
Abstract
Malfunction of myocardial mitochondria plays a crucial role in the development of cardiovascular disorders, especially hypertrophic and dilated cardiomyopathies. Cardiac troponin I (cTnI) is an important structural protein and essential to contraction and relaxation of cardiomyocytes. Recent studies suggest that mutated cTnIR193H could function as a regulatory molecule for other cell functions. This study was to determine whether mutated cTnI could contribute to mitochondrial dysfunction of cardiomyocytes. Primary cardiomyocytes were transfected with cTnIR193H adenovirus with empty vector as control. Mitochondrial structure and function were evaluated in the cells 72 h after transfection. Transmission electron microscopy examination showed mitochondria in the cardiomyocytes with R193H mutation displayed broken cristae, vacuolation, and mitophagy. Mitochondrial function studies revealed a significant decrease in complex I activity, ATP and reactive oxygen species levels, and oxygen consumption rate compared with controls. Western blot analysis demonstrated that expressions of mitochondria-related genes, including ND5 (ubiquinone oxidoreductase chain 5), LRPPRC (a leucine-rich protein of pentatricopeptide repeat family), and PGC-1α (PPARG co-activator 1 alpha), were significantly downregulated in R193H mutation cardiomyocytes compared with the control. Swelling and broken cristae were observed in the mitochondria of cardiomyocytes from cTnIR193H mutation transgenic mice with decreased mitochondrial function, not from the littermate control mice. The data from the present study demonstrated that mitochondrial structure and function were significantly impaired in cardiomyocytes with cTnIR193H mutation, suggesting that cTnI might be critically involved in maintaining the structural and functional integrity of myocardial mitochondria.
Introduction
Cardiac troponin I (cTnI) is a structural protein in sarcomeric thin filament, and is essential to contraction and relaxation of cardiomyocytes. cTnI inhibits cardiac muscle contraction and actin-activated myosin ATPase activity by binding to actin-tropomyosin (Tobacman, 1996; Pan et al., 2016). Mutations of cTnI could lead to impaired relaxation of cardiomyocytes, cardiac diastolic dysfunction, and cardiomyopathies, including hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and restrictive cardiomyopathy (RCM) (Morimoto, 2007; Kimura, 2015; Cristina et al., 2016; Pan et al., 2016). It is believed that cTnI mutations could lead to hypersensitivity of myofilaments to Ca2+ (Davis et al., 2007; Dadson et al., 2017 ), resulting in diastolic dysfunction in RCM and HCM. However, it is unclear why mice with cTnI mutations and RCM could develop myocardial stiffness and interstitial fibrosis.
A recent study showed that mutated cTnIR193H could interact with HDAC1 with decreased expression of phosphodiesterase 4D (Zhao et al., 2020), suggesting that mutated cTnI could function as a regulatory molecule for other cell functions. Heart is a high energy-consuming organ, and requires an adequate and continuous supply of ATP for normal function (Osterholt et al., 2012). About 95% ATP production is from mitochondrial oxidative phosphorylation, with the remainder from glycolysis (Lopaschuk, 2017). Mitochondria produces ATP by metabolizing a variety of energy substrates through electronic transport chain (ETC), including fatty acids, glucose, lactate, ketones, and amino acids (Lopaschuk, 2017). Typically, 60% of cardiac energy need is from the metabolism of fatty acids, with the remaining 40% coming from the metabolism of other substrates (De Jong and Lopaschuk, 2017). Complex I, also known as NADH-ubiquinone oxidoreductase, is the largest and arguably the most complex component of the ETC, as well as the origin of electron transfer in the chain and essential for ATP synthesis (Zhu et al., 2016a).
Abnormalities in mitochondrial energy balance, production, or propagation would result in significant cardiac pathology (Murphy et al., 2016). Defects in mitochondrial complex I could lead to interruption of the electron transfer process with reduced ATP production, resulting in cardiomyopathy (Piekutowska-Abramczuk et al., 2018). Alterations in mitochondrial function have been increasingly recognized as a contributing factor to cardiovascular disorders, especially HCM and DCM (Murphy et al., 2016). It is unclear whether cTnI could be involved in mitochondrial function. Therefore, this study was to test the hypothesis that could impair the integrity of myocardial mitochondria structure and function.
Materials and Methods
Cell culture and transfection
Primary cardiomyocytes from newborn C57BL/6 mice were cultured in DMEM with 10% fetal bovine serum (Hyclone, New Zealand) for 48 h. Adenoviruses with mutant cTnIR193H-GFP (R193H) and negative controls with GFP (Control) (from Shanghai Jikai Gene Technology Co., China) were transfected into the cells at a multiplicity of infection (MOI) of 40. An MOI of 40 was used for transfection in this study based on the findings that transfection with an MOI of 40 yielded the best adenovirus transfection efficiency in cardiomyocytes72 h after transfection compared with an MOI of 20 and 80. All cell experiments were performed 72 h after transfection. Transfection efficiency was evaluated with Western blot analysis for Flag expression and FACSCanto flow cytometry (Becton Dickinson) for GFP-positive cells. No primary cardiomyocytes isolated from cTnIR193H mutation transgenic (TG) mice were used because of their poor survival in vitro culture system and the fact that only about 20% mutant cTnI was displayed in the TG mice heart (Li et al., 2013).
Confocal microscopic analysis
Cells were stained directly in culture medium with 50 nM Mitotracker® Red (Molecular Probes) for 1 h, and then counterstained with Hoechst 33342 (Beyotime, China) for 2 min. After washing with phosphate-buffered saline (PBS) twice, the cells were imaged with a confocal microscope (NIS Nikon) at Children's Hospital of Chongqing Medical University.
ATP assay
Cardiomyocytes were prepared for ATP assay using ATP Assay Kit (Beyotime Biotechnology, Beyotime Institute of Biotechnology, Haimen, Jiangsu, China; #S0026) as per manufacturer's instruction.
Reactive oxygen species measurement
Cardiomyocytes were prepared in microplate using PBS with the total volume of 50 μL. Cellular reactive oxygen species (ROS) level was measured with ROS ELISA assay kit (MEIMIAN, China; #MM-43700M1) as per manufacturer's protocol. The samples were read at 450 nm with a microplate reader (BioTek).
Complex I activity assay
Mitochondrial respiratory chain complex I activity in cardiomyocytes was determined using microplate assay kit (Solarbio Science and Technology Co., Beijing, China; #BC0515) as per manufacturer's protocol. The samples were read at 340 nm with a microplate reader (BioTek). The values were normalized with protein concentration for each sample.
Mitochondrial stress test
Cardiomyocytes were seeded in Seahorse XFe24 cell microplate at a density of 1.5 × 105 cells per well and cultured at 37°C with 5% CO2 and room air for 48 h, and then transfected with adenoviruses for 72 h. Cell mitochondrial oxidative phosphorylation level was assessed using direct measurement of oxygen consumption rate (OCR) with Seahorse XFe Cell Mito Stress Test kit (Agilent) with a Seahorse XFe24 Extracellular Flux analyzer as described (Dai et al., 2018), with minor modifications on the concentrations for oligomycin (2.5 μM), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (2 μM), and antimycin (0.5 μM). The OCR values were normalized with protein concentration in each sample.
Western blot analysis
Total protein was prepared from cardiomyocytes with a protein extraction kit (Beyotime) as per manufacturer's instructions. Protein concentration was quantified with Bradford assay (KeyGen, China). Western blot analysis was then performed to determine the levels of mitochondria-related proteins using specific primary antibodies against ND5 (Biorbyt, Britain; 1:500, #orb318883), Actin (Bioss, China; 1:1000, #bs-0061R), LRPPRC (Absin, China; 1:1000, #abs105907), PGC-1α (Bioss; 1:1000, #bs-1832R), and Flag (GenScript, China; 1:1000, #A00187), and corresponding secondary antibodies (Proteintech, 1:5000; #SA00001-1, #SA00001-2). The protein bands were detected using LI-COR Odyssey imaging system and analyzed with Bio-Rad Quantity One 1-D Analysis Software.
Animals
All animal experiments were conducted in accordance with the “Guide for the Care and Use of Laboratory Animals of the United States National Institutes of Health.” The experimental protocols were reviewed and approved by the Animal Care and Use Committee of Chongqing Medical University (License number: SYXK YU 2012-0001), Chongqing, China. The α-MHC-cTnIR193H TG mice (with C57BL/6 background) were generated by our laboratory. By crossing wild-type (WT) C57BL/6 mice with TG mice, both TG mice and littermate WT mice at the age of 1–2 and 3–4 months of age were used for experiments. Mouse genotypes were confirmed with PCR as described and TG mice contained 20% mutant cTnI in the heart (Li et al., 2013). Cardiac function was evaluated with echocardiography under general anesthesia with isoflurane using MyLabTM Twice with SL3413 Scan-head (Esaote, Italy) 1–2-month-old mice (n = 8 per group) and 3–4-month-old mice (n = 9 per group); all measurements were carried out and analyzed using at least three contraction cycles.
Histological examination
Hearts were collected from mice of 1–2 and 3–4 months of age (n = 3 per group), and fixed with 4% paraformaldehyde for 48 h. After dehydration with gradient alcohol and xylene, the hearts were embedded in paraffin and cut into 5 μm thick slices for hematoxylin and eosin and Masson staining. The cardiac structure and fibrosis were examined with a microscope.
Transmission electron microscopy
Mouse left ventricle was cut into small pieces and mechanically minced into small blocks of 1 mm3 in volume (n = 3), and then fixed in 2.5% glutaraldehyde phosphate buffer (pH 7.4) overnight at 4°C. Cultured myocardiocytes were collected and fixed with 2.5% glutaraldehyde for 2 h, rinsed three times in 0.1 mol/L PBS (pH 7.4), and postfixed in 2% osmium tetroxide. All samples were then further prepared for transmission electron microscopy (TEM) examination as described using Hitachi-7500 system (Japan) (Zhu et al., 2016b).
Statistical analysis
All data were expressed as mean ± standard deviation, and analyzed using independent samples t-test with the SPSS 20.0 software package. A p-value of <0.05 was considered statistically significant.
Results
Structural damage was present in the mitochondria of cardiomyocytes with R193H mutation
Cardiomyocytes were transfected with vectors for cTnIR193H-GFP with vector for GFP as control. The cells were collected for experiments 72 h after transfection. Flow cytometry analysis showed that the transfection efficiency was over 80% (Fig. 1A). Western blot analysis showed significant expression of Flag in the cells, confirming successful transfection of the vector for cTnIR193H (Fig. 1B). R193H cardiomyocytes had about 60% mutant cTnI 72 h after transfection (data not shown). Laser confocal microscopic examination demonstrated that expression of mutant cTnIR193H in primary neonatal cardiomyocytes resulted in fragmented mitochondria in the cells after 72 h of transfection. Most cardiomyocytes with mutant R193H presented in short-squat shape, while the control cells were mostly long spindle shaped (Fig. 1C). TEM analysis showed that mitochondria displayed significant structural damage, including swelling, broken cristae, and vacuolation, as well as mitophagy in cardiomyocytes with R193H mutation, while the mitochondria appeared normal in control cells (Fig. 1D). Of note, there was no difference in cell viability between the cells without transfection (blank) and cells transfected with control vector (GFP, control, data not shown), suggesting that transfection itself had no toxicity to the cells.
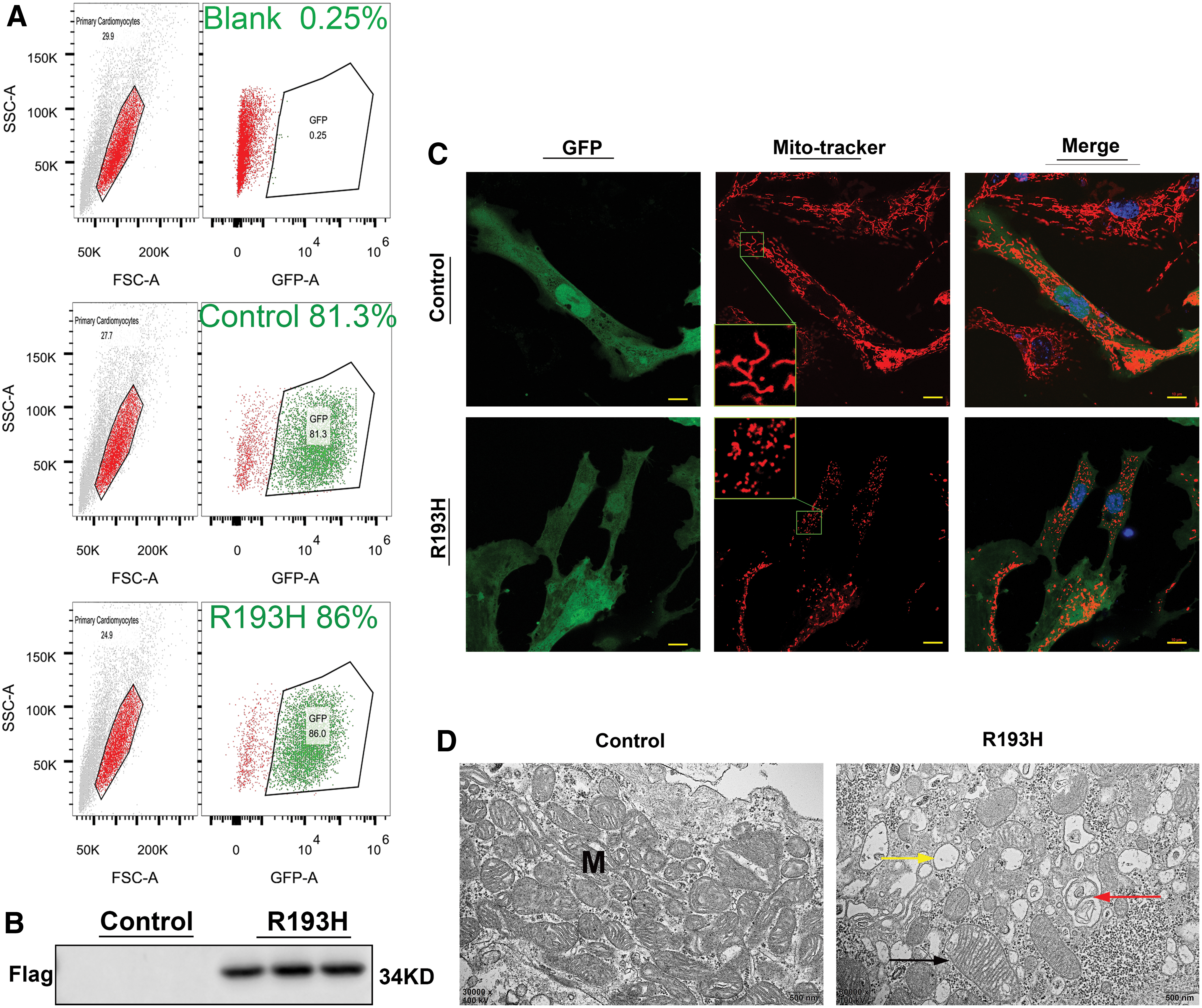
Morphological changes in R193H cardiomyocytes. Primary neonatal cardiomyocytes were transfected with vectors for cTnIR193H-GFP with vector for GFP as control. The cells were collected for experiments 72 h after transfection.
Mitochondrial function was decreased in cardiomyocytes with R193H mutation
We analyzed complex I activity, ATP and ROS production, and OCR as the function of mitochondria, and observed that complex I activity and ATP and ROS levels were all significantly decreased in R193H cardiomyocytes compared with the controls (Fig. 2A). The basal respiration, maximum respiration, spare respiratory capacity, ATP-linked respiration, and proton leakage were all significantly decreased in R193H cardiomyocytes compared with the controls (Fig. 2B, C). Of note, overexpression of normal cTnI in cardiomyocytes had no significant effect on ATP level compared with the control (Supplementary Fig. S1).
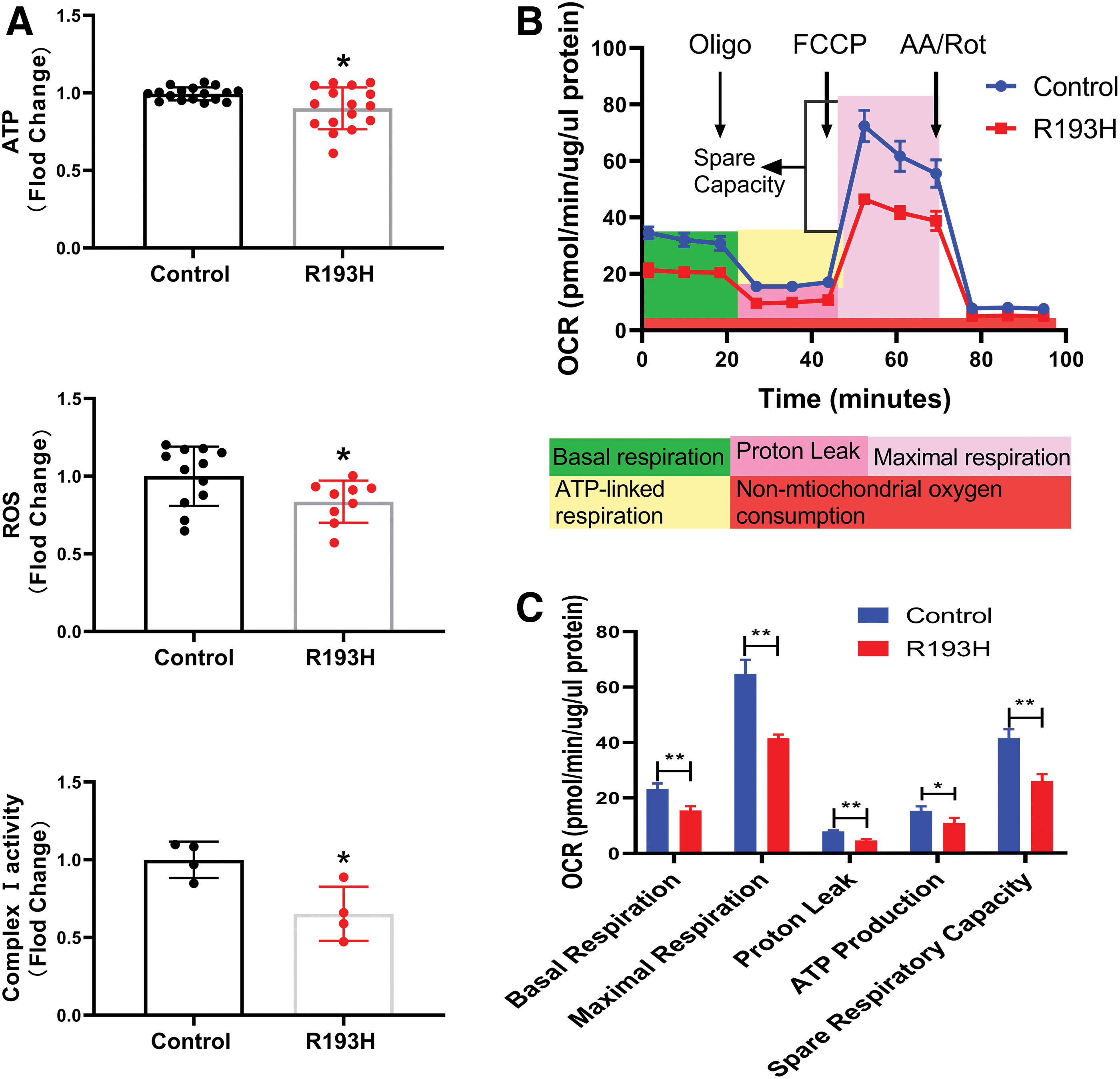
Mitochondrial function was significantly decreased in R193H cardiomyocytes. Complex I activity, ATP and ROS production, and OCR in cardiomyocytes were evaluated as the function of mitochondria.
The levels of mitochondria-associated proteins were significantly decreased in cardiomyocytes with R193H mutation
Western blot analysis showed that the expressions of mitochondria-related proteins, including ND5, LRPPRC, and PGC-1α, were significantly decreased by twofold to threefold in the mutant R193H cardiomyocytes when compared to the controls (Fig. 3), while the expression of ND5 or LRPPRC genes remained unchanged in cardiomyocytes transfected with normal cTnI, or empty control vector (data not shown).
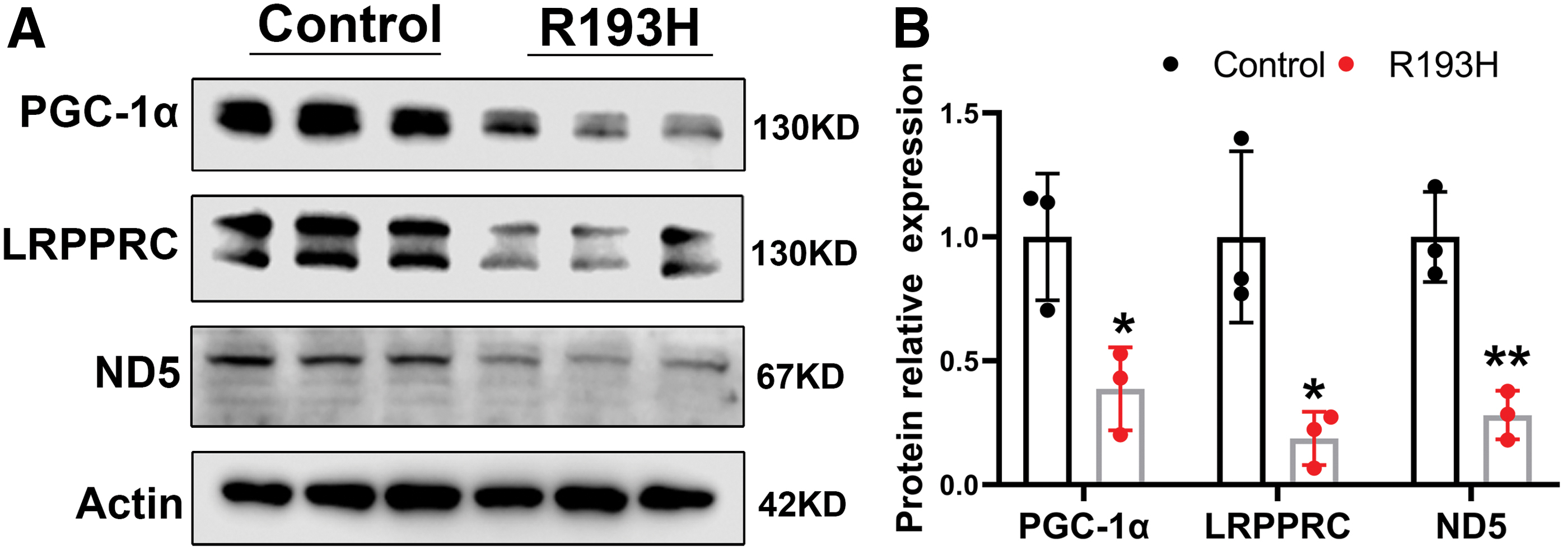
Levels of mitochondrial energy-related proteins were significantly decreased in R193H cardiomyocytes.
Structural damage was present in myocardial mitochondria in mice with cTnIR193H mutation
To determine if cTnI mutation could lead to myocardial mitochondrial abnormalities, myocardial mitochondrial structure was evaluated in α-MHC-cTnIR193H TG mice. Histopathological evaluation of the hearts from 1–2- to 3–4-month-old TG mice showed no myocardial hypertrophy or fibrosis (Fig. 4A) with normal cardiac function compared to sex- and age-matched littermate WT controls (Supplementary Fig. S2; Supplementary Table S1). TEM analysis demonstrated significant mitochondrial swelling without change in the number of mitochondria in the hearts of 1–2-month-old TG mice, and significant cristae damage of myocardial mitochondria in 3–4-month-old TG mice compared with age- and sex-matched WT mice (Fig. 4B), confirming that structural abnormalities indeed existed in the myocardial mitochondria in mice with cTnI mutation. Of note, no difference in the number of myocardial mitochondria was observed between mice with cTnI mutation and age- and sex-matched WT mice (Fig. 4C, D). Cardiac mitochondrial complex I activity was also significantly decreased in the mice with cTnI mutation at the age of 5–6 months compared with age-matched WT mice (Fig. 4E).
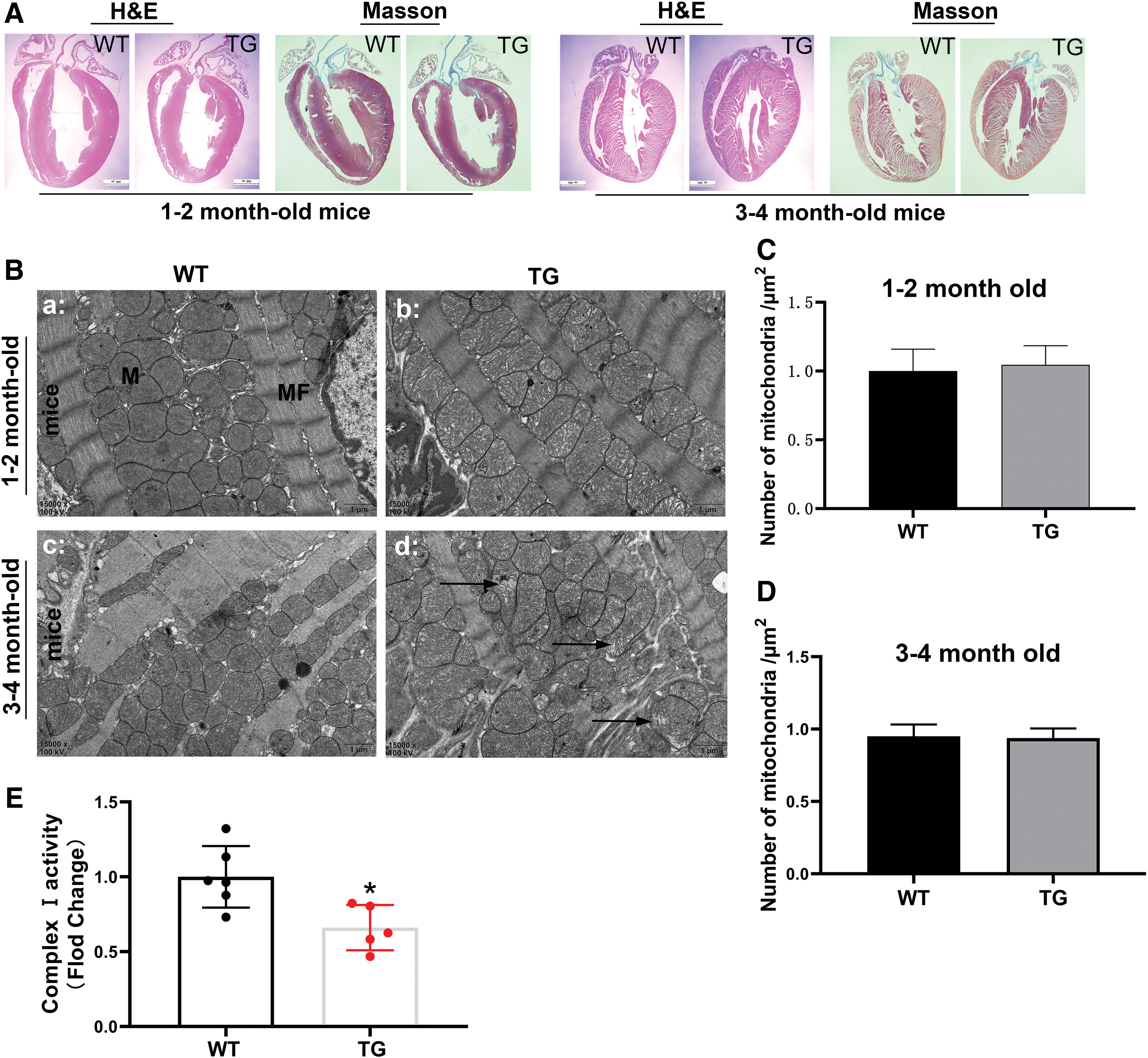
Structural damage was present in myocardial mitochondria in mice with cTnI mutation. To determine cTnI mutation could lead to myocardial mitochondrial abnormalities, myocardial mitochondrial structure was evaluated in α-MHC-cTnIR193H TG mice.
Discussion
In this study, we demonstrated for the first time that cTnI mutation resulted in significant structural damage, and substantially attenuated the expressions of mitochondria-related proteins, including ND5, LRPPRC, and PGC-1α, with significant reduction of mitochondrial complex I activity and ATP and ROS levels, as well as OCR in R193H cardiomyocytes. Myocardial mitochondrial abnormalities were also observed in mice with cTnI mutation without changes in cardiac structure and function. The data from this study support the concept that cTnI mutation significantly impairs the structure and function of mitochondria in cardiomyocytes.
Traditionally, cTnI is considered a structural protein in sarcomeric thin filament, and plays a key role in cardiac function mechanically (Tobacman, 1996). One of the important findings in this study was that structural and functional abnormalities were present in cardiomyocytes with cTnI mutation and that significant myocardial mitochondrial damage existed in mice with cTnI mutation with normal cardiac structure and function. These findings raised an exciting possibility that cTnI could also function as a regulatory protein in other cellular compartments, and significantly contribute to the structural and functional integrity of myocardial mitochondria. Recent studies showed that cTnI was expressed in the nucleus of rat mesenchymal stem cells during the early phase of their cardiac differentiation, although the role of cTnI in the nucleus of the cells remains largely unknown (Asumda and Chase, 2012). It was reported that cTnI was involved in the regulation of chromosomal stability and cell polarity during early Drosophila embryogenesis (Sahota et al., 2009). cTnI was also shown to be involved in protein transports across the nuclear envelope (Asumda and Chase, 2012). Our previous studies have demonstrated that mutant cTnIR193H could enter the nucleus and interact with HDAC1 to suppress the expression of phosphodiesterase 4D, and that cTnI could interact with other subtypes of histone proteins in cardiomyocytes (Zhao et al., 2019, 2020), indicating a potential role for cTnI in epigenetic modification in the nucleus, and subsequent cell functions. The data from this study demonstrated that cTnI mutation resulted in significant interruption of mitochondrial structure and function, leading to reduced ATP level and interruption of cellular energy metabolism. Thus, cTnI could regulate cellular function through various mechanisms beyond contraction and relaxation of cardiomyocytes.
Mutations or deficiency of cTnI is associated with dysfunction of cardiomyocytes and development of cardiomyopathies, especially RCM, with significant restrictive filling pattern with myocardial stiffness and subsequent diastolic dysfunction (Kostareva et al., 2009; Mogensen et al., 2015; Dadson et al., 2017). Although increased sensitivity of myofilaments to Ca2+ was proposed to potentially contribute to diastolic dysfunction of RCM in mice with cTnI mutations (Davis et al., 2007; Dadson et al., 2017), the mechanism(s) for restrictive filling pattern with myocardial stiffness in mice with cTnI mutations and RCM has yet to be clearly defined. Mitochondria is a critical source of ATP for the hearts; thus, mitochondrial damage could impair mitochondrial function, energy generation, and cell function (Sasarman et al., 2010). It is known that alterations of mitochondrial function significantly contribute to the development of various forms of cardiovascular disorders, especially HCM and DCM (Murphy et al., 2016). However, it is unclear if mitochondrial dysfunction could contribute to the pathogenesis of RCM as well. One of the characteristic features for the mice with cTnI mutation is the presence of RCM with diastolic dysfunction at 6 months of age or older (Du et al., 2008). The data from this study showed that cardiomyocytes with cTnIR193H displayed significant mitochondrial damage with decreased expression of mitochondria-related proteins and substantial reduction of mitochondrial function. Significant structural abnormality was present in the hearts of mice with cTnIR193H mutation at 1–2 and 3–4 month of age without cardiac fibrosis or cardiac dysfunction (both systolic and diastolic), suggesting that myocardial mitochondrial abnormalities occurred before the development of RCM. This finding might provide the evidence that supports the concept that cardiac stiffness with diastolic dysfunction in mice with cTnIR193H mutation and RCM could be due to myocardial mitochondrial dysfunction. Owing to high ATP demands, cardiomyocytes have the highest density of mitochondria of any human cell (Murphy et al., 2016). In this study, no difference in the number of myocardial mitochondria was observed between mice with cTnI mutation and age- and sex-matched wild-type mice, suggesting that impaired myocardial mitochondrial function, not mitochondrial number, could be critical for diastolic dysfunction in mice with cTnIR193H mutation and RCM. Since only about 20% mutant cTnI is present in the mice heart with cTnIR193H mutation, and about 60% mutant cTnI exists in R193H cardiomyocytes, it could be much more likely to demonstrate the potential effect of mutant cTnI on mitochondrial function using R193H cardiomyocytes in vitro. Future study is needed to determine if the level of cTnI content could be a key factor to cardiac mitochondrial function.
It is unclear how cTnI mutation leads to myocardial mitochondrial dysfunction. Data from our previous studies showed that mutant cTnIR193H could enter the nucleus and interact with HDAC1 and that cTnI could interact with many subtypes of histone proteins in cardiomyocytes (Zhao et al., 2019, 2020). It was reported that HDAC5 could attenuate PGC-1α expression through interaction with the promoter region of mitochondria-related protein PGC-1α gene, leading to mitochondrial dysfunction in cardiomyocytes (Czubryt et al., 2003). Data from this study demonstrated that expression of PGC-1α, LRPPRC, and ND5 protein significantly decreased in cTnIR193H mutation cardiomyocytes. PGC-1α can form a complex with LRPPRC, regulating the expression of genes associated with gluconeogenesis and mitochondrial respiration, as well as the stability and handling of mature mRNAs, such as ND5. (Harmel et al., 2013; Edgett et al., 2016). Therefore, it is possible that cTnIR193H mutation could decrease the expression of PGC-1α through interactions with HDAC5 or other HDACs, leading to myocardial mitochondrial dysfunction. Further studies are needed to investigate the mechanism on how cTnI regulates myocardial mitochondrial structure and function, and to address the important question, “how myocardial mitochondrial dysfunction contributes to RCM.” Future studies are also needed to determine if other cTnI mutations could impair myocardial mitochondrial structure and function similar to cTnIR193H mutation.
Conclusions
In summary, we demonstrated for the first time that mitochondrial structure and function were impaired in cardiomyocytes with mutant cTnIR193H. The data might provide a new mechanism for the development of cardiac diastolic dysfunction and RCM in mice with cTnI mutation.
Footnotes
Authors' Contributions
Concept development: J.L., Y.G., W.Z., and J.T. Experiment design: J.L. and W.Z. Experiment performance: J.L. and Y.G. Data analysis: J.L., L.L. Article preparation and revision: J.L., Z.L., W.Z., B.P., and J.T.
Disclosure Statement
No competing financial interests exist.
Funding Information
The study was supported by research grants from National Natural Science Foundation of China (Grant Number: 81974030).
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.