
Abstract
Objective:
Listeria monocytogenes is a foodborne pathogen found in a wide variety of environments. Subtype characterization of L. monocytogenes isolates from listeriosis outbreaks that have occurred over the last three decades has suggested that a number of outbreaks were caused by a small number of L. monocytogenes epidemic clones (ECs). In this study we compared the prevalence, ecology, and phylogenetic position of outbreak-associated isolates and non-outbreak–associated isolates to probe the evolutionary and ecological characteristics of outbreak-associated L. monocytogenes subtypes, including those representing previously described ECs.
Methods:
Multilocus sequence typing data for isolates from 15 listeriosis outbreaks in Europe and North America were generated and compared, using a phylogenetic framework, with 180 isolates representing a local sampling of diverse sources, including human sporadic cases.
Results:
Isolates from 15 listeriosis outbreaks represented eight sequence types (STs). STs corresponding to previously designated ECI (ST1 and ST93) and ECIa (ST29) represented isolates from eight outbreaks. ST17 (corresponding to ECII) was involved in two outbreaks in the United States (1998 and 2002). No other STs were involved in multiple outbreaks. While ST1 was the most common ST among sporadic human cases and non-human listeriosis–related isolates, ST29 was rare among non-human listeriosis–related isolates and was significantly overrepresented among isolates from human listeriosis outbreaks and sporadic cases as compared to isolates from other sources in our local sampling.
Conclusions:
STs associated with outbreaks (and representing previously designated ECs) appear to differ in their ecology. While association of ECI with multiple human listeriosis outbreaks appears to reflect strain abundance across environments, ECIa seems to represent an L. monocytogenes EC that appears to be overrepresented among outbreaks and sporadic cases and thus may have increased transmission potential.
Introduction
Listeria monocytogenes is a common environmental bacterium and a facultative intracellular human and animal pathogen (Gray et al., 2006). The source of almost all human infections appears to be contaminated foods (Farber and Peterkin, 1991); mainly, the immunocompromised, the elderly, unborn children, and neonates are at risk of acquiring listeriosis (Schlech, 2000). Severe manifestations of listeriosis include septicemia, encephalitis, meningitis, and spontaneous late abortion (Vazquez-Boland et al., 2001). Less severe manifestations include gastroenteritis (Dalton et al., 1997) and may often go undiagnosed as listeriosis. Phylogenetic and population genetic studies have shown that L. monocytogenes consists of three (Rasmussen et al., 1995) and possibly four (Ward et al., 2008) major phylogenetic lineages, including two lineages (designated as lineages I and II) that represent the most common human- and food-associated lineages (Gray et al., 2004). These lineages represent the major subdivisions of L. monocytogenes as a species (comparable to subspecies) and should not be confused with ECs, which are groups of closely related strains on the population level. While the first report of L. monocytogenes lineages (Piffaretti et al., 1989) assigned serotype 1/2b and 4b isolates to lineage I and serotype 1/2a and 1/2c isolates to lineage II (the nomenclature followed here), others (Doumith et al., 2004) have called the lineage containing serotype 1/2b and 4b isolates lineage II and the lineage containing serotype 1/2a and 1/2c isolates lineage I.
Subtype characterization of L. monocytogenes isolates from listeriosis outbreaks that have occurred over the last three decades has suggested that many outbreaks were caused by a small number of L. monocytogenes ECs (Kathariou, 2002, 2003). Specifically, four L. monocytogenes ECs have been reported to date, designated ECI, ECII, ECIII, and ECIa (ECIa has also been referred to as ECIV) (Chen et al., 2007). At least one EC (ECI) has been associated with multiple outbreaks in Europe and North America, including a coleslaw-associated outbreak in the Maritime Provinces of Canada in 1981 (Schlech et al., 1983), a soft cheese–associated outbreak in Switzerland (1983–1987) (Boerlin et al., 1996), and a Mexican-style cheese–associated outbreak in California (1985) (Linnan et al., 1988). Another EC, ECIII, has been identified as the likely causative agent in one outbreak and one listeriosis case, which occurred approximately 12 years apart and could be traced back to a single strain that seems to have persisted in a food-processing facility that was responsible for both disease events (Kathariou, 2002; Olsen et al., 2005). Almost every listeriosis outbreak has been traced back to a single molecular subtype or a group of closely related subtypes; however, multiple distinct molecular subtypes (including isolates representing ECI and ECIa) were isolated from patients linked to an outbreak in Philadelphia in 1987 (Schwartz et al., 1989; Bibb et al., 1990).
Although pulsed-field gel electrophoresis and other banding pattern–based subtyping methods provide sensitive subtype discrimination of L. monocytogenes (Borucki et al., 2004a), DNA sequencing–based subtyping methods such as multilocus sequence typing (MLST) are valuable tools to probe the evolution of bacterial pathogens and, hence, to identify clonal complexes. Several MLST schemes have been developed for L. monocytogenes, ranging from classic MLST schemes that target seven housekeeping genes, which are assumed to evolve relatively slowly and are therefore suitable for phylogenetic reconstruction (Salcedo et al., 2003; Ragon et al., 2008), to the so-called multivirulence locus sequence typing scheme (Zhang et al., 2004; Chen et al., 2005, 2007), targeting virulence and virulence-associated genes, which are assumed to evolve relatively rapidly. Regardless of the genes targeted, MLST studies, as well as multilocus enzyme electrophoresis studies (Piffaretti et al., 1989; Bibb et al., 1990), always separate L. monocytogenes into two common lineages; lineage III (Rasmussen et al., 1995) is reported in fewer studies, as isolates in this lineage are sometimes not represented in these studies. In our study reported here, a previously described MLST scheme that uses partial sequences of four housekeeping genes, two virulence genes, and one stress response gene (Nightingale et al., 2005) was used. Although MLST is well suited to infer evolutionary relationships between strains, it is generally not discriminatory enough for outbreak investigations (Maiden et al., 1998).
Over the years, several studies on L. monocytogenes outbreak strains have been published using other subtyping methods besides MLST (Borucki et al., 2004a) and multivirulence locus sequence typing (Chen et al., 2007), such as multilocus enzyme electrophoresis (Piffaretti et al., 1989; Bibb et al., 1990), single-nucleotide polymorphism–based multilocus genotyping (Ducey et al., 2007; Ward et al., 2008), and multiple-locus variable number tandem-repeat analysis (Sperry et al., 2008). However, our understanding of the ecology and evolution of outbreak-associated L. monocytogenes is still limited. Therefore, we performed MLST of isolates associated with 15 major human listeriosis outbreaks and compared the MLST types for these outbreak-associated isolates with MLST data for 180 non-outbreak–associated isolates from various clinical and environmental sources, using a phylogenetic framework. Comparing the prevalence, source distribution, and phylogenetic position of outbreak-associated isolates and non-outbreak–associated isolates specifically allowed us to probe the evolutionary and ecological characteristics of outbreak-associated L. monocytogenes subtypes.
Materials and Methods
Isolates
A collection of human and food isolates representing 15 major human listeriosis outbreaks was assembled (Table 1). All isolates were obtained from the Centers for Disease Control and Prevention (Atlanta, GA). For eight outbreaks, both human and food isolates were included in our study. MLST data for all of these isolates have not previously been reported and can be found on PathogenTracker (
PFGE types refer to unique combined AscI and ApaI profiles as described in Fugett et al. (2006, 2007).
Sequence data of these isolate have been obtained from the Broad institute (see Materials and Methods section).
Serotype 4bx is a rare serotype described by Mclauchlin (1986, 1991). It is characterized by a weak reaction with O factors V/VI and VI, but a strong reaction with factor VII.
This isolate represents a sporadic human case.
EC, epidemic clone; ST, sequence types; PFGE, pulsed-field gel electrophoresis; NA, not available.
MLST data for outbreak strains were compared to previously reported MLST data for 180 L. monocytogenes isolates collected in the New York state (den Bakker et al., 2008). These isolates were obtained from six different sources: (i) sporadic human clinical cases (56 isolates; 4 human clinical isolates reported in Nightingale et al., 2005, were removed, as they were likely associated with outbreaks), (ii) foods (30 isolates), (iii) animal clinical cases (30 isolates), (iv) urban environments (30 isolates), (v) ruminants without clinical symptoms and farm environments (21 isolates), and (vi) natural environments (13 isolates) (Nightingale et al., 2005; den Bakker et al., 2008).
MLST
All outbreak isolates were characterized using a previously described MLST scheme (Nightingale et al., 2005). This scheme includes DNA sequences of partial open reading frames for seven genes: four housekeeping genes (gap [569 bp], prs [663 bp], purM [714 bp], and ribC [639 bp]), two virulence genes (inlA [771 bp] and actA [561 bp]), and one stress response gene (sigB [666 bp]). Sequences of the MLST loci for outbreak isolates HPB2262 and FSL R2-503 (Table 1) were obtained from genomic sequences available at the Broad Institute (
Phylogenetic analyses
To compare the sequence types (STs) of the outbreak strains with the STs found among the 180 isolates representing a local sampling of diverse sources in the New York state, a neighbor-joining tree based on concatenated sequences was constructed using PAUP* 4.010 (Swofford, 2002). While previous studies (Nightingale et al., 2005; den Bakker et al., 2008) have shown that the loci used in this study have been subject to homologous recombination, which may hamper proper phylogenetic inference with methods like neighbor joining, the same studies also have shown that recombination is relatively rare in lineage I, the phylogenetic lineage that contains most outbreak strains. The phylogenetic relationships inferred by neighbor joining for lineage I should therefore be close to the true phylogeny. To assess the robustness of the clustering in this tree, a bootstrap analysis (Felsenstein, 1985) consisting of 2000 pseudo-replicates (replicates generated through sampling of the original dataset with replacement) was performed.
Statistical analysis
To assess whether outbreak-associated STs and ECs are significantly overrepresented among isolates from sporadic human clinical cases and outbreaks (as compared to isolates from other sources included in the local sampling, i.e., foods, animal clinical cases, urban environments, farm environments, and natural environments), categorical statistical analyses were performed using the R-package for statistical computing (
Results and Discussion
Characterization of 36 L. monocytogenes isolates representing 15 human listeriosis outbreaks in the United States, Canada, and Europe showed that (i) the majority of outbreak-associated isolates cluster into a well-supported clade within L. monocytogenes lineage I, and (ii) isolates from all but one outbreak represent STs also found among isolates from our local (New York state) sampling, indicating that most human outbreak–associated STs appear to be present in a variety of sources. Overall, our data indicate that human outbreak–associated L. monocytogenes STs previously designated as “epidemic clones” seem to be widely distributed. Some of these STs appear to represent subtypes that could be characterized as ecological generalist. Their common involvement in human outbreaks may, thus, reflect their relative abundance in diverse environments. Other outbreak-associated STs seem to be verrepresented among outbreaks and human clinical cases and may thus potentially have unique virulence characteristics.
The majority of outbreak strains cluster into a well-supported clade within L. monocytogenes lineage I
The outbreak-associated isolates characterized here represented eight distinct STs: six STs that belong to lineage I and two STs that belong to lineage II. ST29 was the most common ST among the isolates from 15 outbreaks. This ST was found among isolates from five outbreaks: the 1979 and 1983 outbreaks in Boston, Massachusetts (Fleming et al., 1985; Ho et al., 1986); the 1988–1990 outbreak in England (McLauchlin et al., 1991); the 1997 outbreak in Italy (Aureli et al., 2000); and one of the two strains associated with 1987 outbreak in Philadelphia (Schwartz et al., 1989), an outbreak that included multiple subtypes. Isolates from some of these outbreaks have previously been designated as ECIa, indicating that ECIa is represented by ST29. While Chen et al. (2007) excluded the isolates from the milk-associated outbreak in Boston in 1983 from ECIa based on a single-nucleotide difference in one locus (lisR), our data suggest that this outbreak was indeed caused by a subtype closely related to strains previously designated as ECIa. To our knowledge, our analyses reported here represent the first report that indicates that isolates from the 1997 outbreak in Italy represent ECIa. This finding is interesting as the human disease symptoms in this outbreak were febrile gastrointestinal illness (Aureli et al., 2000) and not systemic infections as was the case in the other outbreaks associated with ECIa. We therefore conclude that ECIa/ST29 represents a widely distributed clonal group that has the ability to cause both invasive illness and febrile gastroenteritis.
ST1 was the second most common ST among the isolates from 15 outbreaks. This ST was found among isolates from four outbreaks: the 1976 outbreak in Anjou, France (Carbonelle et al., 1978); the 1983–1987 outbreak in Switzerland (Boerlin et al., 1996); the 1985 outbreak in Los Angeles, CA (Linnan et al., 1988); and also represented one of the two strains associated with 1987 outbreak in Philadelphia, PA (Schwartz et al., 1989). ST93, which was only found among isolates associated with one outbreak (i.e., the coleslaw-associated outbreak in the Maritime Provinces of Canada in 1981), differed by a single nonsynonymous substitution in ribC from ST1. ST1 and ST93 can therefore be considered to belong to the same clonal complex (Feil, 2004) with ST93 representing a single-nucleotide variant of ST1. ST1 was also more common among the 180 isolates from our local sampling (representing 23 isolates) as compared to ST93, which was not found in our local sampling. As isolates from the five outbreaks that were linked to ST1 and ST93 were previously designated as ECI, we conclude that the ST1/ST93 clonal complex represents ECI. The third most common ST among outbreak-associated isolates was ST17; specifically, this ST was found among isolates from two outbreaks, including a hotdog and packaged deli meat–associated outbreak in the United States in 1998 (CDC, 1999; Graves et al., 2005) and a sliced deli meat–associated outbreak in the northeastern United States in 2002 (CDC, 2002). Isolates from these two outbreaks were previously designated as ECII (Kathariou, 2002).
In addition to the STs associated with ECIa (ST29), ECI (ST1 and ST93), and ECII (ST17), which all belong to L. monocytogenes lineage I, we also identified two additional lineage I STs that were associated with a single outbreak each. Specifically, serotype 4b isolates from the Mexican cheese–associated outbreak in North Carolina in 2000 (CDC, 2001; MacDonald et al., 2005) represented ST24, whereas serotype 1/2b isolates from the chocolate milk–associated outbreak in Illinois in 1994 (Dalton et al., 1997), which only involved gastrointestinal symptoms, represented ST33. Neither of these two STs are associated with previously reported L. monocytogenes ECs.
The MLST data for lineage I–associated isolates from 13 listeriosis outbreaks, which included isolates representing three previously reported ECs, allowed us to further probe the phylogeny of outbreak-associated L. monocytogenes strains. Within lineage I, one moderately supported clade containing mainly isolates with serotype 1/2b (bootstrap value 79%; designated clade B in Fig. 1) and a well-supported clade (bootstrap value 99%; designated clade A in Fig. 1), which includes predominantly serotype 4b isolates and all lineage I outbreak–associated STs, were found. These findings suggest that outbreak-associated L. monocytogenes lineage I strains represent a distinct evolutionary subgroup of lineage I strains, which predominantly includes serotype 4b strains, consistent with the long-standing observation that most human listeriosis outbreaks are caused by serotype 4b strains (Farber and Peterkin, 1991). Although a number of individual clades within the lineage I A clade show good (100%) bootstrap support (e.g., ECI, ECIa, and EC II clades; Fig. 1), no good bootstrap support (<70%) was found for the phylogenetic relationships between these clades in lineage I A. Hence, the phylogenetic relationships between ECI, ECIa, and ECII remain unresolved. This is consistent with a lack of good bootstrap support (>70%) for the phylogenetic relationships between ECI, ECII, and ECIa that was also reported by Chen et al. (2007). The lack of well-supported phylogenetic signal for the basal relationships in lineage I A thus does not appear to originate from an analytical artifact, but likely is due to a short period of rapid diversification in the past (Jackman et al., 1999) that gave rise to the main clonal complexes within this lineage.
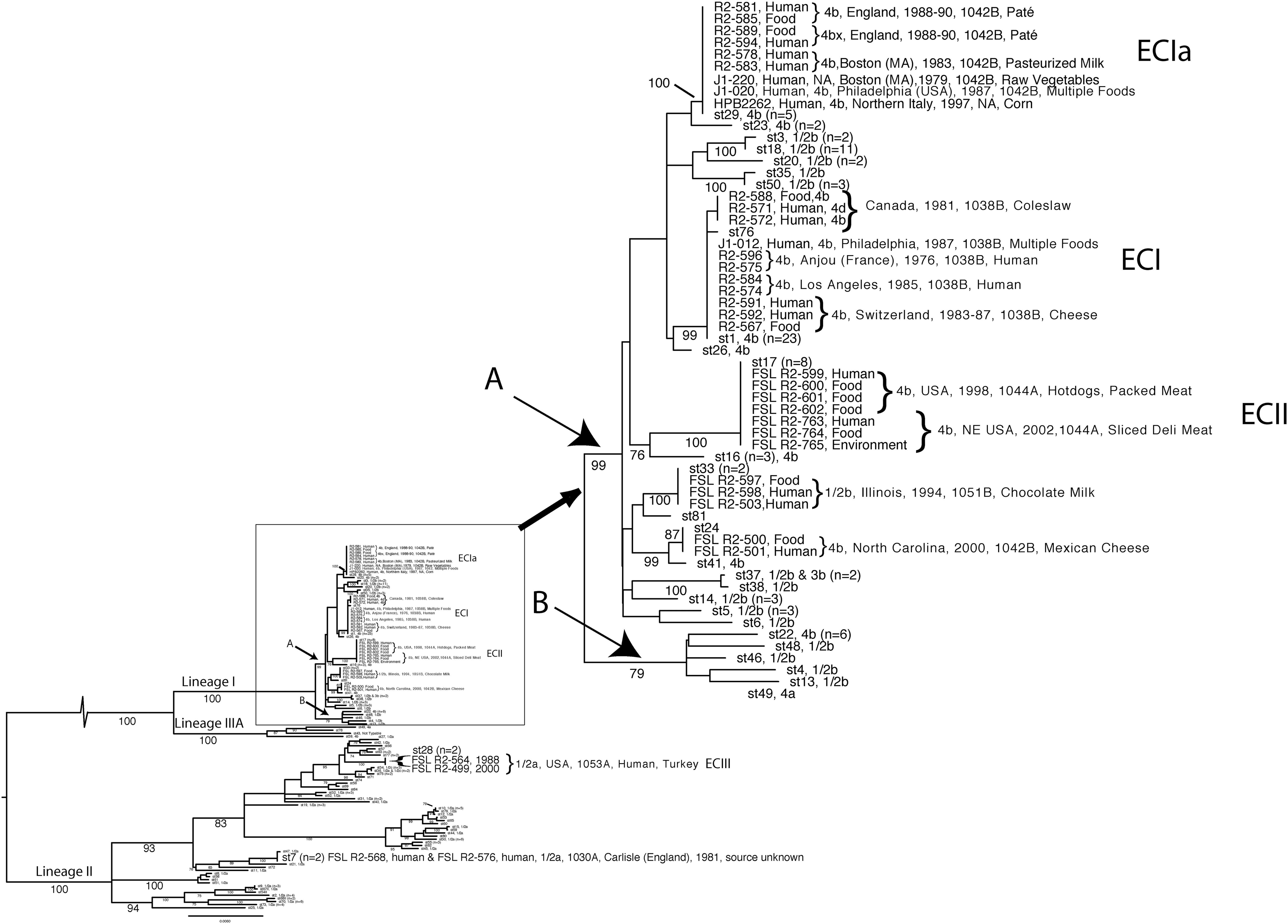
Neighbor-joining tree representing the phylogenetic relationships between 81 sequence types (STs) found among 180 isolates from the New York state and STs found in outbreak-associated isolates. Lineage I has been magnified to give a more detailed picture of the phylogenetic relationships in this clade. The phylogenetic reconstruction is based on the concatenated partial sequences of actA, gap, inlA, prs, purM, ribC, and sigB loci. Values on the branches indicate bootstrap support based on 2000 bootstrap replicates. Bootstrap values <70% are not shown. Branch-lengths are proportional to uncorrected dissimilarity (p-) distances. The serotype associated with the locally found STs is indicated after the ST designations. The frequency of STs that were found more than once in the local sampling is indicated within parentheses (e.g., n=). A and B refer to the two main well-supported clades in lineage I.
In addition to the STs found among outbreak-associated isolates that group into lineage I, we also found two STs (ST7 and ST28) among outbreak-related isolates that belong to lineage II. Each of these STs represented isolates from one outbreak. While isolates involved in the 1981 outbreak in Carlisle (England) (McLauchlin et al., 1986) represented ST7, isolates from a sliced deli meat–associated outbreak in the United States in 2000 (CDC, 2000) represented ST28. Consistent with full genome sequencing data (Orsi et al., 2008), which showed that isolates from this 2000 outbreak were virtually identical to isolates from a 1988 case linked to hot dogs produced in the same plant, we also found that the 1988 human isolate also represented ST28 (CDC, 1989). Our phylogenetic analysis shows that these two outbreak-associated STs in lineage II are only distantly related (Fig. 1), consistent with previous observations based on multilocus virulence gene typing (Chen et al., 2007). There, thus, is no evidence so far of any lineage II STs that are involved in multiple human listeriosis outbreaks, even though serotype 1/2a (a common serotype in lineage II) isolates are generally a predominant among sporadic clinical cases, worldwide (Parihar et al., 2008).
Human outbreak–associated STs appear to be present in a wide variety of sources and have a wide geographic distribution
To probe the ecology of outbreak-associated STs and associated ECs, we compared the STs found among outbreak isolates to STs observed among 180 L. monocytogenes isolates from a diversity of sources, including foods, sporadic human clinical cases, animal cases, natural and urban environments, and farm environments. Interestingly, ST1, which represents most ECI isolates and was found among four outbreaks, also represented the most abundant ST in our local sampling (23/180 isolates were ST1). ST1 (ECI) was found in all six source-categories, including urban, farm, and natural environments (Table 2). ST93, the other ST associated with ECI, was not found among the 180 L. monocytogenes isolates representing a local sampling of isolates in New York state. Our MLST data as well as two other pulsed-field gel electrophoresis–based subtyping studies (Borucki et al., 2004b; Fugett et al., 2007) clearly support that ECI (particularly, ST1) represents a common L. monocytogenes clonal complex (which has been associated with outbreaks in both Europe and North America) and that isolates in this clonal complex are present in a variety of habitats. Consistent with these observations, statistical analyses (Fisher exact test) found no evidence that ECI (i.e., ST1 and ST93) is overrepresented among outbreaks (as compared to its prevalence among isolates from our local sampling) or overrepresented among human sporadic isolates (as compared to non-human–associated isolates from our local sampling; Table 2). Therefore, ECI should be considered an ecological generalist and is a highly successful clone from an evolutionary perspective, as previously suggested by Yildirim et al. (2004). Its common association with human listeriosis outbreaks appears to represent its general abundance (and thus frequent human exposure); however, it cannot be excluded that this general abundance is due to unique virulence characteristics.
The EC designation or the individual outbreak associated with a given ST is indicated in parentheses.
The p-value given is for the Fisher's exact test comparing the number of outbreaks to the total number of non-human listeriosis–related isolates in local sampling.
Human clinical refers to isolates from sporadic human listeriosis cases.
The p-value given is for the Fisher's exact test comparing the number of sporadic human clinical isolates to the total number of non-human listeriosis–related isolates in local sampling.
NS, not significant.
Overall, 5 of the 180 L. monocytogenes isolates from our local sampling represented ST29 (ECIa); ST29 was far less common among these 180 isolates as compared to ST1 (ECI), which represented 23 isolates. Specifically, ST29 (ECIa) represented four isolates from sporadic human listeriosis cases as well as one isolate from an urban environment (a storm drain next to a sidewalk). Statistical analyses showed that ECIa is significantly (p < 0.001, Fisher exact test; Table 2) overrepresented among outbreaks (as compared to its prevalence among isolates from our local sampling) and is also significantly (p = 0.03) overrepresented among human sporadic isolates (as compared to non-human–associated isolates from our local sampling). These findings suggest that ECIa is overall overrepresented among human isolates and its frequent association with human listeriosis outbreaks and cases may be due to unique virulence (or transmission) characteristics rather than frequent exposure. Future phenotypic and/or genomic evidence for increased virulence and transmission will be necessary to truly elucidate possible unique characteristics associated with this clonal complex.
ST17 (ECII) was the second most common outbreak-associated ST among the 180 L. monocytogenes isolates from our local sampling (8 out of the 180 isolates). Specifically, ST17 represented five isolates from sporadic human listeriosis cases as well as one isolate each from foods, animal clinical cases, and farm environments, indicating that this ST is found in a variety of sources. Statistical analyses found no evidence that ST17 is overrepresented among outbreak-associated isolates (as compared to its prevalence among isolates from our local sampling) or overrepresented among human sporadic isolates (as compared to non-human–associated isolates from our local sampling). This indicates that the relatively recent association of ECII with two human listeriosis outbreaks in the United States (in 1998 and 2002) may represent the fact that ECII is a common environmental subtype. While one study showed no increased cell-to-cell spread for isolates with ribotype DUP-1044A (a ribotype exclusively associated with ST17) as compared to other lineage I isolates (Nightingale et al., 2006), further phenotypic characterization of ST17 isolates will be needed to determine whether this clonal group has unique virulence and transmission characteristics.
Among the STs only associated with a single outbreak, both ST28 (representing the sliced turkey–associated outbreak in the United States in 2000) and ST24 (representing the Mexican-style cheese–associated outbreak in North Carolina in 2000) were only found among human clinical isolates from our local sampling (two and one isolate, respectively; Table 2). ST33 (representing the chocolate milk–associated outbreak in Illinois in 1994) and ST7 (representing the outbreak in Carlisle, England, in 1981) were not found among human clinical isolates in our local sampling, but both were found twice among non-human disease–associated isolates in our local sampling (Table 2). Statistical analyses found no evidence that any of these four STs associated with single outbreaks are overrepresented among human sporadic cases in our local sampling, despite their presence in the local sampling area, suggesting that these STs may not show enhanced virulence. Overall, these observations are consistent with previous studies that have suggested that the subtypes associated with listeriosis outbreaks in the United States in 2000, North Carolina in 2000, Illinois in 1994, and England in 1981 were not involved in other outbreaks (Fugett et al., 2006; Chen et al., 2007).
STs associated with human listeriosis outbreaks (including STs representing ECI, ECIa, and ECII) were found in the sample of isolates of geographically restricted origin (New York state). This observation is consistent with findings reported by Ragon et al. (2008), who showed that STs associated with three outbreaks representing ECI, ECIa, and ECII were also represented among a local sampling of French isolates. Combined with the observation that both ECI and ECIa strains have been involved in human listeriosis outbreaks in both North America and Europe, these findings confirm the findings of previous studies (Piffaretti et al., 1989; Bibb et al., 1990; Buchrieser et al., 1993) that many outbreak-associated subtypes show a global distribution pattern, including outbreak-associated clonal complexes that are found in Europe and North America.
Conclusions
Our data support the finding of previous studies that a number of distinct L. monocytogenes subtypes have been associated with human listeriosis outbreaks worldwide (Kathariou, 2002). Only three clonal groups have been responsible for multiple outbreaks though. These three clonal groups represent three of the four previously proposed L. monocytogenes ECs: ECI, ECIa, and ECII. Further, our data suggest that association with multiple outbreaks of ECI represents the general abundance of this clonal group across different environments. While this clonal group is probably an evolutionarily successful ecological generalist, it cannot be excluded that this clone has advantageous virulence or transmission properties that make it more successful. ECIa is the only clonal group that is clearly overrepresented among isolates from outbreaks and sporadic human clinical cases as compared to isolates from non-human listeriosis–related cases in our local sampling. This indicates that this clonal group has a high probability of causing disease when exposure occurs, and suggests that this clonal group has advantageous virulence or transmission properties that remain to be determined. Additional studies on the ecology, distribution, genomics, and phenotypic characteristics of outbreak-associated L. monocytogenes are needed to characterize L. monocytogenes ECs. In particular, studies on large isolate sets from diverse sources are needed. Such studies should include intraserotype comparisons of ST diversity and will help to further clarify the distribution and ecology of L. monocytogenes subtypes. Future comparative studies on genomic and phenotypic characteristics of ECIa and ECI strains, which represent the clonal groups most commonly associated with human listeriosis outbreaks, will provide a particular opportunity to better understand the ecology and evolution of human disease–associated L. monocytogenes clonal groups.
Footnotes
Acknowledgments
We would like to thank Art Turko, Natasha Brooks, and Kimberly Alexander for their help with the collection of the sequence data for the outbreak-associated isolates; Xavier Didelot and Doug Call for their help with the definition of the term EC and Teresa Bergholz for her helpful comments on the article. Support for this project was provided by USDA Special Research Grants to M.W. (grant nos. 2004-34459-14296, 2005-34459-15625, and 2006-34459-16952).
Disclosure Statement
No competing financial interests exist.