
Abstract
Abstract
Precipitation of carbonate minerals can play important roles in geological carbon sequestration and engineered processes for mineral carbonation. Influences of temperature, solution composition, and the presence of a solid substrate on the nucleation and precipitation of magnesium carbonate minerals were examined in a set of batch experiments. Conditions studied are relevant to full-scale geological carbon sequestration systems. Aqueous phase analysis by inductively coupled plasma mass spectrometry quantified the extent of precipitation. X-ray diffraction analysis was conducted to identify solids. Temperature significantly affected the identity of the solid obtained. At 25°C and 60°C the solids were magnesium carbonate minerals, and at 100°C the solid phase was identified as brucite [Mg(OH)2]. Although magnesite (MgCO3) was predicted to be the most thermodynamically stable magnesium carbonate phase, no magnesite precipitated and instead metastable magnesium carbonate phases formed. Evolution of dissolved concentrations was consistent with precipitation of these metastable phases. Presence of the magnesium silicate forsterite had no measurable effect on the rate or extent of precipitation. Mineralization in geological systems is likely to also be controlled by ionic strength, pressure, and mineralogy of the host formation.
Introduction
Divalent cations may also be provided via the dissolution of magnesium-rich minerals like olivine and brucite in engineered reactors for ex situ mineralization (Goff and Lackner, 1998; O'Connor et al., 2005; Zhao et al., 2010). Analyses indicate that ex situ mineralization may have major environmental impacts due to the scale of mining required to provide ore as a source of magnesium and other carbonate-forming cations (Gerdemann et al., 2007). Combined with geochemical and economic considerations, this suggests that ex situ mineralization may be part of an integrated solution to carbon sequestration but will not stand alone in stemming anthropogenic CO2 release.
The MgO-CO2-H2O system can produce a number of solid phases (Hanchen et al., 2008). Depending on the pH and concentrations of carbonate and Mg, the thermodynamically most stable phase will be either magnesite (MgCO3) or brucite [Mg(OH)2]. In practice their production is often preceded or usurped by metastable phases (Bender and Sprague, 1965; Pokrovsky, 1998; Hanchen et al., 2008; Zhao et al., 2010). Metastable phases include the hydrated and basic Mg-carbonates nesquehonite (MgCO3·3H2O) and hydromagnesite [(MgCO3)4·Mg(OH)2·4H2O]. Relatively low temperatures and CO2 partial pressures, including ambient conditions, favor the formation of nesquehonite (Davies and Bubela, 1973; Ferrini et al., 2009; Ballirano et al., 2010). Higher temperatures favor hydromagnesite (Davies and Bubela, 1973). The transformation between these two phases has been demonstrated to occur at 40°C (Bender and Sprague, 1965), though a more recent real-time X-ray diffraction (XRD) study found nesquehonite to be stable up to 100°C (Ballirano et al., 2010). Production of magnesite requires, at a minimum, elevated temperature and CO2 partial pressure (Deelman, 2001; Hanchen et al., 2008).
The objective of this study was to examine the effects of temperature, solution composition, and substrate presence on the formation of magnesium carbonate minerals in aqueous systems at ambient pressure and atmospheric CO2 composition. Although experiments were not performed at the high pressures and CO2 fugacity that will be encountered in carbon sequestration systems, the variables of temperature, solution composition, and substrate presence can impact precipitation reactions that may occur during carbon sequestration. The impacts of these variables on magnesium carbonate precipitation may be different at higher pressure, especially due to the simultaneous low pH and high carbonate concentrations that can be achieved when high PCO2 is present; however, the investigation of these variables at ambient conditions can still provide information regarding magnesium carbonate formation processes. A better understanding of the variables affecting magnesium carbonate precipitation can provide insights into the role of carbonate mineral formation in carbon sequestration strategies.
Experimental Protocols
Batch experiments
Experiments were performed in 250 mL glass reactors with Teflon-lined plastic caps that behaved as closed systems (I-CHEM). Reactors were loaded with 200 mL of magnesium chloride and sodium bicarbonate solutions (Fisher Scientific) to the concentrations given in Table 1. These solutions were prepared under the ambient atmosphere with a CO2 partial pressure of 10−3.4 bar before being added to the reactors and sealed. Control experiments containing either only magnesium chloride or only sodium carbonate were also performed. The pH was allowed to drift over time. Experiments were carried out in ovens monitored by digital thermometers and maintained at 25°C±4°C, 60°C±4°C, and 100°C±4°C. Reactors were loaded under the ambient atmosphere and operated as sealed vessels without pressurization. Temperature was controlled over the course of 7 days, during which pH measurements were taken at the time of mixing and at 1, 3, and 7 days. At those same intervals 10 mL aqueous subsamples were extracted and filtered (0.22 μm; VWR International), and the filtrates were acidified with 0.1 mL concentrated nitric acid. If enough precipitate was available for XRD characterization after 7 days of reaction, the solids were filtered (0.45 μm; Fisher Scientific) and allowed to air-dry before analysis. In selected experiments 0.1 g of ground forsterite (Mg2SiO4) was included to act as a potential substrate for magnesium carbonate nucleation. Forsterite from Ward's Scientific was ground with an agate mortar and pestle and sieved to the 106–250 μm size fraction.
SI with respect to magnesite, determined as described in the text.
Actual initial SI with respect to magnesite was determined by using equilibrium constants provided by the SUPCRT92 software package using the slop98 thermodynamic database.
Type 1 and type 2 methods of setting composition are described in the text.
For some experiments a solid was observed, but insufficient material was available for identification using X-ray diffraction.
SI, saturation index.
Approaches to determining the magnesite saturation index
The saturation index (SI) of the initial solution with respect to the magnesium carbonate magnesite was an important factor evaluated in this study. Saturation indices were calculated with Equation 1:
Here Q is the ion activity product and Ksp is the equilibrium constant for magnesite dissolution. For the calculations of the SI that were used to prepare experiments, a Ksp for MgCO3(s) of 10−5.17 was obtained from Lide (2000). The concentration of
The amount of inorganic carbon that could partition to the headspace of the reactors was negligible (<2.5% of the total added), and the headspace PCO2 could increase only as high as 0.003 bar. The equilibrium constants used in SI calculations are compiled in Table 2. The conditional acid–base equilibrium constants reported for nonzero ionic strength solutions take activity coefficients into account:
Values of K at 25°C are from the MINEQL+ database, and values at higher temperatures were calculated using the van't Hoff equation with ΔHr values from the MINEQL+ database.
K′ is the conditional equilibrium constant based on concentrations of species and is specific to a particular ionic strength at 25°C. This particular ionic strength value (I=0.945 M) is determined for the experiment having method 1 composition at 25°C and ambient pressure and target SI for magnesite of 1. Activity coefficients were calculated with the Davies equation.
Values were calculated using SUPCRT92 with the database dslop98. Since SUPCRT92 can only calculate K values in the single-phase regions of fluid water, K values reported for 100°C are actually those calculated for 99°C.
K′ is the conditional equilibrium constant based on concentrations of species and is specific to a particular ionic strength, as described in notation b. Activity coefficients were calculated with the Davies equation.
logKsp at 25°C from Lide et al. (2000).
logKsp from Lide et al. (2000) corrected for elevated temperature using the van't Hoff equation with ΔHr values from the MINEQL+ database.
Activity coefficients (γHCO3- and γCO32-) were calculated with the Davies equation (Davies, 1962) and standard values of Ka1 (10−6.35) and Ka2 (10−10.33) were employed. The Davies equation is appropriate when ionic strength is lower than 0.5 M. Some precedent exists for applying the Davies equation in situations where I>0.5 (Pokrovsky, 1998).
In the calculations to set experimental solution compositions, the equilibrium constants were adjusted for temperature using the van't Hoff equation with the Ksp value from Lide (2000) and the Ka1 and Ka2 values from the MINEQL+ database (Schecher and McAvoy, 2007). Subsequent to these initial calculations, saturation indices for magnesite were calculated using the initial compositions and the equilibrium constants from SUPCRT92 (Table 2). Because of large differences in the equilibrium constants calculated using SUPCRT92 (Johnson et al., 1992) and those calculated from Lide (2000) and the MINEQL+ database, the difference in calculated saturation indices is quite large (Table 1). In the presentation of experimental results, the SI used to set up experiments will be referred to as the target SI.
At standard temperature, the Ksp for magnesite varied by three orders of magnitude between the Lide (2000) value and that obtained from SUPCRT92. As temperature increases, the van't Hoff corrections using Ksp for magnesite from Lide (2000) evolve in the same direction as SUPCRT92 Ksp, with both predicting a decrease of just over an order of magnitude (Table 2). Using information regarding the molar volume changes of reactions, SUPCRT92 also provides a means of calculating equilibrium constants at nonstandard pressures. Although all experiments in this study were performed at ambient pressure, differences in equilibrium constants with increasing pressure to values relevant to geological carbon sequestration are relatively small. For example, the logKsp value for magnesite decreased from −8.04 to −7.94 at 25°C and from −9.39 to −9.31 at 95°C when increasing the pressure from 1 bar to 100 bar. Consequently, the primary effects of increasing PCO2 would be to decrease the solution pH and increase the dissolved inorganic carbon concentration.
Setting initial composition
Two types of initial compositions were employed in this study. In the first, referred to as type 1, the Mg concentration was set to 0.1 M, the theoretical initial pH to 8, and the target SI was achieved by addition of sodium bicarbonate (Eqs. 1 and 2). In the second approach, referred to as type 2, the MgCl2:NaHCO3 ratio was fixed at 1:2. The alkalinity added to the reactors is the same as the NaHCO3 concentration, and this 1:2 ratio of magnesium to alkalinity is the same as would result from the dissolution of the magnesium silicate mineral forsterite. Charge balance (Eq. 5) and Equation 1 were used to calculate the required initial composition to generate the target SI.
Analysis methods
Aqueous concentrations of Mg were determined by inductively coupled plasma mass spectrometry (Agilent 7500 ce) with Sc, Li, and Ga internal standards. A pH meter with a glass electrode (Fisher Scientific; Accumet) was used to measure pH, using automatic temperature correction when necessary. To prepare for XRD, solids were ground in an agate mortar and pestle and flattened onto a glass slide. XRD scans were run from 10 to 60° of 2θ with a 0.04° 2θ step size on a Rigaku Geigerflex DMAX/A diffractometer using Cu-Kα X-rays.
Equilibrium speciation calculations
A series of equilibrium speciation calculations were performed to predict the compositions of aqueous solutions in equilibrium with different Mg-containing minerals. Speciation calculations were executed separately from the experiments and provide context for examining the results. These calculations used the equilibrium constants determined with the SUPCRT92 software package using the slop98 thermodynamic database (Johnson et al., 1992) that are included in Table 2. The chemical equilibrium software program MINEQL+ Version 4.6 was employed for speciation calculations (Schecher and McAvoy, 2007). Since the initial MgCl2 and NaHCO3 concentrations were selected based on a closed system using Equations 1 and 5, the speciation calculations were also performed for closed systems.
Results
Effect of temperature
The measured loss of Mg ions from solution was interpreted as a proxy for extent of solid precipitation, since all observed solids bore Mg and no other pathway for loss of Mg existed. Higher temperature corresponded to more and faster precipitation of Mg-containing solids. At 25°C, little loss of Mg was observed even when the solution was supersaturated with respect to magnesite. However, experiments with the same SI at 60°C and 100°C indicate more overall loss of Mg (Fig. 1). Regardless of SI, experiments at 25°C show no significant decrease in Mg concentration, whereas some decrease is seen at 60°C (Fig. 2). At 100°C the most loss is observed, peaking at 86% loss at SI=2 and no initial forsterite (Fig. 3).
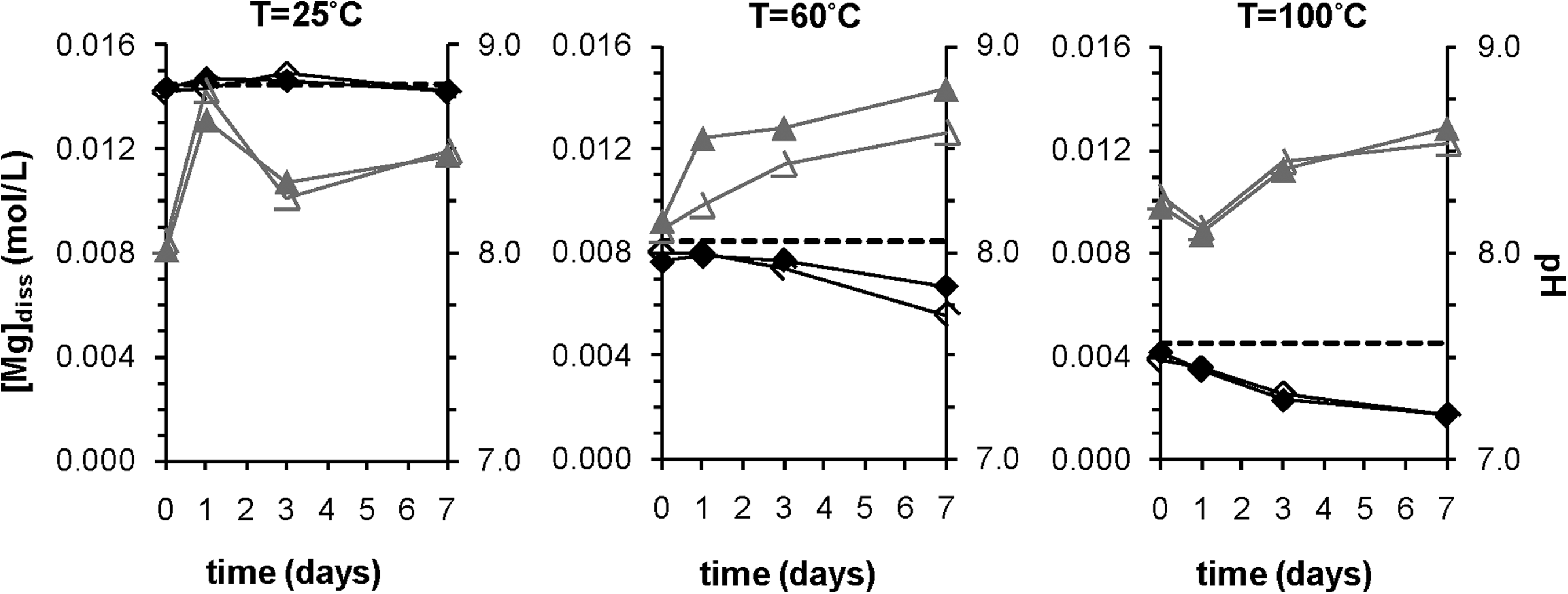
Effect of temperature on dissolved magnesium and pH. Data from duplicate experiments are shown. Black diamonds represent Mg concentrations, and the dashed line is the initial dissolved Mg concentration. Gray triangles represent pH. All three subplots are for type 2 compositions with a target magnesite saturation index (SI) of 1 and no forsterite.
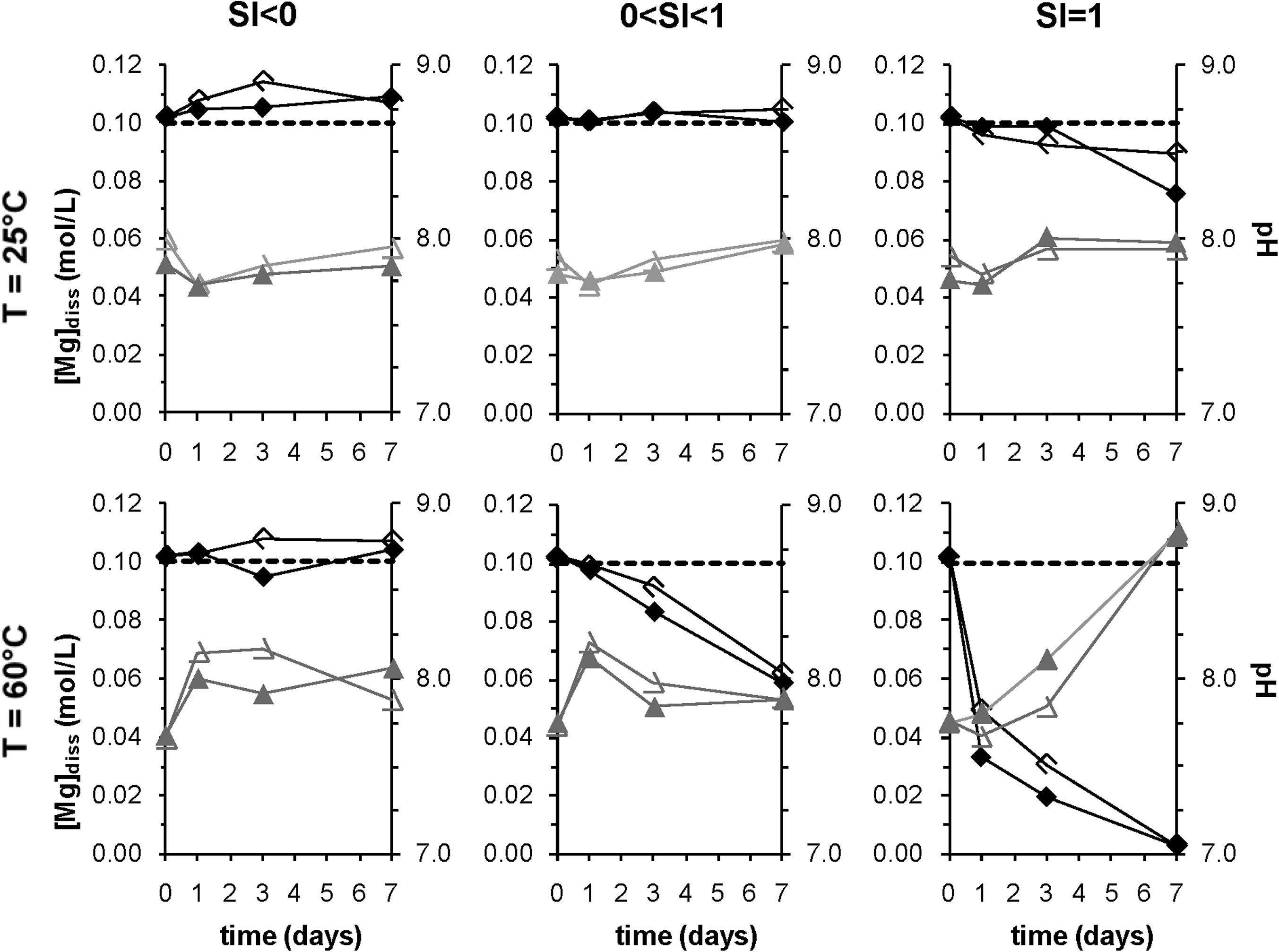
Effect of SI on dissolved magnesium and pH. Within each color the two curves are replicates of the experiment. Black diamonds represent Mg concentrations, and the dashed line is the initial dissolved Mg concentration. Gray triangles represent pH. All experiments were for type 1 compositions and had no forsterite. Data from 100°C experiments are not included, as explained in the text.
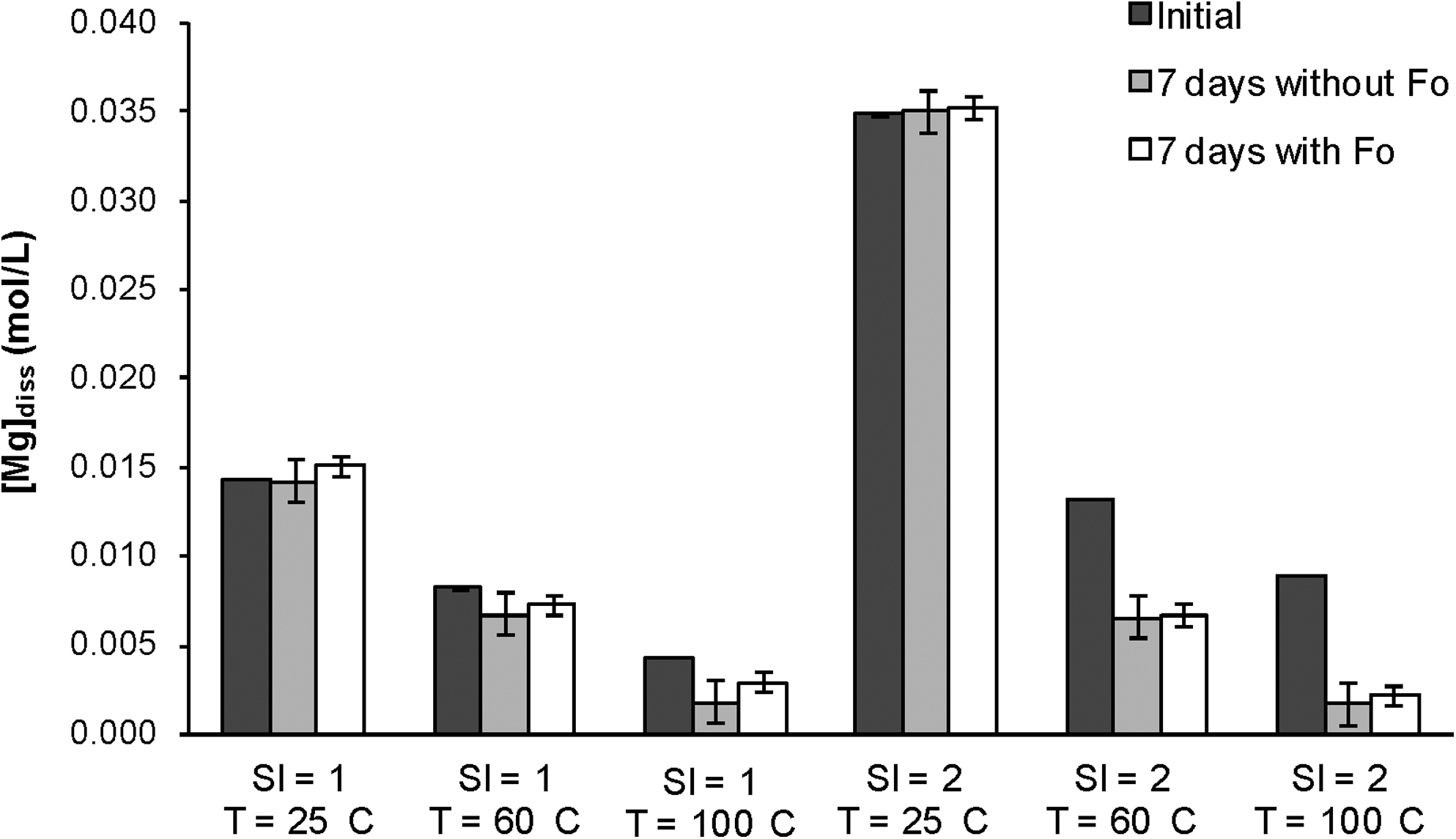
Effect of forsterite presence on dissolved magnesium. Dark gray bars represent the initial Mg concentration. Light gray and white bars represent the Mg concentration after 7 days without and with forsterite substrate, respectively. Error bars are one standard deviation.
The pH rarely changed by more than one unit (Fig. 1). In cases where little precipitation was observed, pH did not vary significantly. Trends in pH might indicate whether the reactors behaved as open or closed systems (i.e., whether the reactors were sealed tightly). At elevated temperature, the decreased solubility of CO2 would drive it out of solution, causing an increase in pH if the systems were open. The control experiments that contained just NaHCO3 generally had very stable pH values, which indicated that the reactors were usually well sealed. In some experiments at 100°C, loss of solution was observed concomitantly with a continual rise in Mg concentrations, which indicated that these particular reactors were not completely sealed; consequently, the 100°C data are not presented in Fig. 2.
Temperature plays a significant role in the identity of the solid precipitated (Fig. 4). At low temperature (25°C) nesquehonite (MgCO3·3H2O) formed, whereas at 60°C hydromagnesite [(MgCO3)4·Mg(OH)2·4H2O] dominated the XRD signal. Both are Mg-bearing minerals and sequester inorganic carbon. At 100°C magnesium hydroxide [Mg(OH)2] formed. At elevated pressures relevant to either in situ or ex situ mineral carbonation, the fugacity of CO2 would make magnesium hydroxide formation unfavorable, so its formation is not discussed further (Marini, 2007).
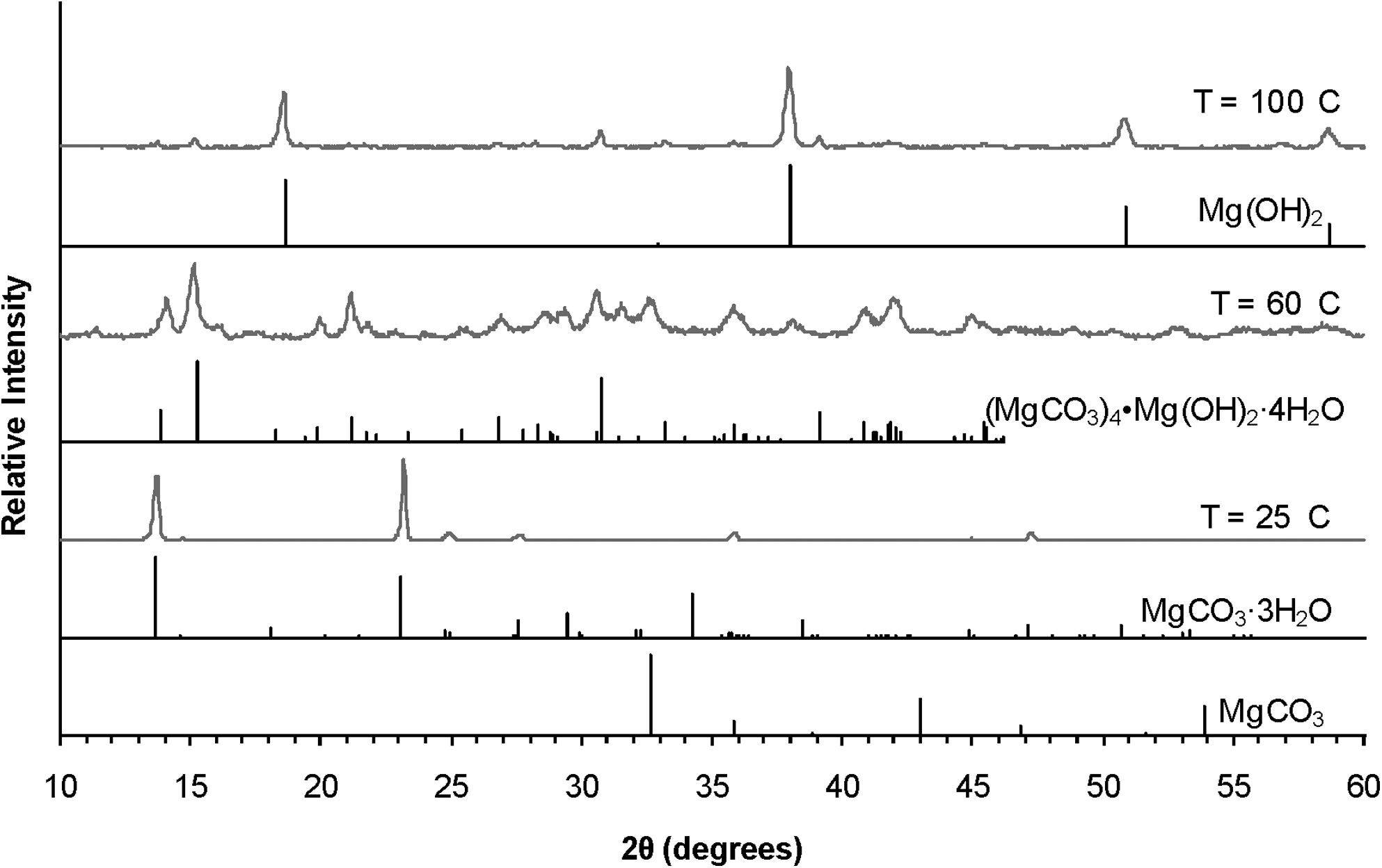
X-ray diffraction patterns of solids produced in experiments at different temperatures. Gray curves are patterns of solids from experiments. For reference standard powder diffraction files are included for the indicated species in black: (MgCO3, magnesite, 08-0475; MgCO3·3H2O, nesquehonite, 20-0669; Mg(CO3)4(OH)2·4H2O, hydromagnesite, 25-0513; Mg(OH)2, brucite, 98-000-0021). All experiments had type 1 compositions at a target magnesite SI of 1 and no forsterite. The intensity of the patterns has been normalized to present all patterns with the same maximum intensity.
Effect of SI
When undersaturated solutions were prepared (SI<0), precipitation was never observed (Fig. 2). Higher saturation indices uniformly resulted in more extensive decreases in Mg concentration over time (Fig. 2). The saturation indices referred to here are those calculated using equilibrium constants from Lide (2000) and MINEQL+. Separate calculations made using constants from SUPCRT92 indicated that all of the solutions studied were initially supersaturated with respect to magnesite (Table 1). However, because magnesite precipitation is kinetically hindered at low pressures and the relatively low temperatures of the experiments, supersaturated solutions may persist without any precipitation of magnesite. At a given temperature, SI does not seem to affect the identity of the solid precipitated (Fig. 5).
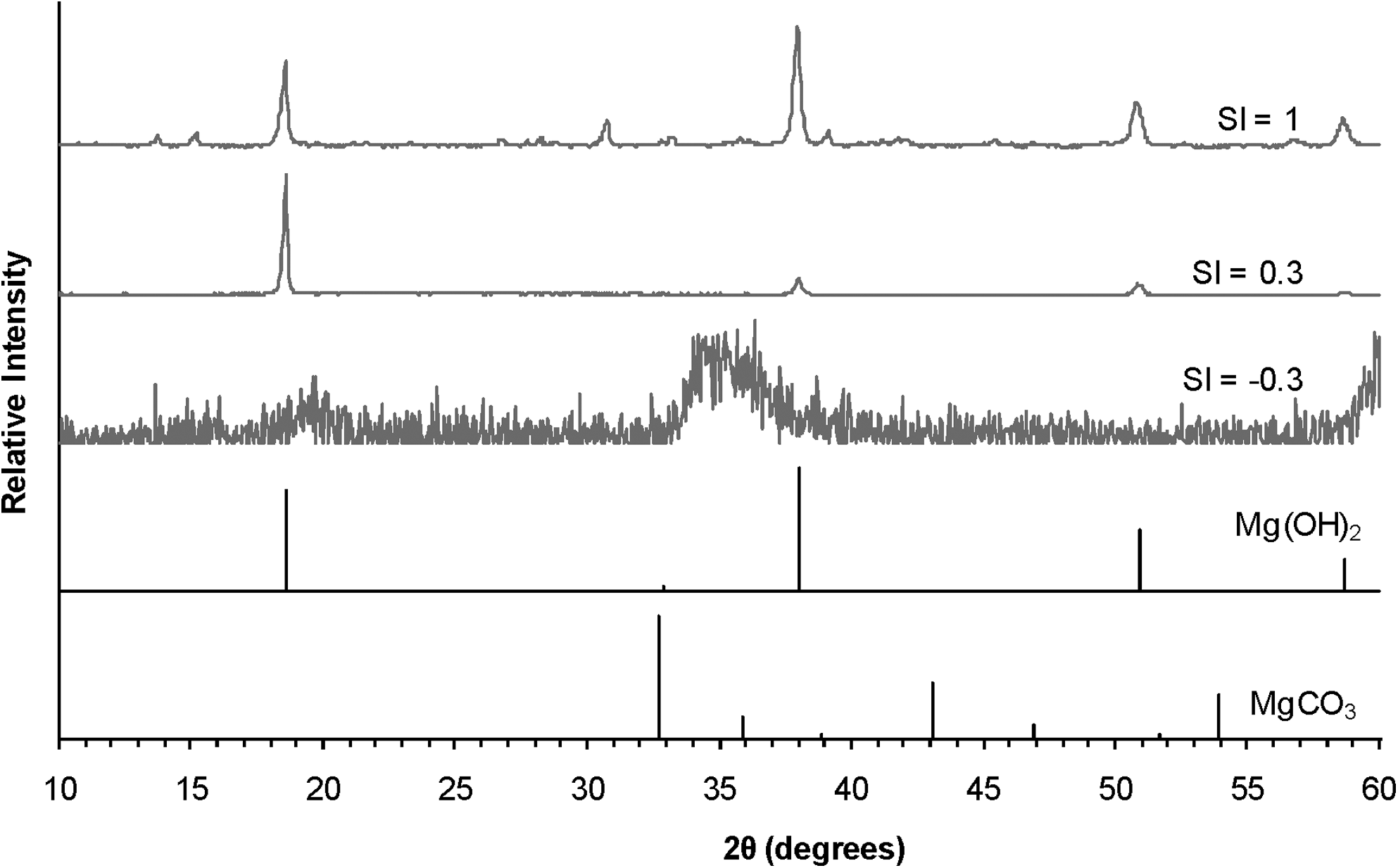
X-ray diffraction patterns of solids generated in solutions with different initial saturation indices with respect to magnesite. Gray patterns are for solids from the experiments. Standard powder diffraction files for the indicated species are also included (MgCO3, magnesite, 08-0475; brucite, 98-000-0021) in black. All experiments were performed at 100°C, without forsterite substrate, and have type 1 composition. The intensity of the patterns has been normalized to present all patterns with the same maximum intensity. The SI=1 pattern appears to have a small amount of non-Mg(OH)2 phase, likely hydromagnesite (see reference pattern in Fig. 4).
Effect of presence of forsterite
The presence of forsterite does not seem to play a significant role in the amount of precipitation observed. At every combination of temperature, SI, and type of initial composition, the Mg concentration after 1 week was not significantly altered regardless of whether a forsterite substrate was present (Fig. 3).
Discussion
Effect of temperature
The extent and rate of precipitation increased with increasing temperature. Decreasing Mg concentrations in solution are assumed to indicate precipitation because no other pathway for Mg loss existed and all precipitated solids bore Mg. Higher temperature corresponded to a higher rate of precipitation (Fig. 1).
The increasing loss of Mg at elevated temperature can be interpreted as a kinetic, rather than thermodynamic, effect because it is observed in experiments that were run at the same target SI (Fig. 1). Although the actual SI may be different from the target SI, the degrees of supersaturation were similar for solutions with the same target SI regardless of the equilibrium constants used to calculate the SI. Previous studies have also concluded that temperature plays a significant role in the precipitation of solids from the MgO-CO2-H2O system (Giammar et al., 2005; Hanchen et al., 2008; Saldi et al., 2009). Results from our present study corroborate previous studies that only nesquehonite precipitated below 40°C and that higher temperatures resulted in hydromagnesite formation (Bender and Sprague, 1965; Sayles and Fyfe, 1973; Hanchen et al., 2008). That magnesite was not observed in the present experiments is consistent with a number of studies that demonstrated the difficulty of precipitating magnesite at low temperatures and ambient CO2 pressures (Giammar et al., 2005; Hanchen et al., 2008). Hanchen et al. (2008) observed that at 120°C hydromagnesite initially formed and later transformed into magnesite.
Although magnesite is calculated to be the thermodynamically most stable phase (Fig. 6), it did not form, and instead nesquehonite and hydromagnesite formed. This inconsistency between the predicted and experimental results suggests that, for the experimental conditions outlined in this study, the precipitation of metastable minerals is preferred over the most thermodynamically stable phases. The initial formation of metastable phases is consistent with the Ostwald step rule, which suggests that the least stable solid phase will form first because of its higher nucleation rate (Stumm and Morgan, 1996).
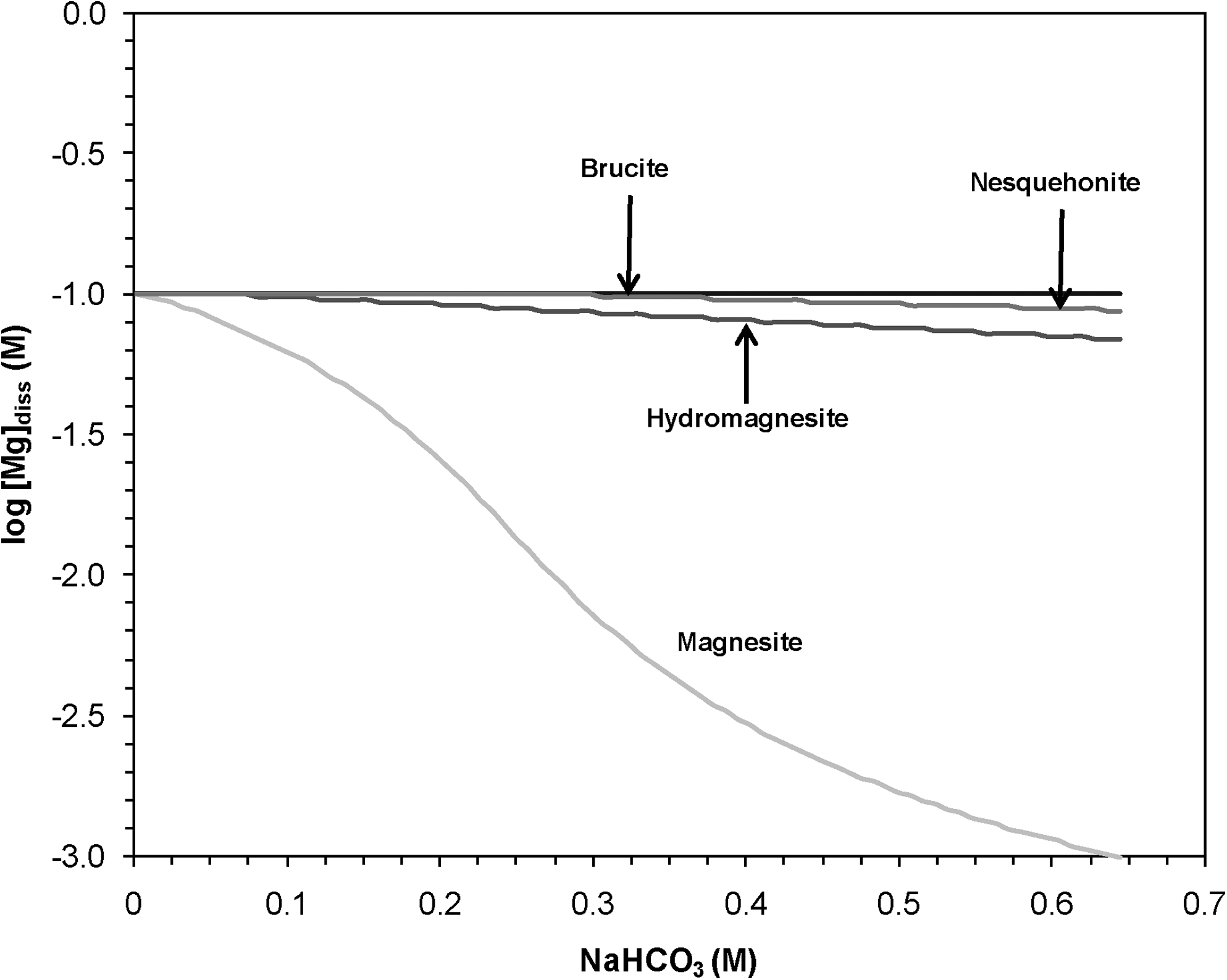
Calculated dissolved Mg concentrations for a system containing 0.1 M Mg and titrated with inorganic carbon in the form of NaHCO3 when the solution equilibrates with different Mg-bearing minerals at 25°C. All equilibrium constants used in these calculations were determined with the SUPCRT92 software package using the slop98 thermodynamic database.
Effect of solution composition
At target magnesite saturation indices below zero, no precipitation was observed. At target saturation indices above zero a precipitate formed, although the precipitated solid was not magnesite. At 25°C, when the target SI for magnesite was 1 and the actual SI calculated using SUPCRT92 was 2.90, nesquehonite precipitated instead of magnesite; the initial solution was supersaturated with respect to nesquehonite (SInesquehonite=0.20). Similarly at 60°C for a target magnesite SI of 1 (calculated to be 3.48 using SUPCRT92), hydromagnesite precipitated instead of magnesite; at these conditions the initial solution was highly supersaturated with respect to hydromagnesite (SIhydromag=7.35), so the precipitation of hydromagnesite was not surprising. Our experiments indicate that higher SI does result in greater overall precipitation (Fig. 2). SI, however, did not affect the species of solid observed (Fig. 5), indicating a different control on speciation.
Calculations of the predicted solubilities of different Mg-bearing solids with increasing addition of inorganic carbon illustrate that magnesite precipitation is favored over the formation of other solids at 25°C (Fig. 6). When the solution is at equilibrium with magnesite, the dissolved Mg concentration is always lower than when the solution is in equilibrium with other Mg-bearing solids. Hydromagnesite and nesquehonite are metastable phases with relatively high dissolved Mg concentrations. Similar calculations for 60°C also indicated that magnesite would be the most energetically favorable solid phase at equilibrium. Although the initial SI for hydromagnesite was actually higher than that of magnesite at 60°C, as the solution composition changes with precipitation, magnesite quickly becomes the less soluble phase. The higher initial SI of hydromagnesite may cause it to precipitate first and to then persist even once the solution reaches conditions that favor magnesite.
When the speciation calculations for 25°C are restricted to only allow nesquehonite precipitation, the experimental results are consistent with predictions (Fig. 7). With increasing total inorganic carbon added as NaHCO3, nesquehonite will precipitate from a 0.1 M MgCl2 solution. At the highest NaHCO3 loading, which corresponded to a target SImag of 1, the model not only predicts nesquehonite precipitation, but the experimentally measured dissolved Mg decreases with time and the final values are in close agreement with calculated nesquehonite solubility. Therefore, using the appropriate solubility-controlling solid, equilibrium calculations are capable of predicting mineral precipitation and dissolved Mg concentrations.
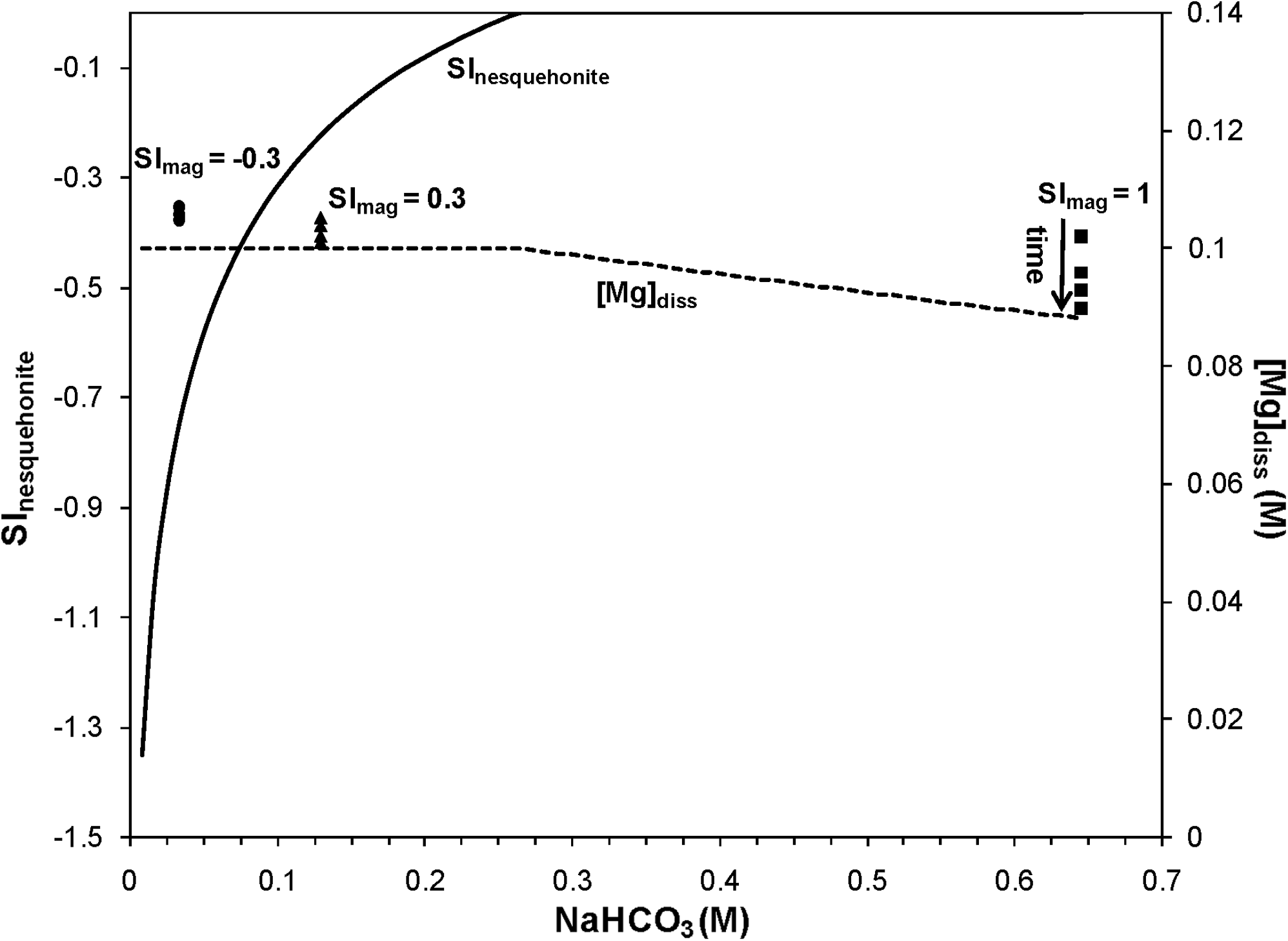
Measured (points) and calculated (dashed line) dissolved Mg concentrations as controlled by equilibrium with nesquehonite at 25°C and ambient pressure. For reference, the solid line is the calculated SI for nesquehonite as a function of the added NaHCO3 concentration. Calculations were made using equilibrium constants determined with the SUPCRT92 software package with the slop98 thermodynamic database. Data are included from experiments having method 1 composition and different target saturation indices for magnesite (SImag). For the experiment with SImag=1, the dissolved Mg concentrations decreases with time as indicated by the arrow.
Effect of presence of forsterite
The presence of forsterite could provide a surface onto which Mg-carbonates can precipitate through a heterogeneous nucleation process. A previous study observed the precipitation of magnesite crystals on forsterite particles from solutions that were supersaturated with respect to magnesite at 95°C (Giammar et al., 2005). In the present study the forsterite substrate did not result in any acceleration of magnesite precipitation or increase in the amount of solid that formed.
Implications for carbon sequestration
The formation of magnesium carbonate minerals after the injection of CO2 into geological formations or introduction into ex situ carbonation reactors will depend on the experimental variables examined in this study as well as on the CO2 pressure. Within the temperature range of this study, carbonate mineral precipitation can proceed rapidly and the dissolution of Mg-containing minerals will also be faster than at ambient temperature. Magnesium silicate dissolution will be the rate-limiting step in the overall process of forming magnesium carbonate minerals from their reaction with CO2 (Gerdemann et al., 2007; Prigiobbe et al., 2009). The selection of appropriate equilibrium constants for both dissolution–precipitation and acid–base reactions are critical to predicting whether or not solids should precipitate and which solids should precipitate. The large differences in the equilibrium constants used in the first calculations of this study and the subsequent ones using SUPCRT92 illustrate the magnitude of differences in predicted solution saturation for calculations made using different constants. Use of consistent calculation approaches and equilibrium constants is critical to allowing comparison among the results of different studies. Even with appropriate equilibrium constants, the formation of metastable phases like nesquehonite and hydromagnesite may be observed instead of magnesite. Both of these metastable minerals still sequester carbon, and nesquehonite, in particular, if formed ex situ, could have downstream industrial uses (Ferrini et al., 2009). The consideration of activity coefficients is also critical in making speciation calculations.
The importance of temperature, equilibrium constant selection, and formation of metastable phases to magnesium carbonate precipitation that were observed at ambient pressure in this study can also impact magnesium carbonate precipitation at the higher CO2 pressures that will be encountered during carbon sequestration. At high pressures the rates of reactions will still increase with temperature. At higher PCO2 the initial pH of the solutions will be lower than at ambient conditions, which will accelerate the dissolution of magnesium silicate minerals (Giammar et al., 2005), but silicate mineral dissolution should still be the rate-limiting step in the overall conversion process of magnesium silicates into magnesium carbonates. While equilibrium constants will be different at higher pressure, the differences between values at 1 bar and 100 bar pressure are small relative to the differences that occur with changes in temperature or to the order of magnitude differences between constants from different sources. The greater pressures encountered during in situ carbon sequestration will make magnesite more favorable than in the experiments of the present study, and it is possible that metastable phases will not form at higher pressures.
Conclusions
Three important factors that can influence magnesium carbonate precipitation were examined. Temperature affected the extent of precipitation as well as the identity of the solid. Higher temperatures resulted in faster and more extensive precipitation, but the solids that formed were not the most thermodynamically stable ones. The degree of solution saturation with respect to magnesium carbonate influenced whether or not precipitation was observed and, to some extent, how extensive the precipitation was. Saturation indices are highly sensitive to the equilibrium constants used, which in turn depend on the reference from which they are taken and the method of temperature correction. The presence of an initial forsterite substrate did not significantly affect magnesium carbonate precipitation experiments. The precipitation of magnesium carbonate minerals has potential applications to carbon sequestration via in situ mineralization or ex situ engineered reactors.
Footnotes
Acknowledgments
This work was supported by the Consortium for Clean Coal Utilization at Washington University in St. Louis. We appreciate the comments of five anonymous reviewers that helped in the revision of this article. We thank Alexis Boleda for assistance with selected experiments, Patty Wurm for help with inductively coupled plasma mass spectrometry operations, and Abhas Singh for helpful discussions.
Author Disclosure Statement
The authors state that no conflicting financial interests exist.