
Abstract
Hepatocellular carcinoma (HCC) is an aggressive disease with a high degree of tumor heterogeneity. Genetic lesions of mTOR-related genes, including TSC2 and hyperactivation of mTOR signaling, are common in HCC. However, the association of genetic alterations with hepatocarcinogenesis remains unclear. In this study, continuous truncating mutations occurred within or upstream of the TSC2 Rap_GAP domain in clinical HCC samples. To elucidate whether hyperactivation of mTOR signaling in HCC is caused by TSC2 truncating mutations, HCC cell models carrying the TSC2 deletion (CRISPR/Cas9) or the TSC2 truncating mutation (mutagenesis) were established. Our findings showed that either TSC2 deletion or TSC2 mutant could lead to TSC2 loss-of-function and hyperactivation of mTOR signaling. Furthermore, hyperactivation of mTOR signaling was relieved by rapamycin. Immunohistochemistry of clinical samples confirmed frequent TSC2 loss in HCC. Thus, our study revealed that genetic alterations cause TSC2 loss of function and result in the hyperactivation of mTOR, and high frequency of TSC2 truncating mutations around RAP_GAP domain may be one of the reasons for the hyperactivation of mTOR in HCC patients.
Introduction
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related deaths worldwide (Zhu et al., 2014; Roayaie et al., 2015; Kulik and El-Serag, 2019; Yang et al., 2019). More than 80% of HCC cases are diagnosed at advanced stages, and few of them are considered candidates for resection (Roayaie et al., 2015; Kulik and El-Serag, 2019). Treatment options for advanced HCC are limited (Lopez et al., 2006), especially following the failure of sorafenib (Zhu et al., 2014). In addition, HCC is a human cancer with widespread molecular heterogeneity and somatic genetic diversity (Guichard et al., 2012; Huang et al., 2012; Kan et al., 2013).
mTOR is a member of the PI3K family of protein kinases and is the core component of mTORC1 that regulates cell growth, mRNA translation, and metabolism. TSC2 is a well-known tumor suppressor that forms a complex with TSC1 and negatively regulates the mTOR signaling pathway by activating Rheb, a small GTPase, as a GTPase-activating protein (Inoki et al., 2003; Zhang et al., 2003; Huang and Manning, 2008). Notably, aberrant activation of the mTOR pathway in HCC is common, ranging from 15% to 41% of cases (Villanueva et al., 2008), and is largely related to genetic alterations (Hennessy et al., 2005; Huynh et al., 2015). TSC2 has an intermediate mutation frequency (2–20%) (Lawrence et al., 2014) in human cancers, and TSC2 loss of function is frequent in HCC cell lines, patient-derived HCC xenografts, and patients with HCC (Huynh et al., 2015).
TSC2 has three domains (Fig. 1C). The C-terminal region of TSC2 is highly conserved across species and contains a GAP domain (Inoki et al., 2003). TSC1 is important for stabilization and localization of the TSC1/2 complex (Inoki et al., 2003), but it is not required directly for GAP activity, as TSC2 alone inhibits S6K1 and 4EBP1. More mutations are found in TSC2, and a relatively high frequency of mutations occur in its C-terminal Rap_GAP domain, thus confirming the importance of TSC2 and its Rap_GAP domain (Ito and Rubin, 1999; Hodges et al., 2001; Inoki et al., 2003; Henske et al., 2016). However, the influence of genetic alterations in TSC2, especially around the GAP domain, in HCC tumorigenesis remains unclear.
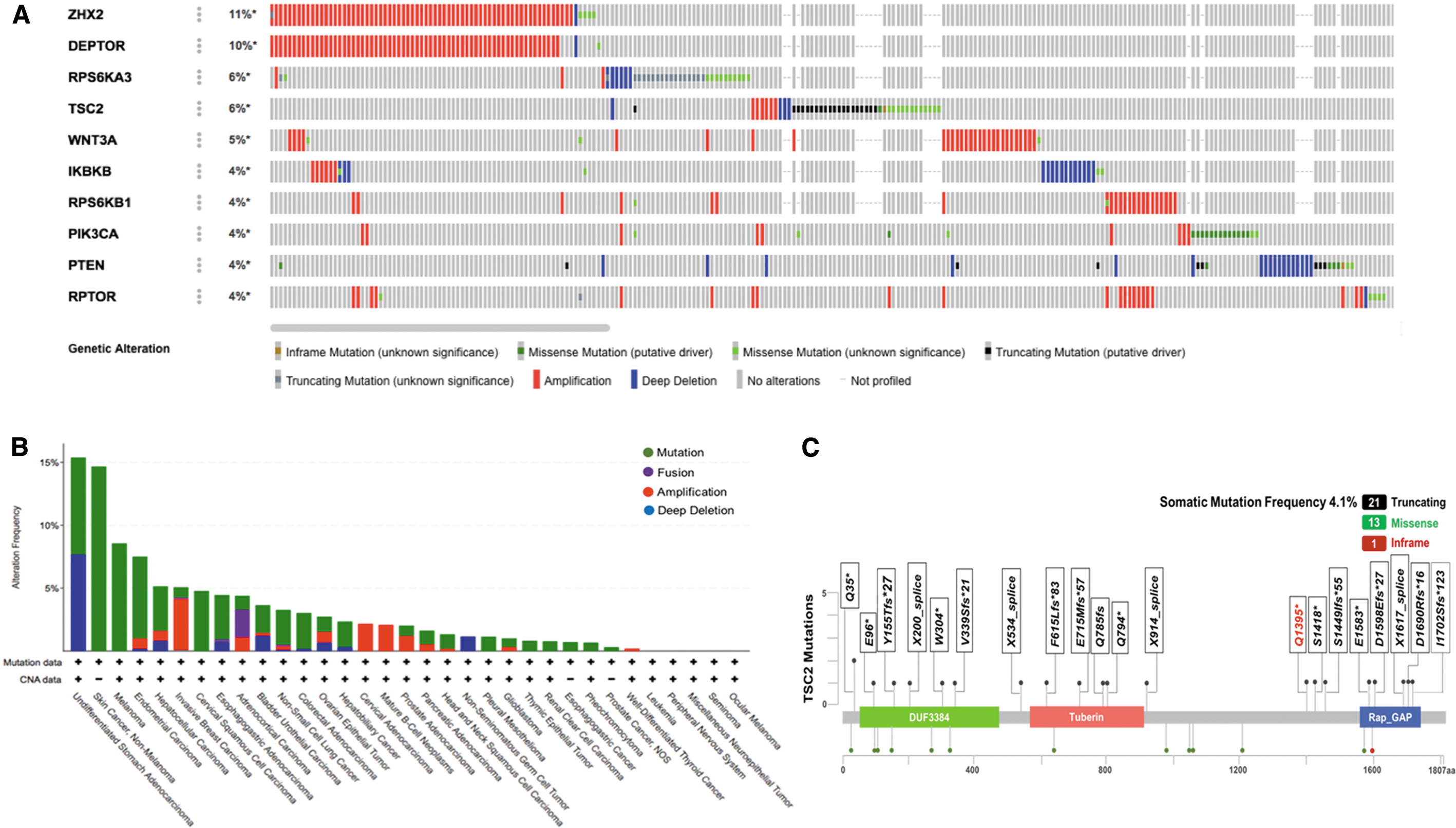
Public dataset-based analysis of genetic alterations for TSC2 and genes in mTOR signaling pathway. Data mining was accomplished using cBioPortal for Cancer Genomics, a data portal available at
Through bioinformatics analyses, we recently identified frequent continuous truncating mutations within or upstream of the functional region of the Rap_GAP domain in clinical HCC samples. In the present study, we created a TSC2 mutation cell model based on an HCC patient who had a truncating mutation upstream of the Rap_GAP domain. We investigated the role of this mutation in TSC2 loss of function and its association with the hyperactivation of the mTOR pathway. We further evaluated the TSC2 expression in HCC clinical samples and aimed to identify and confirm some genetic changes involved in HCC tumorigenesis.
Materials and Methods
Plasmids
pSpCas9(BB)-2A-GFP (px458) (#48138) was purchased from Addgene (Cambridge, MA). The TSC2 target Cas9 guide sequence was listed in Supplementary Table S1. The px458 backbone was digested by BbsI (NEB). sgRNAs were annealed by slow cooling from 95°C down to 10°C. Annealed sgRNA primer mix was ligated to the digested px458 backbone using T4 DNA ligase (NEB). Flag-tagged TSC2 (#14129) was purchased from Addgene.
Cell lines
The HepG2 cell line was obtained from the American Type Culture Collection (ATCC, Rockville, Maryland). The cell line was authenticated by examination of morphology and growth characteristics and was confirmed to be mycoplasma free. TSC2 knockout colonies were confirmed by western blot and Sanger sequencing after single cell sort (BD FACSMelody™ Cell Sorter) with GFP.
Antibodies
Antibodies specific for the following proteins were used as primary antibodies: TSC2 (sc-893, 1:2000 for Western blotting (WB) and 1:100 for Immunohistochemistry [IHC]), Santa Cruz; Phospho-p70 S6 Kinase (Thr389) (9206, 1:1000), S6 Ribosomal (2317, 1:1000), Phospho-S6 Ribosomal (Ser235/236) (2211, 1:1000 for WB and 1:100 for IHC), 4EBP1 (9644, 1:1000), Phospho-4E-BP1 (Ser65) (9451, 1:1000 for WB and 1:100 for IHC), Flag (9451, 1:1000), Cell Signaling, Inc.; and Beta IV Tubulin (ab179509, 1:5000), Abcam.
Western blotting
Cell lysates containing 50–100 μg of protein were prepared and subjected to 10% sodium dodecyl sulfate–polyacrylamide gels. Proteins were transferred to Polyvinylidene Fluoride (PVDF) membranes, which were blocked in 5% nonfat milk for 1 h and incubated overnight with primary antibodies. The membranes were incubated at room temperature for 1 h with anti-rabbit/mouse IgG HRP-linked secondary antibody (1:5000, Cell Signaling, Inc.).
Site-directed mutagenesis
Site-directed mutagenesis of the TSC2-Mutation vector was accomplished following the protocols from the Site-Directed Mutagenesis System (Thermo Fisher Scientific). The primers, including the mutation site, were designed as following: Forward: 5′-CTGAGGTTAAGGCCCGGTCATAGTCAGGGACCCTGGACGGG-3′; Reverse, 5′-CCCGTCCAGGGTCCCTGACTATGACCGGGCCTTAACCTCAG-3′.
Xenogeneic transplantation
Inject cells (100 μL, 5 × 107/mL) into the left flanks of 6-week-old NOD/SCID/IL-2Rγ(null) (NSG) mice (Jackson Laboratories) subcutaneously. Twenty male NSG mice were assigned to four different groups randomly, five mice in each group. Tumor growth was monitored every 2 or 3 days. At 31 days after injection, NSG mice were sacrificed and tumor volume and weights were measured. The experiment was repeated three times, and a total number of 60 mice were used. All experiments were conducted in accordance with accepted standards of animal care and approved by the Institutional Animal Care and Use Committee of the Second Hospital of Jilin University (Number of permit: KT202103006).
Tissue microarray samples
Three hundred four formalin-fixed tissue microarray (TMA) samples were purchased from Alenabio, Inc (China, Xi'an). Brief clinical and pathological characteristics of the subjects are presented in Table 1. All had histologically confirmed HCC with information on tumor stage (Tumor–Node–Metastasis) and grade based on the American Joint Committee on Cancer (AJCC) stage system.
Tissue Microarray Sample Characteristics
HCC, hepatocellular carcinoma.
The commercialized microarray was determined as Not Human Subjects for the reason that we do not have any identified patients' information. This project was determined as Not Human Subjects Research and approved by the Ethics Committee of the Second Hospital of Jilin University. The approval letter is submitted as additional file.
Immunohistochemistry
Antigens were retrieved in retrieval buffer (Dako) for 12 min. ABC detection system (Vector Laboratories, Burlingame, CA) was used for immunostaining. Tissues were incubated with specific primary antibodies at 4°C overnight. Goat anti-mouse/rabbit IgG (Invitrogen, 1:200) was used as the secondary antibody. Staining was followed by ABC detection system (VECTASTAIN Elite ABC). H-scores were calculated by the multiplication of dominant intensity pattern of staining (1, weak; 2, moderate; 3, intense) and the percentage of positive tumor cells per sample (0–100%). Thus, the H-scores range from 0 to 300.
Statistical analyses
Mean, standard deviation, and median values were used to summarize continuous variables. Samples were evaluated for the distribution using the one-sample Kolmogorov–Smirnov test. In samples with normal distributions, the two-tailed t-test was used to compare mean variation between two groups. In samples with non-normal distributions, the Mann–Whitney U test was used to compare the median variation between two groups. One-way analysis of variance (ANOVA) was used to test for differences among at least three groups, two-way ANOVA was used to compare the influence of two categorically independent variables on one dependent variable. All data were analyzed with SPSS (version 24; IBM, Armonk, NY).
Results
Continuous TSC2 truncating mutations occur frequently within or upstream of the Rap_GAP domain
Many genes in the mTOR pathway are dysregulated in cancer, leading to the hyperactivation of this pathway (Zoncu et al., 2011). Among this set, loss-of-function mutations of tumor suppressors, such as phosphatase and tensin homolog (PTEN), TSC1/2, and neurofibromin 1/2 (NF1/2), are the most common (Zoncu et al., 2011).
In the present study, the mutation rates and patterns were analyzed in 87 mTOR-related genes from the KEGG gene set of four HCC studies. TSC2 was among the top four mutant genes in the list, in which 86 genes had at least one mutation (Fig. 1A). TSC2 was mutated in 28 of 33 different cancers, and HCC ranked fifth (Fig. 1B). The somatic mutation frequency was 4.1%, with 13 cases of missense mutations, 21 cases of truncating mutations and 1 case of an in-frame mutation (cBio,
Establishment of TSC2 knockout and TSC2 mutant HCC cell models
To investigate the association of the TSC2 truncating mutation, especially upstream of the Rap_GAP domain, with tumorigenesis and development of HCC, we used several approaches to generate cell models. First, we compared the basal TSC2 protein expression in HepG2 cells and Huh7 cells with that in other cancer cell lines. Figure 2A showed that TSC2 was highly expressed in HepG2, MDA-MB-231, DU145, and PC3 cells but poorly expressed in Huh7 cells. Second, we used CRISPR/Cas9 to knock out TSC2 in HepG2 cells, and the TSC2 knockout (TSC2 KO) cells were confirmed by Sanger DNA sequencing and western blotting (Fig. 2B). Third, we selected a truncated mutation (Q1395*) from the continuous truncating mutations upstream of the functional region of the Rap_GAP domain and use site-directed mutagenesis to mutate the Gln (CAG) to a stop codon (TAG) in a wild-type (WT) TSC2 expression vector (Fig. 2C, D).
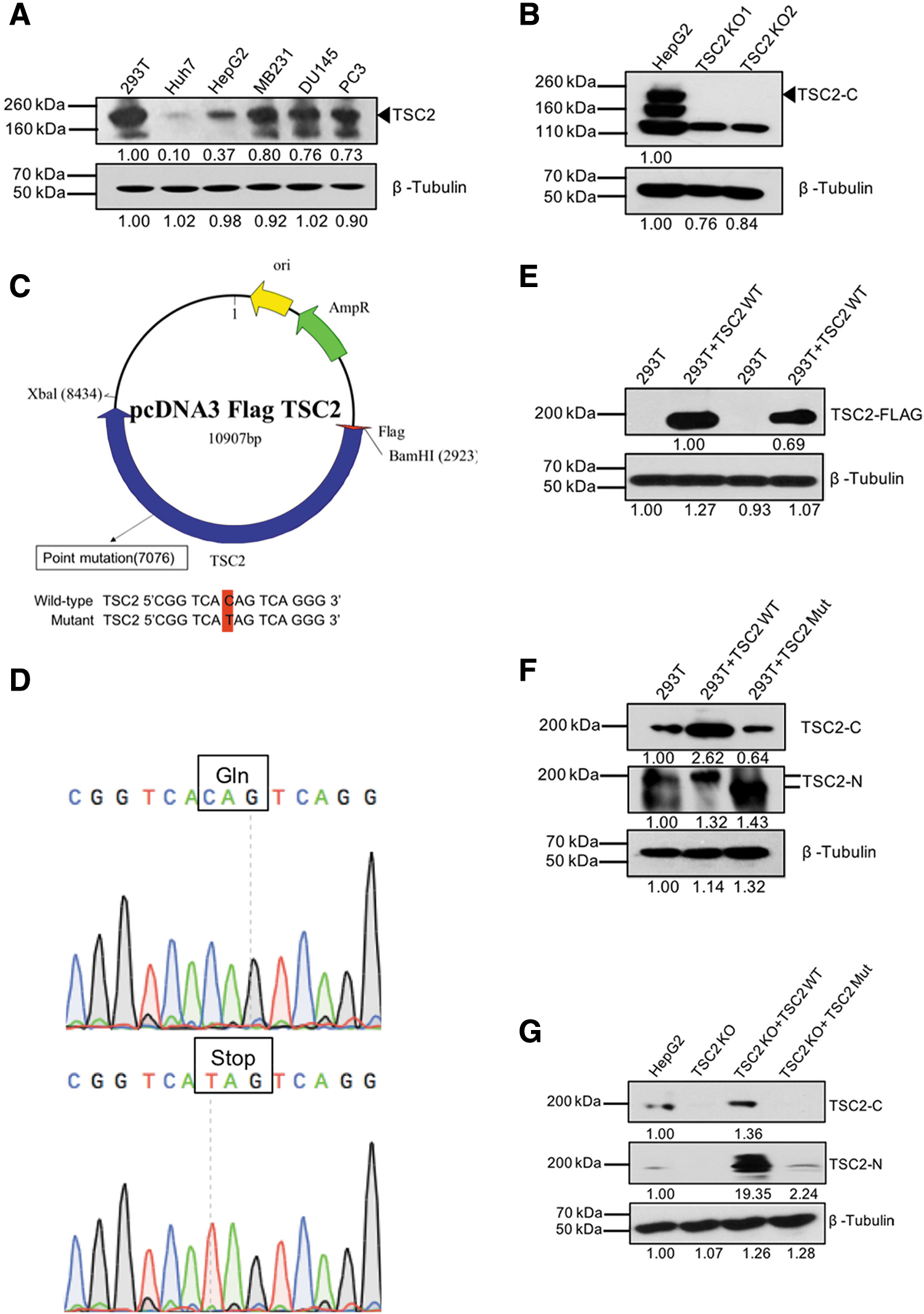
Establishment of TSC2 knockout and TSC2 mutant HCC cell models.
The efficiency of the mutant vector was confirmed in the HEK293T cell line (with endogenous TSC2; Fig. 2E, F) and TSC2 KO HepG2 cells (Fig. 2G). We identified the expression of TSC2 with an N-terminal but not C-terminal antibody (Fig. 2G), indicating that the C-terminal region of TSC2 was lost, while the N-terminal region was expressed normally. Next, HepG2 cells were transfected with pcDNA3.0 control vector, whereas TSC2 KO cells were transfected with pcDNA3-Flag- WT TSC2 or mutant TSC2 vector to set up TSC2 KO+TSC2 WT, and TSC2 KO+TSC2 Mut stable cell models.
TSC2 loss of function promotes cell proliferation and invasion
Functional loss of tumor suppressors plays a pivotal role in tumorigenesis. To determine the function of TSC2 in cell growth, we carried out several cell proliferation experiments. The cell growth was facilitated in TSC2 KO cells compared with WT HepG2 cells, and cell growth was inhibited following TSC2 overexpression (TSC2 KO+TSC2 WT) (Fig. 3A, B). However, tumor suppressor activity was largely reduced once TSC2 was mutated, as cell proliferation was higher in TSC2 mutant cells than in WT HepG2 cells, but lower in TSC2 KO cells. Furthermore, TSC2 overexpression and TSC2 mutant play opposite role in cell growth and colony formation (Fig. 3C). Separately, we also identified that TSC2 mutant cells displayed a stronger clone formation ability (Fig. 3D), thereby suggesting that the N-terminal region of TSC2 is partially required for suppression of cell proliferation.
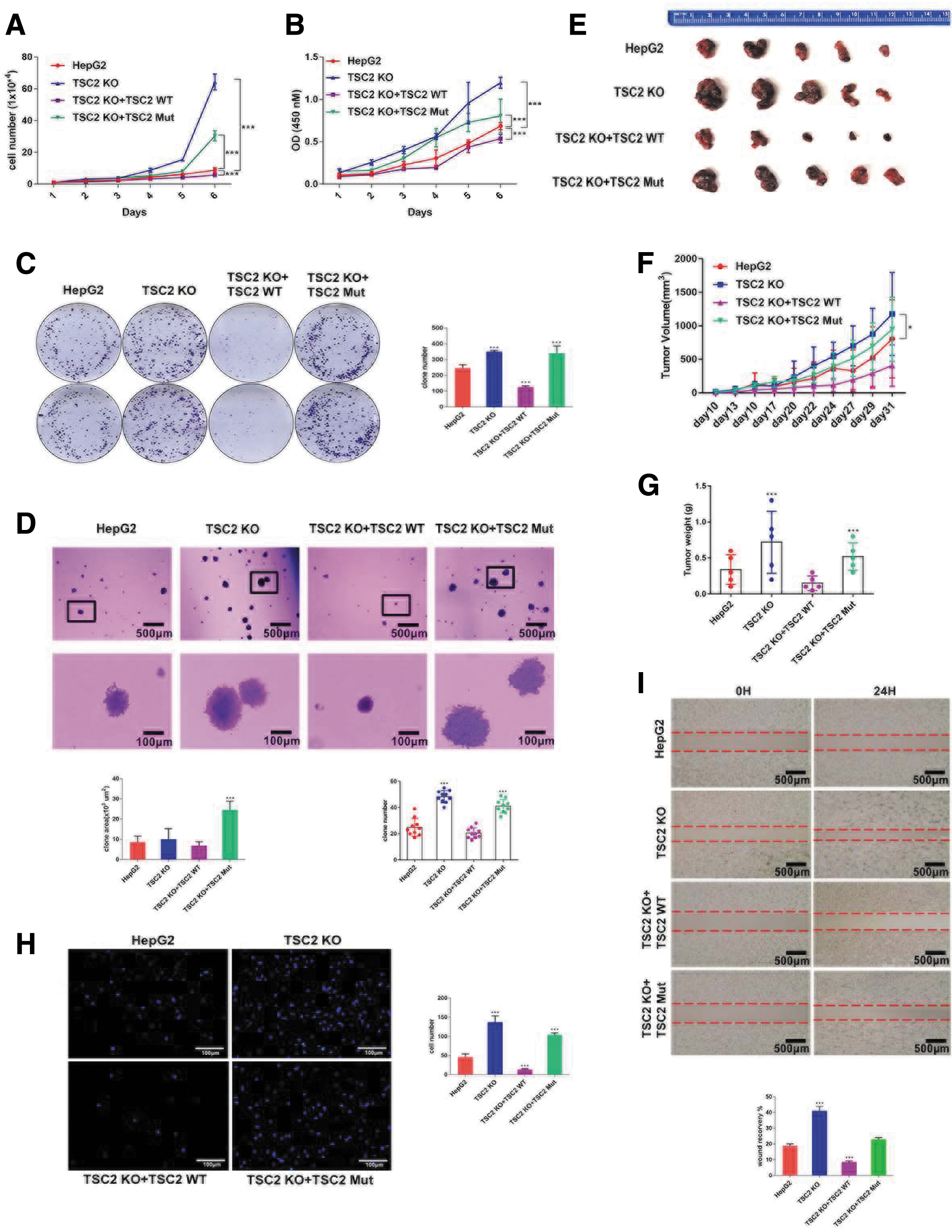
TSC2 loss of function promotes cell proliferation and invasion.
To confirm these results in vitro, we injected WT HepG2, TSC2 KO, TSC2 KO+TSC2 WT, or TSC2 KO+TSC2 Mut HepG2 cells into NGS mice subcutaneously. Xenograft tumor growth was increased and tumor weights were higher in mice with TSC2 KO or TSC2 mutant cells compared with WT HepG2 cells, while tumor growth was strongly inhibited in mice with TSC2 overexpressed cells (Fig. 3E–G).
We also tested the effects of TSC2 on cell migration by transwell assays (Fig. 3H) and scratch assays (Fig. 3I). TSC2 KO significantly accelerated wound closure 24 h after scratch wounds in HepG2 cells. In contrast, the 24-h wound recovery of TSC2 overexpressed cells was less than 10%. Consistent with scratch assays, migrated cells were increased significantly following TSC2 KO. The number of migrated TSC2 mutant cells increased two-fold relative to that in WT HepG2 cells, whereas only a few cells per foci were transferred through the membrane when TSC2 was overexpressed.
TSC2 loss of function leads to the hyperactivation of the mTOR pathway
The PI3K-Akt-mTOR pathway not only participates in many metabolic processes but also plays an essential role in the regulation of cell size by controlling biochemical synthesis of proteins, nucleic acids, and lipids (Schmelzle and Hall, 2000; Gingras et al., 2001; Saran et al., 2015). The TSC complex accepts signals from nutrients, energy, and growth factors, regulate Rheb, and abolishes the activation of rapamycin complex 1 (mTORC1) (Saran et al., 2015). We observed changes in cell morphology and cell size after TSC2 KO. Cells were characterized by the large nuclei with conspicuous and vacuolated nucleoli (Fig. 4B), and these changes were also observed in TSC2 mutant cells (Fig. 4B), indicating that the loss of function of either TSC2 or the Rap_GAP domain may affect the reception of growth factor signals by TSC2.
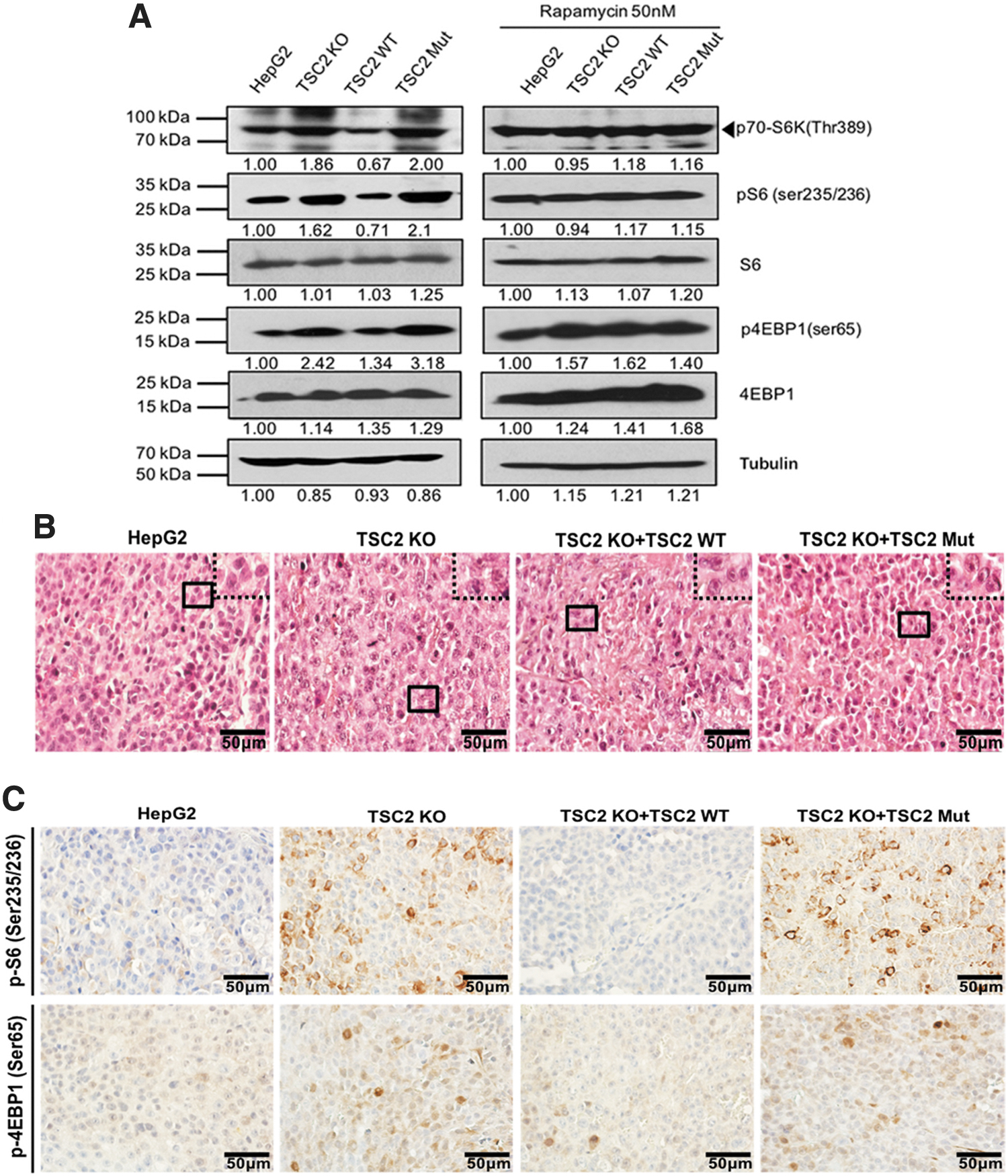
Loss of function of TSC2 leads to hyperactivation of mTOR pathway.
Hyperactivation of mTOR has been reported in various cancers (Zoncu et al., 2011; Grabiner et al., 2014). Phosphorylation of 4EBP1 releases eukaryotic translation initiation factor 4E (eIF4E) to promote mRNA translation, whereas the activation of S6K1 stimulates ribosome biogenesis and the phosphorylation of downstream substrates, including S6 (Jeno et al., 1988). Of note, the main upstream regulators of mTORC1 are the TSC1/2 complex, therefore, TSC2 loss of function may cause dysregulation of the signaling network.
During the development of anticancer therapies targeting mTOR, rapamycin and its analogs showed great promise and eventually became U.S. Food and Drug Administration (FDA)-approved anticancer agents (Saran et al., 2015). Our data show that mTOR was hyperactivated following TSC2 KO, but this phenotype was partially relieved after treatment with rapamycin. Expression of p-S6K1, p-S6, and p-4EBP1 were dramatically upregulated after TSC2 KO (Fig. 4A).
Conversely, the mTOR pathway was suppressed if TSC2 was overexpressed (Fig. 4A). However, treatment with 50 nM rapamycin abolished the differences in p-S6K1, p-S6, and p-4EBP1 expression (Fig. 4A), suggesting that TSC2 functions upstream of mTORC1, while rapamycin antagonizes the dysregulation of the mTOR pathway after TSC2 loss of function. Likewise, we found overexpression of mTOR downstream proteins in the TSC2 mutant group (Fig. 4A). IHC data from xenograft tumors showed that either TSC2 KO or TSC2 mutant increased p-S6 and p-4EBP1 expression levels (Fig. 4C), which was consistent with the in vitro data, showing that TSC2 loss of function results in hyperactivation of the mTOR pathway.
TSC2 loss is common in HCC clinical samples
Proteins are essential for the translational and post-translational regulation. However, protein expression level and functions of proteins cannot be predicted by genomic analysis (e.g., DNA copy number and mRNA expression) (Akbani et al., 2014), and both factors vary between different tumors and subtypes.
To determine the correlation between TSC2 and tumorigenesis, we analyzed its protein expression profile in different cancers using data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) Confirmatory/Discovery dataset (Supplementary Fig S1). We found that protein expression levels of TSC2 are higher in normal tissues than in tumor tissues, with the exception of breast cancer, which indicates loss of TSC2 in clinical cancer samples. Notably, low TSC2 expression is associated with more aggressive tumor behavior and poor outcome in uterine corpus endometrial carcinoma. To detect TSC2 status in HCC, we performed IHC analysis in HCC clinical samples. A total of 240 HCC tissues and 64 normal tissues were used to test TSC2 protein expression (Table 1).
We defined normal tissues with H-scores ranging from 100 to 300, and HCC tissues ranging from 0 to 300. These included 37 samples with high TSC2 expression (H-score = 200–300) in normal and 149 low TSC2 expression (H-score = 0–100) in tumors, suggesting that low TSC2 expression is common in HCC tumor tissues (Fig. 5A). In addition, TSC2 loss in primary HCC (Grade I/T1) was not obvious compared with that in aggressive HCC (Grade II/III/IV; Fig. 5B, C). These findings indicate an association between TSC2 loss and tumorigenesis and HCC pathogenesis.
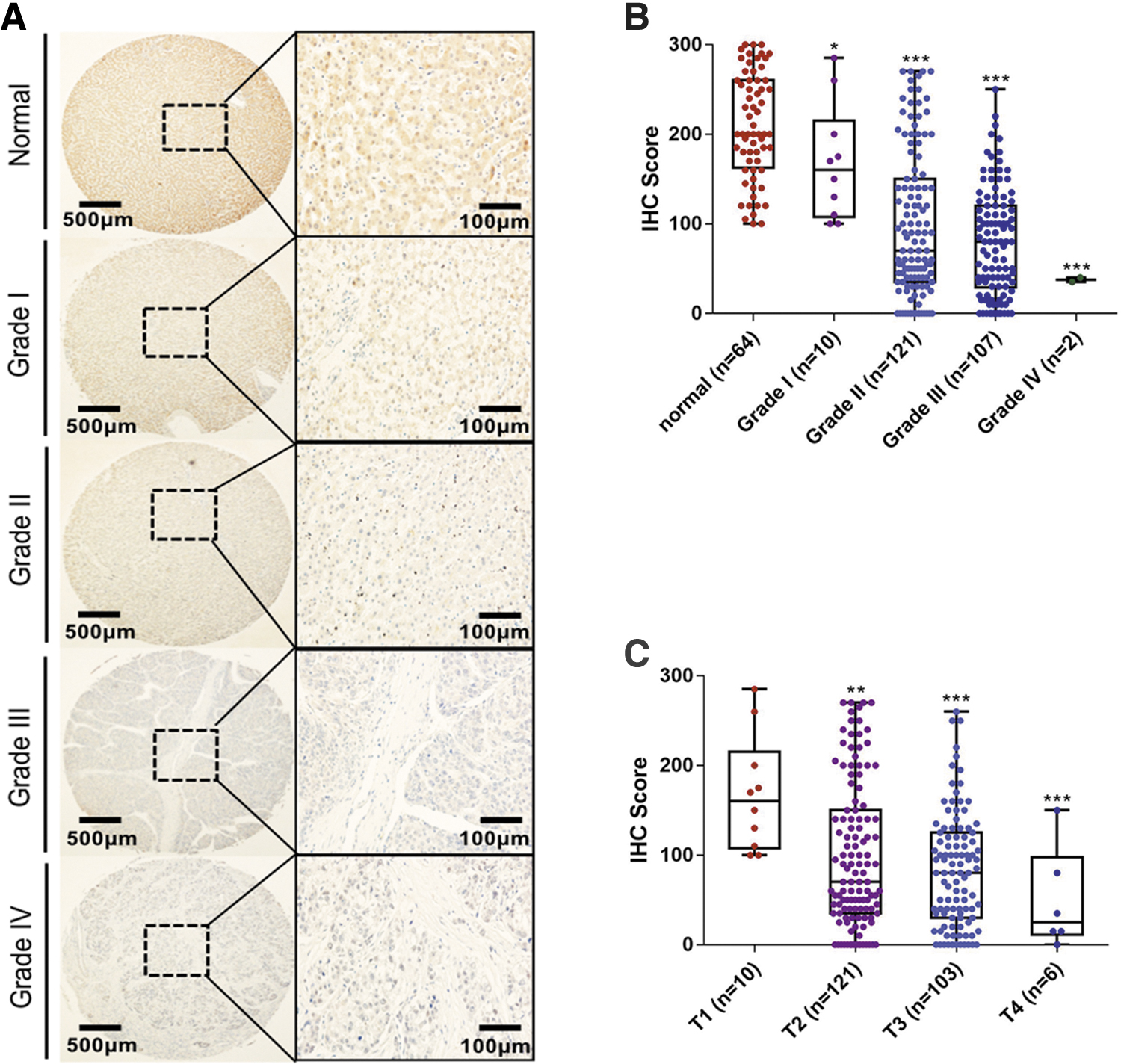
TSC2 loss was common in HCC clinical samples.
Discussion
A loss of function in tumor suppressors, including TSC, and the hyperactivation of mTOR signaling have been reported in HCC-related studies (Villanueva et al., 2008; Huynh et al., 2015), but the molecular mechanisms remained unclear.
Our data analysis demonstrates that multiple pivotal mTOR signaling pathway-related genes show frequent mutations in HCC, and tumor suppression genes have a relatively high frequency. A previous study found many different types of TSC2 mutations in HCC, including nonsense, missense, in-frame deletions, and splices (Ho et al., 2017). However, we found that truncating mutations were the major genetic alterations in clinical HCC samples. Furthermore, this kind of premature termination of translation is always around the Rap_GAP domain of TSC2, illustrating the mechanisms of inactivation by mutation of tumor suppressor genes in carcinogenesis.
In human genetics, a single loss-of-function mutation can result in obvious phenotypic effects, and mutations in pleiotropic genes cause accumulated invisible effects (Fuller et al., 2019). Data from this study indicated that a single truncating mutation of TSC2 can promote proliferation in HCC, suggesting that loss function in the Rap_GAP domain is likely to have a discernible phenotypic effect in HCC. Our in vitro and in vivo data showed that both TSC2 KO and TSC2 mutants caused the loss of function in TCS2 and the hyperactivation of the mTOR pathway. Therefore, the increased frequency of truncating mutations upstream of the TSC2 Rap_GAP domain could be one of the reasons for the hyperactivation of mTOR in HCC patients. These data also suggest that the Rap_GAP domain contributes to TSC2-mediated suppression of tumor growth and progression. Locus-specific mutation databases list more than 3000 variants of TSC2 (
In our experiments, the N-terminal region of TSC2 seems to have a limited role in HCC proliferation, thus, it is necessary to verify the relationship between truncating transcription variants and human cancer. In addition, TCS2 loss of function increased the expression of p-S6K, p-S6, and p-4EBP1 expression, which unraveled the underlying molecular mechanism of the hyperactivation of the mTOR pathway in HCC. Of note, once a mutation occurs in the GAP domain of TSC2, it cannot regulate the small GTPase Rheb or downstream S6 and 4EBP1 (Zhang et al., 2003; Yang et al., 2017). Therefore, activation of mTOR in the TSC2 mutant group confirmed the results above.
Cell models carrying TSC2 truncating mutations with hyperactivation of mTOR signaling were sensitive to rapamycin treatment. This may be the reason why cell lines/xenograft models with TSC2 mutations display heightened sensitivity to rapamycin. Detection of genetic events in TSC2 is more likely to identify patients with HCC that benefit from mTOR inhibitors (Ho et al., 2017).
Currently, TSC KO mouse models have demonstrated the crucial role of the TSC1/2 complex in HCC development (Menon et al., 2012; Kenerson et al., 2013) and few studies have focused on the expression of TSC2 in HCCs. Herein, we performed TSC2 IHC assays and found that TSC2 loss is common in tumors and that low TSC2 protein expression is associated with more aggressive tumors. These data may provide a theoretical basis for gene testing of HCC. Unfortunately, we were unable to confirm the correlation of TSC2 expression with the sensitivity to mTOR inhibitors in these patients. As IHC can reflect the TSC2 loss from both genetic and nongenetic mechanisms, it could be superior to DNA sequencing as a predictor for cancer-targeted therapy.
Conclusion
This study provides novel insights into the genetic alteration-dependent regulation of TSC2 in mTOR signaling and the underlying molecular mechanism for TSC2 loss of function of and hyperactivation of mTOR. TSC2 truncating mutations with loss of the Rap_GAP domain represents one of the major mechanisms of TSC2 loss, which is highly associated with tumorigenesis and progression of HCC.
Footnotes
Authors' Contributions
The authors confirm that this article have not been published previously and it is not under consideration for publication elsewhere. The publication is approved by all authors and tacitly or explicitly by the responsible authorities where the work was carried out, and that, if accepted, it will not be published elsewhere in the same form, in English or in any other language, including electronically without the written consent of the copyright holder.
Acknowledgment
The authors thank the Jilin Provincial Key Laboratory on Molecular and Chemical Genetic of the Second Hospital of Jilin University.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the Natural Science Foundation of China (No. 81902484), China Postdoctoral Science Foundation-funded project (2020M670864), and Medical and Health Talents Project of Jilin Province (2019SCZT003).
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.