
Abstract
Autophagy is a critical cytoprotective mechanism that takes a hand in innate or adaptive immune responses. Hypoxia is a common pathophysiological mechanism that can lead to systemic pathological reactions. In recent years, the impact of hypoxia on the central nervous system has attracted more attention. In the past, autophagy was thought to be directly involved in the apoptosis of nerve cells under hypoxia. An increasing amount of evidence shows that the neuroinflammatory response plays an indispensable role in the neural damage caused by hypoxia. There are many mechanisms related to the neuroinflammatory response induced by hypoxia, among which autophagy is an important aspect, but the role of autophagy is still unclear. This article focuses on how autophagy flux of central immune cells is modified under hypoxic conditions, and how this autophagy affects neuroinflammatory response.
Introduction
The process of autophagy is highly conserved from yeast to mammals. During this process, protein complexes are orderly recruited to the phagophore assembly site to form double-membrane vesicles, which are called autophagosomes, and then autophagosomes deliver their content to lysosomes for degradation and recycle (Lippai and Szatmári, 2017). Numerous studies reported that neurons and the neuroglial cells autophagy could be observed in the animal model of intermittent hypoxia. High-intensity autophagy possibly leads to the death of neurons and astrocytes, resulting in cognitive and memory impairment (Xu and Zhang, 2011; Xu et al., 2016). Apart from this, recent studies often suggest a controversial role for autophagy in the neuroinflammation of the central nervous system (CNS), but the possible mechanism remains unclear (Lippai and Szatmári, 2017; Jin et al., 2018).
Neuroinflammation is a cascade of immune responses mediated by innate immune resident CNS glia (primarily microglia and astrocytes). It can be triggered in various injury processes, such as hypoxia, ischemia, infection, and immune response (DiSabato et al., 2016). After ischemic stroke, activated astrocytes secrete proinflammatory cytokines including interleukin (IL)-1β and matrix metalloproteinases, which lead to the blood–brain barrier injury and increase in leukocytes infiltration from the peripheral blood into the CNS, leading to secondary injury (Dabrowska et al., 2019). Under hypoxic conditions, the majority of clinical and experimental data indicate that neuroinflammation has a dual role. For instance, it can remove dead cells and necrotic cell fragments during injury. In turn, excessive inflammatory reactions are also harmful and may lead to increased infarction and decreased neuronal plasticity (Morris et al., 2013).
Microglia, the resident mononuclear macrophage-like cells in the CNS, is vital cellular mediators involved in acute or chronic neuroinflammatory responses (Kierdorf et al., 2013). The polarization of microglia has been described as a functional dichotomy: classical proinflammatory phenotype M1 (promote inflammatory damage) and alternative anti-inflammatory phenotype M2 (repair and protect). Therefore, the balance between proinflammatory and anti-inflammatory response of microglia affects CNS disorders' progress, increased release of associated proinflammatory cytokines from activated microglia can cause neurological dysfunction after ischemic brain injury. In addition to microglia, as the largest population of glial cells, astrocytes, not merely physical support cells, actively regulate the neuroinflammatory reactions (Fig. 1; Colombo and Farina, 2016; Acosta et al., 2017; Neal and Richardson, 2018)..
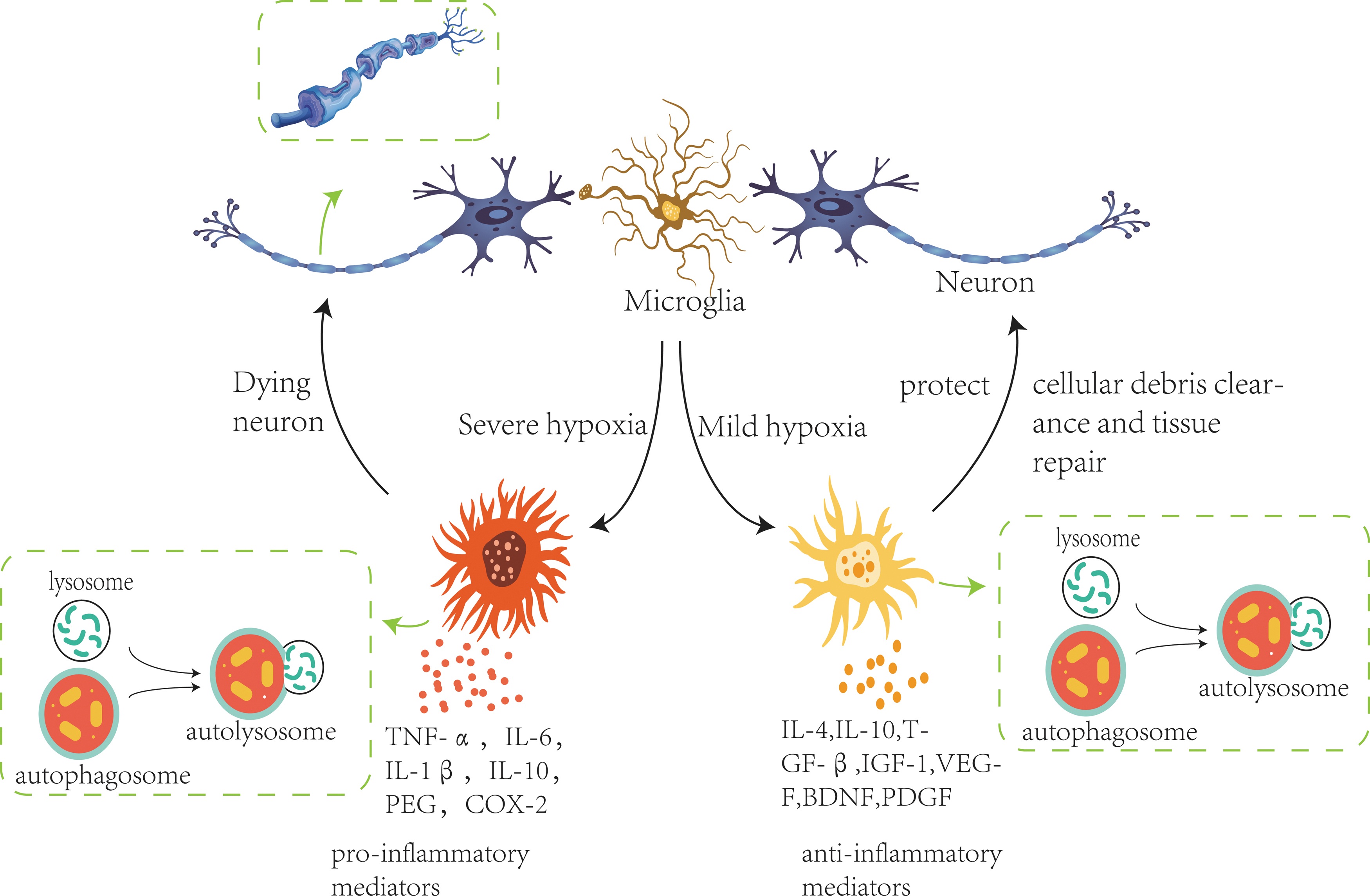
The critical part of central immune cells autophagy in hypoxia-induced neuroinflammatory reactions. Hypoxia activates autophagy in microglia. Appropriate autophagy is mediated by inflammatory mediators (IL-4, IL-10, TGF-β, IGF-1, VEGF, BDNF, PDGF, etc.), removing cellular debris pieces and contributing to tissue repair. Overactivated autophagy exerts a harmful effect by releasing a large number of proinflammatory factors (TNF-α, IL-1β, IL-6, IL-10, PEG, COX-2, etc.), producing a high-intensity neuroinflammatory response. The reason may be related to the duration or intensity of hypoxia. BDNF, brain-derived neurotrophic factor; IL, interleukin; IGF-1, insulin-like growth factors-1; PDGF, platelet derived growth factor; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor.
In this review article, we first elucidated the critical part of autophagy in hypoxia-induced neuroinflammatory reactions and then examined the possible mechanisms of autophagy in central immune cells under hypoxic conditions. Finally, hypoxia-induced central immune cell autophagy on neuroinflammatory response and its possible mechanism were introduced. This article is an attempt to provide a survey of the major central immune cells' autophagy signaling pathways under hypoxic conditions and the autophagic mechanisms, and the role of autophagy in the development and progression of neuroinflammation.
Autophagy Participates in Neuroinflammation Induced by Hypoxia
There are many mechanisms involved in neuroinflammation under hypoxia. Ren et al. (2011) found that percentages of Parkinson's disease (PD)-L1 and PD-L2 in CNS, blood, and the spleen were significantly increased in mice after middle cerebral artery occlusion (MCAO). Mice knocked out of PD-1 exacerbate inflammatory response and stroke outcomes. Propofol has an anti-inflammatory effect, which may attenuate intermittent hypoxia-induced secretion of proinflammatory cytokines in microglia, probably through inhibiting NF-κB and p38 MAPK signaling pathway (Liu et al., 2017). Transcaryophyllene (TC) has been reported to be a cannabinoid receptor subtype 2 (CB2R)-selective agonist, Guo et al. (2014) found that TC significantly inhibits hypoxia-induced production of reactive oxygen species in mitochondria and the activation of NF-κB in microglia through activation of CB2R. WNT-5A emerges as an important means of astrocyte–microglia communication. WNT-5A activates heterotrimeric Gi/o proteins to reduce cyclic AMP levels and to activate a Gi/o protein/phospholipase C/calcium-dependent protein kinase/extracellular signal-regulated kinase 1/2 (ERK1/2) axis, WNT-5A-induced and G protein-dependent signaling to ERK1/2 is important for the proinflammatory microglia response (Halleskog et al., 2012). Aside from the mechanisms already noted, the function of autophagy in hypoxia-induced neuroinflammation has attracted increasing attention. Markers of autophagy can always be found in the brain tissue of acute cerebral ischemia/hypoxia mouse models, which can regulate neuroinflammatory responses by activating or inhibiting autophagy (Zhang et al., 2013). In the following, we focus on the indispensable roles of autophagy during the neuroinflammatory response after hypoxia.
The Mechanism of Hypoxia-Mediated Autophagy of Immune Cells in the CNS
Numerous observations and experimental studies have indicated that autophagy in glial cells can be activated in diseases characterized by hypoxia, for instance, ischemic stroke (stroke), traumatic brain injury (TBI), and sleep apnea hypopnea syndrome (obstructive sleep apnea syndrome), but the specific mechanism is still unclear. We have summarized some relevant mechanisms.
AMPK/mTOR signaling pathway
AMPK/mTOR signaling pathway is a classical autophagy activation pathway in cells, which can be observed in the myocardium (Jia et al., 2019), liver (Tong et al., 2019), pancreas (Jia et al., 2019), lung tissue (Zhang et al., 2019), kidney (Wang et al., 2020b), and CNS (Sun et al., 2018). Protective autophagy is induced by the AMPK/mTOR/UNC-51-like kinase-1 (ULK1) pathway in the CNS, facilitating the proliferation and migration of astrocytes after oxygen–glucose deprivation (OGD), and improves cerebral ischemia reperfusion injury to exert a neuroprotective effect (Zhang and Miao, 2018). Instead, a study conducted by Huang et al. (2020) found that pretreatment with Ginsenoside monomer compound K, which can protect brain OGD and reperfusion (OGD/R) injury, markedly reduced p-AMPK level and increased the p-mTOR level, leading to neuroprotection against OGD/R-induced neural autophagy. Puerarin has been used for the treatment of cardio-cerebrovascular. Wang et al. (2018) found that through the activation of the AMPK/mTOR/ULK1 signaling pathway, Puerarin alleviates the release of inflammatory factors such as IL-6/TNF-α and plays a neuroprotective effect. That is to say, neurons and glial cells autophagy is regulated by the AMPK/mTOR pathway after hypoxia.
Toll-like receptor–ligand signaling pathway
The family of toll-like receptors (TLRs) is a significant type of pattern recognition receptors. By recognizing pathogen-associated molecular patterns, TLRs trigger innate and adaptive immune responses in mammals, including humans (Chang, 2010).
TLRs play a critical role in the regulation of autophagy. As a TLR family member, TLR2-mediated autophagic signaling helps to activate microglial M1/M2 switching and promotes inflammatory response, whereas downregulation of autophagy was observed in TLR2-KO cells (Ma et al., 2020). In a mouse model of white matter hypoperfusion injury, TLR4-dependent autophagy regulates microglial polarization toward a proinflammatory profile through the signal transducer and activator of transcription (STAT)1/6 pathway, thereby promoting neuroinflammatory response. TLR4 deletion protected white matter against ischemic disorganization and facilitated microglial polarization toward an anti-inflammatory phenotype (Qin et al., 2018). Likewise, Zhang et al. (2018) found that after TBI treatment, myeloid differentiation primary response 88 (MyD88)-dependent TLR4 pathway was activated, resulting in microglia activation autophagy and inflammatory response. TLR2 shares a common adaptor molecule with TLR4, MyD88, which is required for their activation. Aside from the MyD88 already noted, TLR4 signals also through toll/interleukin-1 receptor domain-containing adapter-inducing interferon-β adapters. These signaling pathways activate NF-κB and activator protein-1, which are common to all TLRs, contribute to the production of inflammatory cytokines and chemokines (Delgado and Deretic, 2009).
High-mobility group box-1
High-mobility group box-1 (HMGB1) protein is a popular branch of the nonhistone chromosomal binding protein. When hypoxic damage occurs, HMGB1 is translocated into the cytoplasm and then released from nerve cells into the peripheral blood. This process may involve phosphorylation and acetylation (Kim et al., 2008; Xiong et al., 2016; Ye et al., 2019). HMGB1 plays different roles inside and outside of cells. HMGB1 in the cytoplasm is a key autophagy protein that regulates the relationship between protective autophagy and apoptosis in cells. Extracellular HMGB1 acts as a proinflammatory factor and activates microglia and macrophages to amplify inflammatory responses by recognizing TLRs or receptor for advanced glycation endproduct (RAGE) receptors (VanPatten and Al-Abed, 2018). TLR-4 and RAGE are two of the most popular and intensively investigated HMGB1 extracellular receptors. These receptors play a synergistic effect and collaborate with other factors (myeloid differentiation factor-2, cluster of differentiation-14, and lipopolysaccharide) to initiate inflammatory signaling cascades and downstream effects (Andersson et al., 2018). HMGB1 is secreted through the immune cells endoplasmic reticulum, 99% of the released HMGB1 should be in the oxidized form in equilibrium (Tang et al., 2011). Oxidized HMGB1 possesses a cytokine-stimulating function, inducing NF-κB signaling pathway activation, resulting in systemic inflammatory reaction. Inhibiting the expression of HMGB1 and its receptors has the function of reducing inflammatory and preserving neurons from hypoxic–ischemic injury (VanPatten and Al-Abed, 2018).
In addition to directly causing inflammation in the nervous system, HMGB1 is a decisive regulator of autophagy (Pan et al., 2019). During hypoxia and reoxygenation conditions, HMGB1 increases the level of Beclin1 proteins and light chain (LC)3 (Xu et al., 2014). Through the intramolecular disulfide bridge (C23/45) binding to Beclin1, HMGB1 directly interacts with the autophagy protein Beclin1 displacing b-cell lymphoma-2 (Bcl-2), controlling the formation of Beclin1–Bcl-2 complex (Tang et al., 2010). Inhibition of the expression of HMGB1 or block the shuttle between nucleus and cytoplasma of HMGB1 was able to regulate the autophagy process (Cardinal et al., 2009). This phenomenon can be found in the CNS (Ye et al., 2019).
microRNA
More and more evidence indicates that microRNA (miRNA) plays a critical role in regulating autophagy. In the mouse model of intracerebral hemorrhage (ICH), increased expression of miRNA-144 promotes microglia autophagy by downregulating the expression of mTOR. Knockout of endogenous miRNA-144 can inhibit inflammation and improve neurological function in mice with ICH (Yu et al., 2017). This finding is consistent with that in a study by Wang et al. (2017). Again, Hu et al. (2019) found that miR-23b downregulated inositol polyphosphate multikinase (IPMK) and negatively regulated IPMK-mediated microglia autophagy by activating the Akt/mTOR signaling pathway in a rat model of ICH. In conclusion, our current studies indicated that several miRNAs are associated with the CNS autophagic flux, especially under hypoxic conditions. Whether miRNA inhibits or enhances microglia autophagy depends on the different signaling pathways targeted by different miRNA.
Extracellular vesicles
Extracellular vesicles (EVs) are small vesicles that can be detached from cell membranes by endocytosis after cell injury, activation, or apoptosis. They are released by most cell types, including glial cells (microglia, astrocytes, and oligodendrocytes), neurons, and Schwann cells. EVs can be divided into exosomes (30–100 nm), microparticles (100–1000 nm), and apoptotic bodies (500–4000 nm) because of their different diameters (Pan et al., 1985; Fauré et al., 2006; Maybruck et al., 2017; Karnati et al., 2019). The contents of EVs include proteins, lipids, and genetic material such as DNA, mRNA, and miRNA, and mediate intercellular communication (Faille et al., 2012; Rong et al., 2018; Mathieu et al., 2019). At present, EVs released under hypoxic conditions can mediate autophagy, which has attracted increasing attention. Exosomes secreted from adipose-derived stem cells (ADSCs) have been investigated for their therapeutic properties after hypoxic injury. In a rat model of ischemic injury, exosomes secreted by ADSCs enriched in miR-30d-5p prevent inflammation by inhibiting microglia autophagy and promoting microglia phenotypic polarization from M1 to M2 (Figs. 2 and 3; Jiang et al., 2018).
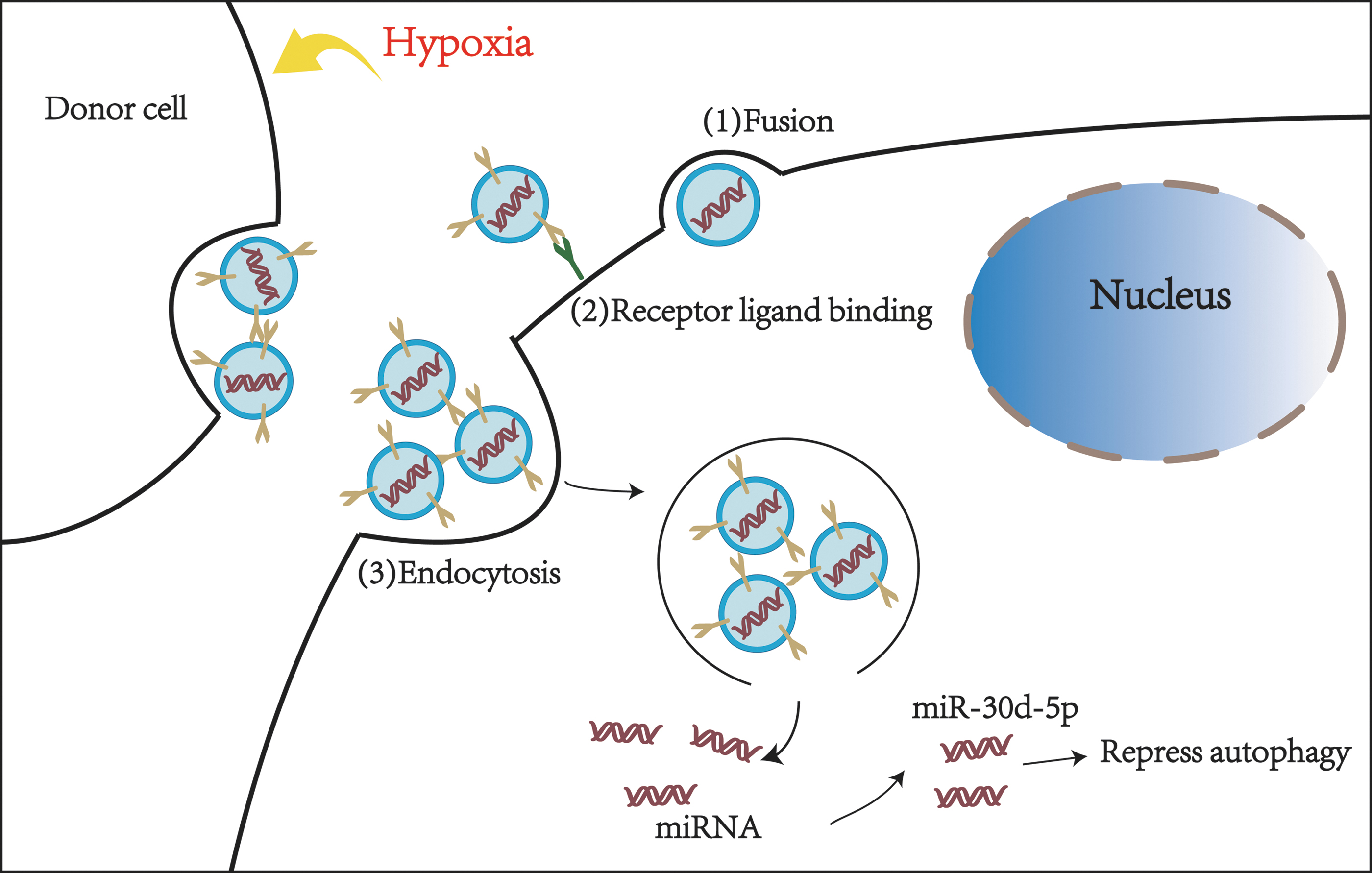
The exosomes released by the donor cells deliver the contents to target cells through three different methods: (1) direct fusion, (2) receptor–ligand binding, (3) endocytosis. After hypoxia stimulation, ADSCs secreted more extracellular vesicles and entered microglia through the mentioned three ways. Extracellular vesicles release miR-30d-5p after entering microglia to inhibit autophagy. ADSCs, adipose-derived stem cells.
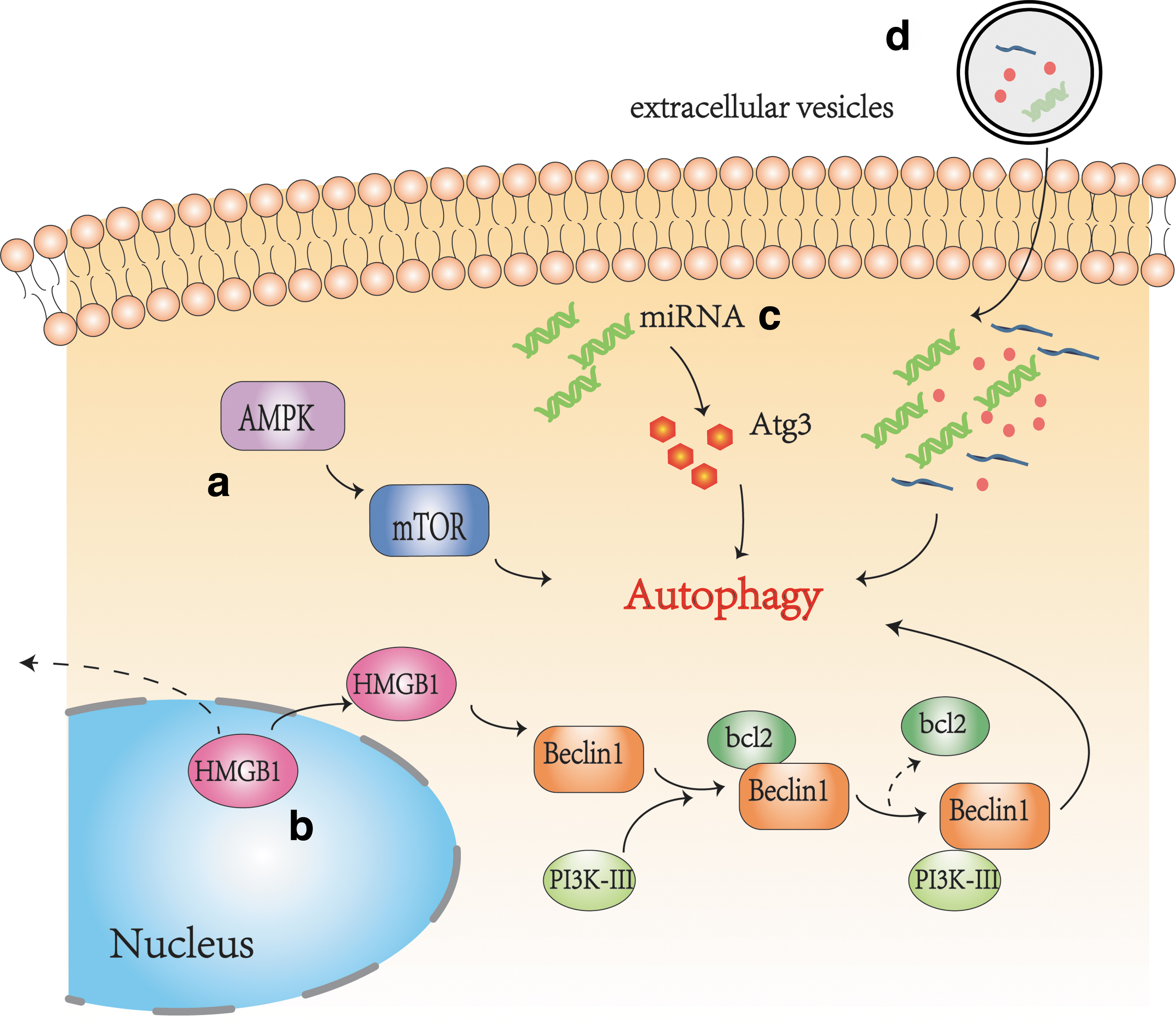
Hypoxia-mediated autophagy of central immune cells (microglia as an example).
The Role of Hypoxia-Induced Glial Cells Autophagy in the Neuroinflammation
At basal state, autophagy engagement with inflammasomes and their substrates supports the production of inflammatory responses by preventing excessive inflammatory responses and tissue damage (Deretic and Levine, 2018). However, under hypoxic environment, influenced by external stimuli, the balance will be broken, leading to different outcomes.
Beneficial effect
The relationship between autophagy and inflammation is complicated, autophagy suppresses inflammation to permit a beneficial inflammatory response is possible (He et al., 2018). Glycogen synthase kinase 3β (GSK-3β) plays a crucial role in cerebral ischemic injury and GSK-3β inhibits autophagic activity in many diseases. Wang et al. (2019) established an MCAO/R model in rats, use chemical inhibitor (SB216763) and GSK-3β siRNA to suppress GSK-3β activation and expression in vivo. The results demonstrated that inhibiting GSK-3β downregulates nod-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome expression by increasing autophagic activity, which reduces cerebral infarct volume. The studies already reviewed illustrate a protective role of autophagy, the enhancement of autophagy in glial cells is considered a treatment strategy for neuroinflammation, and this provides a new idea for the future treatment of hypoxia-associated diseases.
Harmful effect
In contrast to the mentioned studies, autophagy can not only alleviate the neuroinflammatory response but also aggravate the neuroinflammatory response, leading to adverse outcomes. One study conducted by Wang et al. (2020a) has been found that when BV2 microglia exposed to OGD/R, C1q/tumor necrosis factor-related protein-1 (CTRP1), an adiponectin paralog that regulates inflammation, is significantly upregulated. Knockdown of CTRP1 remarkably strengthened OGD/R-induced autophagy and accelerated the inflammatory response in BV2 cells.
Since we have already known the dual effect of autophagy on the neuroinflammatory response, in addition to the mentioned possible molecular mechanisms, the intensity of autophagy may also affect the outcomes of the neuroinflammatory response.
Tian et al. (2010) performed autophagy imaging in mice 1 h and 1, 3, and 6 days after transient MCAO (tMCAO). Western blot analysis showed that the expression of LC3-I and LC3-II peaked 1 day after tMCAO, that is to say, autophagy peaks on the first day after cerebral ischemia and hypoxia, and then the intensity of autophagy decreases. Therefore, we hypothesized that high-intensity autophagy is one of the reasons for aggravating neuroinflammatory response. When autophagy reaches its peak, which may also explain the different effects of autophagy on neuroinflammatory response under different hypoxia or ischemia hypoxia conditions, that is, under severely hypoxic conditions or acute ischemic conditions, high-intensity autophagy aggravates neuroinflammatory response.
Clinical Significance and Prospect
In this review, we show that autophagy is essential for neuroinflammation induced by hypoxic conditions or hypoxic diseases. Autophagy plays an indispensable role in inflammation, immunity, metabolism, and cell survival. A variety of stimuli such as immune and inflammatory signals induce autophagy in macrophages. However, under hypoxic conditions, little is known about the physiological role of autophagy and its signal transduction mechanism in microglia. Microglia is phagocytic immune cells that reside in the CNS and function similarly to macrophages. As the first line of defense against pathogens in the CNS, microglia plays a meaningful role in the initiation and maintenance of neuroinflammation. Neuroinflammation and autophagy are two key cellular processes intertwined that implicated in various diseases in the CNS. These two processes are complicated and tightly linked.
On the one hand, autophagy inhibits neuroinflammation by removing necrotic tissue and debris. On the other hand, autophagy aggravates the development of neuroinflammation. However, the mechanism of relieving autophagy-mediated neuroinflammation to allow beneficial signaling of inflammatory response remains to be explored. Recent studies have found that regulation of microglia activation by modulating mitochondrial autophagy may be conducive to the survival of neuronal cells in PD, and microglia autophagy plays an indispensable role in PD (Li et al., 2019). Therefore, autophagy may be an essential target for regulating neuroinflammation and affecting nervous system injury under hypoxic conditions.
Footnotes
Authors' Contributions
All authors contributed to the concept, design, and writing of this article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (Grant Nos. 81970085 and 81670086) and the Tianjin Science and Technology Plan Project (Grant Nos. 18ZXDBSY00060 and 17ZXMFSY00080).