
Abstract
Bacteriophage engineering is a promising strategy to address multidrug-resistant (MDR) bacterial infections that pose significant challenges to public health due to the overuse of antibiotics. Bacteria can develop resistance mechanisms, such as receptor modification and activation of antiviral defense systems, which further complicates the application of phage therapy. Additionally, long-term phage therapy can result in the production of anti-phage antibodies, which may interfere with treatment. These factors require advanced engineering techniques to improve the efficacy of phages and expand their host range. Recent advances in genome engineering methods, including CRISPR/Cas9, homologous recombination, and other synthetic biology techniques, offer promising solutions to these challenges. By modifying receptor-binding proteins and using high-yield screening methods, researchers can create phages that are better equipped to target MDR bacteria effectively. Furthermore, understanding the intricate interactions between phages and their bacterial hosts is critical to guiding these engineering efforts. Future development perspectives lie in integrating these advanced engineering techniques into clinical practice, potentially putting bacteriophages at the forefront of fighting MDR bacterial infections.
Introduction
Bacteriophages are the most abundant bacterial-infecting viruses in the biosphere due to their diversity and adaptability to various hosts (Mahichi et al., 2009). Bacteriophage therapy, which involves using bacteriophages to combat bacterial infections, is gaining interest due to its efficacy and safety. Phage therapy has shown promising results in various in vivo models and human studies. The use of antibiotics has previously been widely adopted to address bacterial infections, but the emergence of multidrug-resistant (MDR) bacteria poses a significant threat to public health (Kakasis and Panitsa, 2019; Aisha et al., 2021).
Understanding bacteriophages is critical to advancing microbiology research. Bacteriophages for therapeutic purposes must exhibit strong lytic activity and possess a wide host range (Chung et al., 2023). A bacteriophage cocktail is a mixture of different bacteriophages designed to target a broader range of bacteria or to prevent or delay the development of phage resistance. Phage cocktails are commonly employed to broaden the host range and enhance the efficacy of phage therapy (Chan et al., 2013; Ribeiro et al., 2023). While bacteriophage cocktails offer several advantages, they also present certain limitations. Effective phage cocktail creation can be challenging and necessitates a thorough analysis of the phage composition, because strain specificity is necessary for optimum treatment outcomes, a “one-size-fits-all” approach might not work (Mishra et al., 2024). Phage to find efficient phages for every bacterial isolate, time-consuming and tedious screening procedures are required due to the uncertain relative importance of bacterial resistance mechanisms (Kim et al., 2024).
In addition to bacteriophage cocktails, there is another strategy, phage antibiotic synergy. Phage-antibiotic synergy refers to the enhanced effectiveness of bacteriophages (phages) when used with antibiotics, particularly against antibiotic-resistant bacterial strains (Gu Liu et al., 2020). Cefadroxil with ϕSZUT, ϕSZIP1, ϕSZIP2 infecting Salmonella spp., Staphylococcus aureus, Escherichia coli (Iqbal et al., 2020); ϕPT1b with tetracycline/amoxicillin can reduce the level of antibiotic resistance in Escherichia coli (Narulita et al., 2020). Phage-antibiotic synergy holds promise as a therapeutic strategy against MDR bacteria, but it also faces several limitations that can hinder its effectiveness. While some combinations of phages and antibiotics demonstrate synergistic effects, others can exhibit antagonism. The efficacy of PAS is significantly influenced by the host environment. Factors such as pH, nutrient availability, and the physiological state of the bacteria can alter the interactions between phages and antibiotics (Gu Liu et al., 2020; Li et al., 2021).
Bacteriophage genome engineering is an expanding field that allows for the modification and optimization of phages to combat MDR bacterial infections (Cong et al., 2024). These advancements offer promising avenues for tailoring phages to broaden their host range, enhance their lytic activity, and overcome bacterial resistance mechanisms. This highlights the importance of studying and manipulating the bacteriophage genome. Modifying the bacteriophage genome is a new challenge compared to bacterial genome engineering (Islam et al., 2020).
Bacteriophages Genome Engineering
Bacteriophage genome engineering is the process of genetic modification in viruses that infect bacteria (bacteriophages) to improve their efficiency and therapeutic range, especially in the face of antibiotic-resistant bacteria (Jia et al., 2023; Usman et al., 2023). The principles of genome editing involve inducing double-strand breaks (DSBs) at specific sites of DNA. Following this DNA damage, site-specific genomic changes are introduced and cellular DNA repair pathways are activated (Gaj et al., 2016). Given the rise in antibiotic resistance, bacteriophage genome editing holds significant promise for phage therapy in treating bacterial infections. By increasing the host range and efficiency of bacteriophages, this technique can be an effective alternative to traditional antibiotics. So far, some research has been conducted to engineer bacteriophages against MDR strains using various techniques (Table 1).
Previous Works in Bacteriophage Genome Engineering
Cas, CRISPR-associated protein; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeat.
Homologous Recombination
Homologous recombination (HR) in bacteriophages is crucial for genetic manipulation and understanding phage biology. This biological process involves the exchange of DNA between molecules with similar or identical sequences, essential for genome stability and genetic diversity. The T4 bacteriophage is a classical model for studying HR, utilizing it for high genetic exchange, DNA repair, and initiation of DNA replication (Mosig, 1998; Kreuzer, 2000). This process requires coordination between DNA replication and recombination machinery, enabling DNA damage recovery and genetic reorganization necessary for viral survival (Liu, 2010; Kuzminov, 2011). HR in bacteriophages transitions efficiently from recombination intermediates to replication forks. The T4 bacteriophage has evolved to use this transition as a primary means of DNA replication initiation, involving presynaptic filament dynamics and coordinated DNA helicase activity to integrate recombination with DNA replication and repair (Kovacs et al., 2024; Nguyen et al., 2012). These mechanisms highlight the importance of HR in phage biology and its potential for genetic engineering.
In the context of genetic engineering techniques, HR is used to generate recombinant phage with specific gene replacements, deletions or insertions. This process is usually done by introducing plasmids containing homologous sequences into phage-infected bacterial cells. Upon infection, HR will occur between the phage genome and the donor plasmid, resulting in a new phage with the desired genetic modification. Although this method provides an effective way to modify the phage genome, HR efficiency is often low and requires a lengthy screening process to obtain phage progeny with the desired changes (Kovacs et al., 2024; Chen et al., 2019).
The HR process begins with the formation of DSBs in DNA. In bacteriophage T4, the exonuclease activity of the Gp46/Gp47 complex plays a role in resecting DSBs into ssDNA (Kreuzer and Brister, 2010). Once the ssDNA is formed, the RecA protein will bind to the ssDNA and search for homologous sequences on the target chromosome. When homologous sequences are found, strand invasion occurs to form a D-loop structure, which can then be further processed to produce new recombinants (Liu, 2010).
Bacteriophage λ is another example of an HR system that is widely used in research. This system consists of several genes such as exo and bet that function to facilitate DNA exchange by increasing the frequency of recombination. By using the recombination function of bacteriophage λ, researchers can efficiently perform gene replacement in Escherichia coli, allowing genetic manipulation without relying on specific restriction sites (Murphy, 1998; Thomason, 2014).
Recombineering
Recombineering is a genetic technique that utilizes HR to alter the bacterial genome in vivo. This genetic engineering technique employs recombination proteins encoded by bacteriophages to facilitate precise DNA modifications in the bacterial genome, enabling DNA editing without the need for restriction sites, which are essential in conventional genetic recombination. Recombineering uses linear donor DNA, such as PCR products or synthetic oligonucleotides, which are introduced via electroporation to bacterial cells expressing the recombination function (Li, 2023). Bacteriophages, viruses that invade bacterial cells and use host cell machinery to reproduce, encode recombination proteins that enable DNA recombination with homology of 50 nucleotides (nt). This technique allows for gene editing, including gene deletion, gene replacement, gene removal, point mutation and gene modification with tags, reporter fusion, and other constructs (Thomason, 2014; Nafissi and Slavcev, 2014).
Recombineering with bacteriophages has been used in various biotechnology applications, including the development of transgenic vectors, gene manipulation in animals and plants, and clinical applications in humans (Nafissi and Slavcev, 2014). This technique also enables fast, efficient, and accurate in vivo editing of bacterial genomes (Thomason, 2014). The system can use short homologous sequences (30–50 bp) to drive genetic changes, making it possible to manipulate large DNA constructs without the limitations of traditional restriction enzyme methods (Li, 2023).
The recombination function in bacteriophages involves site-specific recombination and HR mechanisms (Menouni et al., 2015). These mechanisms enable the integration of the bacteriophage genome into the host genome through integrase enzymes that orchestrate recombination reactions between two DNA binding sites, attP and attB (Nafissi and Slavcev, 2014). This DNA serves as a target construct to produce the desired genetic change. This process is done through electroporation, where the DNA is introduced into the bacterial cell using electrical impulses. Proteins from bacteriophages such as the λ Red and RecET systems of prophage Rac play an important role in catalyzing this reaction (Sharan et al., 2009). Prophages that are in a latent state can replicate passively with the host until the induction of the lysis process Menouni et al., 2015).
Bacteriophage recombineering of electroporated DNA (BRED) is an innovative technique used to modify bacteriophage genomes in an efficient and simple manner. This method utilizes the recombination ability of modified Mycobacterium smegmatis cells to increase the frequency of recombination, allowing the generation of bacteriophage mutants without the need for complex genetic selection (Marinelli et al., 2008; Wetzel et al., 2021; Jia et al., 2023).
In BRED, phage DNA and recombination substrate (usually a double or single DNA) are introduced into bacterial cells through an electroporation process (Fig. 1A). This process allows the phage DNA and substrate to enter the cell simultaneously, increasing the chance of recombination (Wetzel et al., 2021). BRED has been used for various types of genetic modifications in bacteriophages, including gene deletion, making uncharacterized gene deletions, point mutations, introducing small changes in DNA sequences, foreign gene insertion, adding foreign genes into the bacteriophage genome (Thomason et al., 2009; Lv et al., 2023).
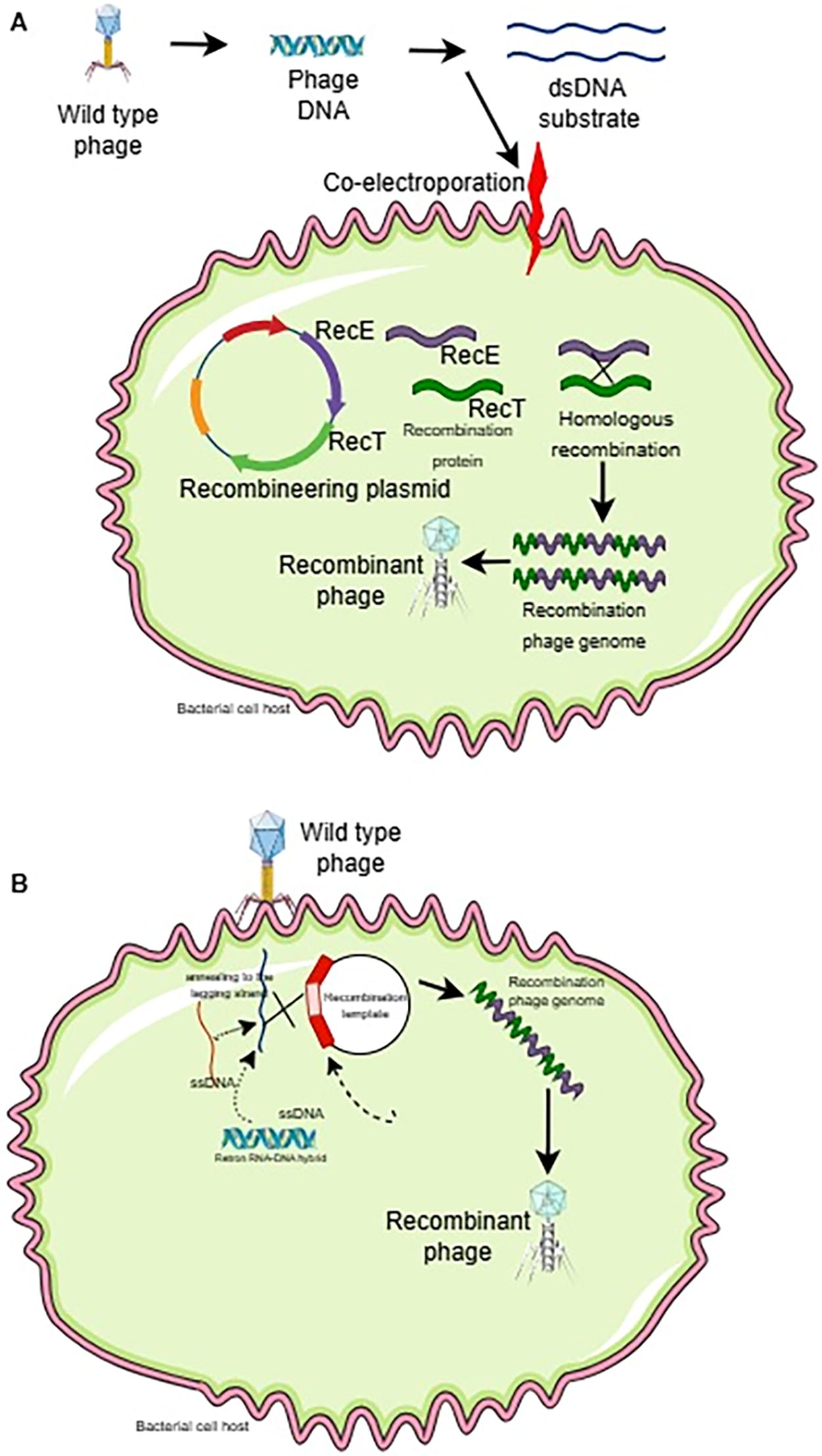
Bacteriophage recombineering with infectious particles (BRIP) is an innovative method for genetic engineering in bacteriophages that separates the electroporation and infection steps of phage (Fig. 1B). This method aims to overcome limitations in traditional recombination techniques that are often ineffective on phage with a lytic life cycle, where positive selection with bacterial growth markers cannot be applied (Isaev et al., 2022; Mahler et al., 2023).
In BRIP, DNA containing genetic modifications is introduced into bacterial cells via electroporation. This allows the DNA to enter the cell while phage infection has not yet occurred. After electroporation, the bacterial cells are then infected with bacteriophages. This utilizes the phage’s ability to perform HR with the DNA that has been introduced (Marinelli et al., 2012; Chen et al., 2019).
CRISPR/Cas
Bacteria and archaea have innate-adaptive immune systems called Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) and CRISPR-associated proteins (Cas) that shield them from viral infections. Six types of Cas proteins have been identified, namely: Cas1, Cas3, Cas8, Cas9, Cas10, Cas12, and Cas13 (Robb, 2019). In genome editing, the most commonly used protein is Cas9, which can cut DNA. CRISPR-Cas9 is unlike transcription activator-like effector nuclease (TALEN) and zinc-finger nuclease (ZFN), which require protein engineering. The mechanism involves a CRISPR gene array consisting of foreign DNA elements integrated between short palindromic repeats that are transcribed into RNA (crRNA) fragments corresponding to the foreign DNA. To help the crRNA associate with Cas9, a bacterial scaffold RNA called trans-activating crRNA (tracrRNA) is also created. This system’s discovery transformed genome editing techniques, which use a guide RNA (gRNA) made up of paired tailored crRNA and tracrRNA sequences to reroute Cas9 to the intended target (Fig. 2; Ran et al., 2013). But for Cas9 to recognize and bind to the target DNA, the protospacer adjacent motif (PAM) of the 50-NGG-30 sequence needs to bind to it successfully. The capacity to multiplex, which enables the simultaneous revision of several genomic regions, is a benefit of the CRISPR/Cas system over alternative genome alteration techniques (Cong et al., 2013; Strich and Chertow, 2019). The sgRNA-Cas9 combination will attach to the target DNA sequence and cleave it if the sgRNA is complementary to it. Researchers can employ a range of sgRNA design tools to create sgRNAs (Cui et al., 2018).
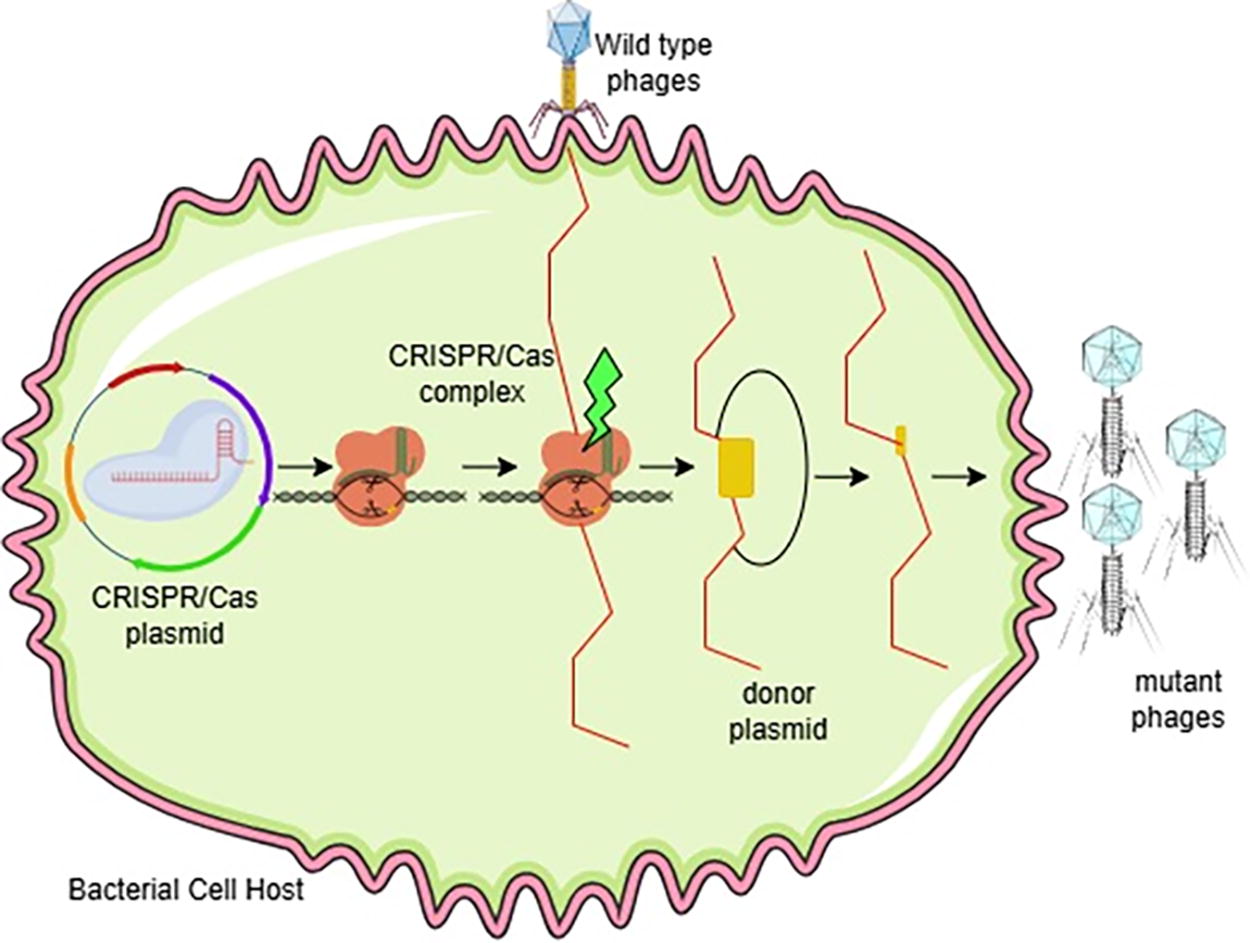
CRISPR/Cas. CRISPR/Cas technology can be used to edit the phage genome. The process starts with infection of bacterial cells by wild-type phages, and then CRISPR/Cas plasmids are introduced into the host cells. The CRISPR/Cas complex then cuts the phage DNA at the location specified by the guide RNA. A donor plasmid containing the desired DNA sequence is then used to repair the DNA cut, resulting in a mutant phage with new properties. Cas, CRISPR-associated protein; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeat.
The CRISPR/Cas system’s initial step involves using nuclease or nickase activity to identify targeted DNA cleavages in the form of double-strand breaks (DSBs/Double Strand Breaks) or single-strand breaks (SSNs/Single Strand Nicks). In the second stage, the targeted cleavage site triggers the DNA repair process, which is typically carried out by the cell’s endogenous DNA damage repair machinery. Two primary pathways are utilized in this stage to accomplish various kinds of genomic alterations (Swartjes et al., 2020; Asmamaw and Zawdie, 2021).
The first approach involves using non-homologous end joining (NHEJ/Non-Homologous End Joining) to repair the cleft region. This method works only with double strands; single strands are unable to trigger the NHEJ pathway. Insertions and deletions (indels and deletion insertions) will be designed into the target region during NHEJ-mediated repair (Ghezraoui et al., 2014; Su et al., 2016; Song et al., 2021). To activate homology-directed repair (HDR/Homology Direct Repair), another pathway needs a repair template donor template that has sequence modifications other than DSB or SSN chosen into the cell. This process necessitates HR between foreign and chromosomal DNA, which is triggered by local DNA cleavage. By substituting the intended HR template for the original sequence, nearly any desired edit can be achieved (Cong and Zhang, 2015; Xue and Greene, 2021).
Characteristic of Engineered Bacteriophages
Engineered phages, similar to wild-type phages, possess a virion consisting of a head (capsid) that contains linear double-stranded DNA (dsDNA), and a tail. The tail’s end has proteins that allow the phage to recognize and attach to specific bacterial host cells (Łobocka et al., 2021). The head diameter (hd) and tail length (tl) are key morphometric parameters used to classify phages. Myophages, which have tails with larger diameters, tend to have larger capsids. Phages form plaques, clear or turbid areas, on a bacterial lawn, with plaque diameter varying based on phage characteristics. The size of the phage head can be correlated to plaque diameter, with larger heads often resulting in smaller plaques due to slower diffusion through the agar (Jurczak-Kurek et al., 2016).
Engineered phages can have distinct tail proteins (Liang et al., 2022). Modifying receptor-binding proteins (RBPs) can alter the host range and infectivity of bacteriophages. Myoviridae phages (with icosahedral heads and contractile tails) possess long tail fibers as RBPs (Jia et al., 2023). Engineered phages can be modified to display cell-penetrating peptides (CPPs) on their surface to enhance internalization into cells (Zhao et al., 2023). Engineered phages have distinct genomic characteristics, with variations in genome size, GC content, predicted gene numbers, and gene density. For example, one study characterized two phages with genome sizes of approximately 111–112 kb and a GC content of around 45% (Liang et al., 2022). The growth of engineered phages may be influenced by regions of heterogeneity (sequence identity <50%) (Jurczak-Kurek et al., 2016).
Application of Engineered Bacteriophages
Applications for engineered phages are numerous and span many different domains. Phages can undergo genetic modification to increase their capacity to eliminate bacterial hosts. Engineered phages can make drug-resistant bacteria susceptible to antibiotics once again. Through genetic engineering, the host range of a phage can be altered. In vivo, engineered phages may have a longer half-life. Endotoxin release can be decreased by genetic manipulation (Lv et al., 2023). It is possible to modify phages to express enzymes that break down antibiotic-resistant bacterial biofilms. In material science and industry, engineered phages are used in a variety of ways, including the production of biomaterials with unique characteristics. For instance, graphene’s stability was enhanced by M13 phages that were modified to express a peptide that binds graphene. Bacteria use quorum-sensing mechanisms to control their pathogenicity and life cycles, which can be altered by modified phages (Pizarro-Bauerle and Ando, 2020). Even in a mixture of several bacterial species, particular bacteria can be identified using engineered phages (Gibb et al., 2021). The efficacy of the engineered phages produced through different techniques vis-à-vis controlling MDR pathogens can be seen in Table 2.
Efficacy of the Engineered Bacteriophages
MDR, multidrug-resistant.
Examples of engineered phages and the modifications made to alter their host range such as T2ppD1. This is an engineered version of phage T2, where the gp37 and gp38 genes (encoding tail fiber proteins) were replaced with those from phage PP01, which specifically targets E. coli O157:H7. As a result, T2ppD1 acquired the host range of phage PP01 and lost the ability to infect the original host of phage T2, E. coli K-12. The tail fiber genes (gp37 and gp38) from phage T2 were swapped out for their counterparts in phage IP008 to create an engineered T2 phage with the same host range as IP008 via the IP008 host range. Compared to phage T2, phage IP008 infects 33% and 7% of environmental E. coli isolates, respectively, indicating a wider host range (Pires et al., 2016). The pb1 portion (long tail fiber protein) of the narrow-host range phage vB STyj5-1 (STyj5-1) was substituted with the corresponding portion from a broad-host range phage vB BD13 (BD13) to develop STB-pb1, an engineered T5-like phage. When compared to STyj5-1, the resultant phage, STB-pb1, showed better absorption rates and a wider spectrum of plaquing hosts (from 20 to 30 strains) (Zhang et al., 2022). PSA phage of Listeria with gp15 mutation. Listeria strain WSLC1033, which is devoid of WTA galactosidase function, can be infected by a Listeria phage PSA with a gp15 mutation (Jia et al., 2023).
Challenges and Perspective
In the context of treating human diseases, bacteriophage engineering offers both views and challenges, particularly in light of the rise of bacteria resistant to antibiotics. The capacity of bacteria to evolve resistance to phages through a variety of defense mechanisms, including altering receptors or targeting phage DNA. Only a few bacterial strains can be infected by the majority of bacteriophages due to their limited host range. Safety issues are raised when phages are introduced into clinical settings, particularly when endotoxins may be released during bacterial lysis (Łobocka et al., 2021; Ranveer et al., 2024).
Because of their varied genomes, bacteriophages are difficult to research and control. Bacteriophages frequently have unique and poorly described genes in their mosaic genomic architecture. High genetic variability results from the increasing complexity of the genomic landscape caused by horizontal gene transfer and recombination processes. Bacteria share genetic material, including genes that confer resistance to antibiotics, with other microorganisms through a process known as horizontal gene transfer. This process involves bacteriophages, which may aid in the development of antibiotic resistance (Sun et al., 2019). Phages have the ability to transduce antibiotic resistance genes from one bacterium to another (Colavecchio et al., 2017). Mobile genetic components such as plasmids, transposons, and genomic islands15 frequently include antibiotic-resistance genes. Through phage-mediated transduction, these mobile genetic components can be passed from one bacterium to another (Ross and Topp, 2015).
While the search results do not directly address the role of genetically engineered phages in transduction, it can be inferred that modifying phages could potentially enhance their ability to transduce specific genes (Penadés et al., 2015). A primary challenge in editing bacteriophage genes using the commonly used CRISPR-Cas9 system lies in the development of bacteriophage resistance. Bacteriophages can acquire resistance to the CRISPR-Cas9 system through various mechanisms, including the emergence of escape mutants and hindering attachment (Jia et al., 2023). Engineered phages can be employed as vehicles for targeted gene delivery and to target particular eukaryotic cells, including tumor cells. HR within the host bacteria is a traditional technique for altering the genes of lytic phages. New strategies, including CRISPR-Cas systems and cell-free synthesis, have emerged to improve the efficiency and precision of phage engineering, offering promising avenues for tailoring phages to broaden their host range and enhance their lytic activity (Bowring et al., 2022).
Regulatory agencies worldwide are in the process of establishing frameworks for phage therapy, including genetically modified phages, but several challenges remain. Navigating the regulatory landscape for phage therapy is difficult (Anomaly, 2020). For extensive medicinal applications, phages need to be generated in large scale under Good Manufacturing Practices certified by regulatory agencies (Pires et al., 2020). Standardized clinical protocols are necessary (Toussaint et al., 2024). Advanced clinical trials are needed to establish the effectiveness of engineered bacteriophages in real-world scenarios.
Footnotes
Authors’ Contributions
R.A.F. and E.N. conceptualized the review topic and framework. R.A.F. conducted the initial literature search and analysis. E.S. and H.S.A. contributed to the literature review by identifying key studies and theories. E.N., E.S., and H.S.A. supervised the review. All authors wrote the initial and final drafts of the article. All authors have critically reviewed and approved the final draft and are responsible for the content and similarity index of the article.
Disclosure Statement
The authors declare no potential conflicts of interest concerning the research, authorship, and/or publication of this article.
Funding Information
No funding was received for this article.