et al., 2018). More recently, the functional role of CXCR2 signaling has been studied with regard to positional migration and myelin formation during development (Tsai et al., 2002; Padovani-Claudio et al., 2006). Discerning the function of CXCR2 signaling on relevant cell types, for example, OPCs, oligodendrocytes, and neurons involved in demyelination and myelin repair is an area of ongoing investigation.
Oligodendroglia and CXCR2 Signaling
How CXCR2 signaling influences oligodendroglia with regard to survival/maturation has been studied using in vitro and in vivo models. Filipovic and Zecevic (2008) found that treatment of cultured human fetal OPCs with recombinant CXCL1 dramatically increased proliferation, whereas blocking CXCL1 signaling reduced OPC proliferation. Within the developing CNS, CXCR2 signaling regulates numbers of OPCs that augments tissue structural integrity while also promoting positional migration of OPCs within the white matter of the mouse spinal cord (Robinson et al., 1998; Tsai et al., 2002).
Moreover, studies from Miller and colleagues (Padovani-Claudio et al., 2006) have shown that germline Cxcr2−/− mice have insufficient numbers of OPCs that persist into adulthood resulting in reduced myelin associated with altered expression of myelin-specific proteins, including proteolipid protein (PLP) and myelin basic protein. Moreover, OPCs derived from these Cxcr2−/− mice have decreased numbers of mature oligodendrocytes when differentiated in culture (Padovani-Claudio et al., 2006). Collectively, these findings offer a compelling argument for an important role for CXCR2 signaling in contributing to OPC maturation.
Given the important role for CXCR2 signaling in affecting OPC responses and myelination during development, identifying whether this signaling pathway was relevant to MS with regard to influencing remyelination became an important area of investigation. Early reports from Raine and colleagues (Filipovic et al., 2003; Omari et al., 2006) revealed expression of CXCR2, on normal and proliferating oligodendrocytes in active MS lesions. In addition, oligodendrocytes were occasionally associated with reactive astrocytes positive for CXCL1. These findings argued for a potential role for the CXCL1:CXCR2 signaling axis in regulating oligodendroglia biology in MS patients and affecting remyelination during disease progression. Within the context of experimental preclinical models of demyelination, there have been conflicting reports on the functional role of CXCR2 in disease initiation, progression, and repair. Numerous reports have clearly shown that CXCR2 is necessary for induction of experimental autoimmune encephalomyelitis (EAE) due to the ability to attract neutrophil to the CNS (Glabinski et al., 2000; Carlson et al., 2008; Kroenke et al., 2008); however, the functional role of CXCR2 on oligodendroglia was not examined. Recent studies from Tiwari-Woodruff and colleagues (Karim et al., 2018, 2019) have shown that treatment of mice that have established EAE with ERβ ligands results in increased remyelination and axonal sparing that correlated with increased expression of the CXCR2 ligand CXCL1. This was associated with increased oligodendrogenesis suggesting that CXCL1-mediated signaling through CXCR2 on oligodendroglia augments remyelination (Karim et al., 2018, 2019).
Conversely, studies using CXCR2 small molecule antagonists in EAE and spinal cord injury showed that blocking CXCR2 induced oligodendrocyte differentiation and promoted recovery arguing that CXCR2 signaling under defined pathological conditions mutes the ability of oligodendroglia to participate in repair (Kerstetter et al., 2009; Marsh and Flemming, 2011). However, in the JHMV model of viral-induced demyelination, CXCR2 appeared to protect oligodendrocytes against apoptosis during the chronic phase of infection as treatment of persistently infected mice in which demyelination was established with an anti-CXCR2 antibody resulted in increased white matter demyelination and a more severe clinical course that was not associated with changes in infiltrating immune cells or viral titers (Hosking and Lane, 2010). In addition, in vitro studies indicated CXCR2 signaling protects cultured OPCs against IFN-γ-induced apoptosis. Collectively, these studies highlight the potentially diverse roles of CXCR2 on oligodendroglia under defined experimental conditions.
In vivo studies examining how CXCR2 signaling affects oligodendroglia survival and remyelination have employed using either small molecule antagonists, antibody targeting of CXCR2 or germline Cxcr2−/− mice, and this raises the possibility that CXCR2 signaling on other cell types, for example, either resident and/or immune cells, in contributing to reported effects. Recently, Ransohoff and colleagues (Liu et al., 2015b) made important strides to better understand how CXCR2 signaling on oligodendroglia affects remyelination in two independent models of demyelination. Using transgenic mice in which Cxcr2 is conditionally ablated in oligodendroglia allowed for the clear demonstration of increased remyelination after cuprizone-mediated demyelination (Liu et al., 2015b). Similarly, there was accelerated remyelination in lysophosphatidylcholine-treated cerebellar slice cultures after CXCR2 deletion on oligodendrocyte lineage cells further supporting that remyelination is enhanced when CXCR2 signaling is silenced.
We recently employed a similar approach by generating Plp-Cre-ER(T); Cxcr2fl/f transgenic mice (Cxcr2-CKO mice) in which Cxcr2 was conditionally ablated upon tamoxifen treatment in adult mice (Marro et al., 2019) (Fig. 1, Fig. 2A, B). Using these mice, we were able to assess how timed ablation of Cxcr2 in oligodendroglia affected disease and remyelination after intracranial infection with the neurotropic virus JHMV. Targeted silencing of Cxcr2 did not impact either clinical disease nor was the ability to control viral replication within the CNS affected. Correspondingly, neither generation of activated virus-specific T lymphocytes nor the ability of these cells as well as myeloid cells to access the CNS was impacted in the absence of CXCR2 signaling on oligodendroglia indicating that this pathway does not influence immune responses after viral infection of the CNS. Evaluation of demyelination in JHMV-infected Cxcr2-CKO mice indicated there were no significant differences in the severity of demyelination between vehicle- and tamoxifen-treated Cxcr2-CKO on day 28 post-infection (p.i.) (Fig. 2C–E).
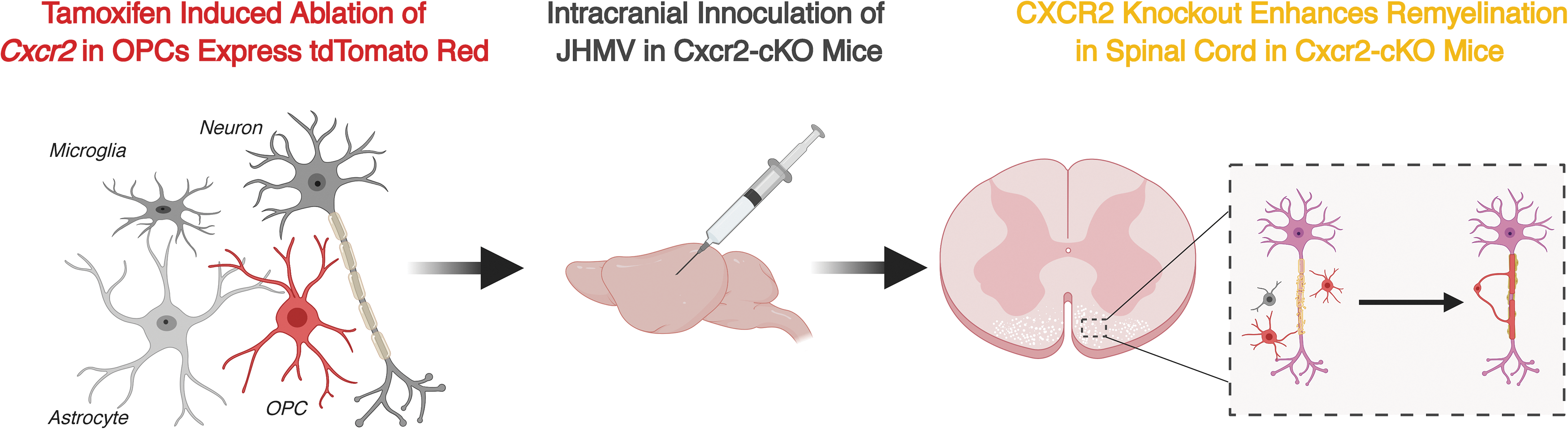
Experimental diagram to investigate how targeted Cxcr2 ablation in oligodendroglia affects disease progression. Tamoxifen-induced ablation of Cxcr2 was specific to OPCs labeled through a tdTomato reporter mouse line before intracranial inoculation with the JHM strain of the mouse hepatitis virus (JHMV). Spinal cord sections were assessed at defined points postinfection for evidence of demyelination and remyelination. OPCs, oligodendrocyte progenitor cells.

Targeted ablation of CXCR2 promoted spinal cord remyelination in JHMV-infected mice.
Although neither neuroinflammation nor demyelination was affected after tamoxifen treatment of Cxcr2-CKO mice compared with control animals, this did not preclude the possibility that remyelination is occurring in the face of ongoing demyelination. Notably, deletion of Cxcr2 resulted in increased spinal cord remyelination as assessed by electron microscopy and g-ratio calculation when compared with vehicle-treated control mice (Fig. 2F–H). Collectively, our findings argue that CXCR2 signaling in oligodendroglia is dispensable with regard to contributing to neuroinflammation, but its deletion enhances remyelination in a preclinical model of the human demyelinating disease MS. Although mechanisms associated with remyelination have yet to be fully defined, our data would argue for an enhanced maturation of OPCs into oligodendrocytes in the absence of CXCR2 suggesting that signaling through this receptor may suppress events influencing this process.
Perspectives
Determining how signaling pathways in oligodendroglia affects both disease progression and remyelination continues to be an area of intense investigation. Strategies to stimulate the maturation of OPCs to myelinating oligodendrocytes are ongoing yet successful translation to effective and sustained therapies has yet to occur. Research from our laboratory and others has shown CXCR2 signaling on oligodendroglia impacts remyelination potentially by augmenting OPC maturation (Liu et al., 2015b; Marro et al., 2019). OPC differentiation into mature myelinating oligodendrocytes is a complex process that is regulated by activation and repression of specific growth and transcription factors as well as epigenetic factors (Tauheed et al., 2016; Santos et al., 2018; Tiane et al., 2019). How suppression of CXCR2 affects these processes will be the focus of ongoing studies as this may offer novel therapeutic approaches for promoting remyelination in patients with demyelinating diseases.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
T.E.L. was supported by funding from the National Institutes of Health R01NS041249, National Multiple Sclerosis Society (NMSS) Collaborative Research Center Grant CA-1607-25040 and The Ray and Tye Noorda Foundation.