
Abstract
Background:
Variants in the human X-linked cyclin-dependent kinase-like 5 (CDKL5) gene have been reported as being etiologically associated with early infantile epileptic encephalopathy type 2 (EIEE2). We report on two patients, a boy and a girl, with EIEE2 that present with early onset epilepsy, hypotonia, severe intellectual disability, and poor eye contact.
Methods:
Massively parallel sequencing (MPS) of a custom-designed gene panel for epilepsy and epileptic encephalopathy containing 112 epilepsy-related genes was performed. Sanger sequencing was used to confirm the novel variants. For confirmation of the functional consequence of an intronic CDKL5 variant in patient 2, an RNA study was done.
Results:
DNA sequencing revealed de novo variants in CDKL5, a c.2578C>T (p. Gln860*) present in a hemizygous state in a 3-year-old boy, and a potential splice site variant c.463+5G>A in heterozygous state in a 5-year-old girl. Multiple in silico splicing algorithms predicted a highly reduced splice site score for c.463+5G>A. A subsequent mRNA study confirmed an aberrant shorter transcript lacking exon 7.
Conclusions:
Our data confirmed that variants in the CDKL5 are associated with EIEE2. There is credible evidence that the novel identified variants are pathogenic and, therefore, are likely the cause of the disease in the presented patients. In one of the patients a stop codon variant is predicted to produce a truncated protein, and in the other patient an intronic variant results in aberrant splicing.
Introduction
C
The CDKL5-related encephalopathy is an X-linked dominant disorder, yet reported predominantly in females, only a few boys have been reported so far. The CDKL5 gene is localized on chromosome Xp22.13 and contains 21 exons. CDKL5 is an ubiquitously expressed protein with highest levels in the brain, thymus, and testes, involved in the brain maturation (Lin et al., 2005; Rusconi et al., 2008). The protein product shuttles in the cytoplasm and nucleus (Amendola et al., 2014). In the nucleus, CDKL5 also participates in the control of nuclear speckle morphology. Interestingly, the nuclear speckles in the interchromatin compartment are dynamic parts involved in the activation of a transcription factor by continuous activation of splicing factors. This mechanism affects pre-mRNA splicing (Ricciardi et al., 2009). CDKL5 and MECP2 seem to share a common molecular pathway and symptoms from Rett syndrome and can also be found in some females with CDKL5-related encephalopathy. But findings from various studies suggest that CDKL5 disorder is an independent entity with its own specific traits (Evans et al., 2005; Fehr et al., 2013). Variants in CDKL5 occur mostly de novo, but germline mosaicism has also been published (Weaving et al., 2004).
Three stages of CDKL5-related epilepsy have been described by Bahi-Buisson et al. (2008). At stage I, brief tonic seizures typically occur around the age of 4 weeks, interictal electroencephalography (EEG) is often normal at this period, but neurological examination reveals overall motor hypotonia and poor eye contact. Seizures usually respond to antiepileptic treatment. This “honeymoon” period may last several months, but EEG might already worsen at that time and motor and mental developments show no progression. Stage II presents with epileptic spasms or tonic seizures between the age of 6 months and 3 years. Infantile spasms and hypsarrhythmia are corticosteroid responsive at least in some cases, and some improvement of social contact and behavior is also reported. Severe mental and motor developmental delay, absent speech, and visual interaction are consistent with the diagnosis of epileptic encephalopathy. At stage III (after 3 years of age), patients have multiple types of seizures: epileptic spasms, tonic seizures, absences, and frequent myoclonic seizures. Some patients do not enter stage III and remain seizure free.
Previously, most of the CDKL5-related encephalopathies were shown to affect mainly females, probably because the studies included mostly females. Male patients with CDKL5 variants have been reported later (Elia et al., 2008; Fichou et al., 2009; Sartori et al., 2009). The clinical severity of the CDKL5 disease for both genders has been described as being very similar (Liang et al., 2011).
Materials and Methods
The study was approved by the Ethics Committee of 2nd Faculty of Medicine, Charles University in Prague and University Hospital Motol. Affected individuals were selected by an experienced epileptologist KŠ after referral to University Hospital Motol. We have a large cohort of patients with severe epilepsy of unknown etiology at our Centre for Epilepsies in University Hospital Motol. Patients were first selected by our epileptologist (K.Š.) and geneticist (M.V.) according to the criteria: negative brain magnetic resonance (MR) and availability of DNA samples from probands and both parents to verify variants. Previously, negative array comparative genome hybridization (CGH) testing is not a necessary condition. Patient's legal representatives signed informed consent.
DNA samples were isolated from peripheral blood using automated magnetic beads technique (iPrep™ PureLink™ gDNA Blood Kit; Invitrogen).
Massively parallel sequencing (MPS) of a custom-designed gene panel for epilepsy and epileptic encephalopathy with 112 related epilepsy genes was performed (HaloPlex; Agilent Technologies, Santa Clara, CA). The panel included ADAR, ADCK3, ADSL, ALDH7A1, ALG13, AMT, AP4S1, ARHGEF9, ARX, ASAH1, ATP1A2, ATP1A3, BRAT1, C10ORF2, CACNA1A, CASK, CDKL5, CLCN4, CPT2, DCX, DEPDC5, DNM1, DOCK7, EEF1A2, FASN, FLNA, FOLR1, FOXG1, GABBR2, GABRA1, GABRB3, GABRG2, GAMT, GCSH, GLDC, GNAO1, GPHN, GRIN1, GRIN2A, GRIN2B, GRIN2D, HCN1, HDAC4, HNRNPU, CHD2, IQSEC2, KCNA2, KCNB1, KCNC1, KCNH5, KCNJ10, KCNQ2, KCNQ3, KCNT1, KCTD7, KIAA2022, MBD5, MECP2, MEF2C, MFSD8, MOCS1, MOCS2, MTOR, NEDD4 L, NRXN1, PANK2, PCDH19, PHF6, PIGA, PIGQ, PLCB1, PNKP, PNPO, POLG, PRIMA1, PRRT2, PURA, QARS, RNASEH2A, RNASEH2B, RNASEH2C, ROGDI, RYR3, SAMHD1, SCN1A, SCN1B, SCN2A, SCN8A, SLC12A5, SLC13A5, SLC19A3, SLC25A22, SLC2A1, SLC35A2, SLC6A1, SLC9A6, SPTAN1, ST3GAL3, STXBP1, SYN1, SYNGAP1, SYNJ1, SZT2, TBC1D24, TDP2, TPP1, TREX1, TSC1, TSC2, UBE3A, WDR45, and ZEB2.
Our designed gene panel targeted coding regions of the genes and adjacent exon-intron boundaries (flanking introns ±50 bp). Data from MPS were analyzed with SureCall software (Agilent Technologies) and NextGENe Software (SoftGenetics LLC, State College, PA). Average read length was 110 bp. 99.1% of the target region was covered at least 10×. Both methods are based on BWA-MEM alignment (Li and Durbin, 2010)
Sanger sequencing was performed with primers designed for exon 7 and 18 of the CDKL5 gene to confirm the novel variants. Parentity was tested using six microsatellite markers.
For confirmation of the functional consequence of the intronic CDKL5 variant in patient 2, a new blood sample for RNA study was taken. Total RNA was extracted from blood mononuclear cells using TRIzol reagent and transcribed into full length cDNA using Cloned AMV First-Strand cDNA Synthesis Kit according to manufacturer's instructions with Oligo (dT)20 as a primer (Thermo Fisher Scientific).
We designed primers for amplifications of cDNA of exons 4-8: forward ACATGAAATTGTGGCGATCA, reverse TACCATCTGGTGGCAACGTA. We expected aberrant splicing, therefore, we amplified exon 7 and adjacent exons to study the splicing changes. PCR products obtained from the cDNA were loaded on 2% agarose gel. The PCR products were sequenced using BigDye Terminator kit v3.1 (Life Technologies, CA). Capillary electrophoresis on the ABI 3130 Genetic Analyzer (Life Technologies) was performed.
Results
Patient 1
The patient is a 3-year-old boy, the first child of healthy, unrelated parents. He was born in the 38th gestational week through caesarean section because of cephalopelvic disproportion. Birth weight was 2750 g. Early postnatal cardiologic examination confirmed Fallot tetralogy. Postnatal adaptation was otherwise normal.
At the age of 4 weeks, he had a few staring spells with jerking of upper limbs and head deviation to the right. EEG was normal. One month later, he presented with a series of short episodes of behavioral arrest and wide eye opening. Right-sided frontocentral spikes were seen on EEG at that time. At the age of 4 months, hypomotor seizures were frequent. When he was 1 year old, cardiosurgical correction of the Fallot tetralogy was performed. In the postoperative period, a series of epileptic spasms appeared with hypsarrhythmia on the EEG. Later he developed myoclonic seizures and tonic seizures.
Seizures have been intractable: valproic acid, topiramate, phenobarbital, vigabatrin, levetiracetam, phenytoin, clobazam, and their combinations were not permanently effective. Adrenocorticotropic hormone was partially effective. Ketogenic diet was administered from the age of 2 to 3 years with a reduction of seizure frequency, but only for a few months. At the age of 3.5 years, tonic seizures in series predominate but myoclonic seizures are present as well.
Psychomotor development was delayed since the first months. Overall hypotonia and poor social and visual contact were observed, as well. From the age of 3.5 years, relatively short lower limbs became apparent. He has no head control; he is not capable of independent sitting. Speech is absent; he performs only some unintelligible vocalizations and has central visual impairment.
Before performing epilepsy gene panel testing the patient underwent repeated brain MR imaging, showing only mild frontal atrophy. The metabolic screening and skin biopsy showed normal results. Targeted screening for DiGeorge syndrome was also negative. Moreover, array CGH was also done and no causal variants were found. The clinical phenotype is summarized in Table 1.
EEG, electroencephalography; MR, magnetic resonance.
Patient 2
This female patient was born from the third, normal pregnancy of healthy, unrelated parents. Delivery and early postnatal period were uneventful. Bilateral tonic seizures of upper limbs followed by clonic jerking appeared at the age of 6 weeks. Initial EEG recordings did not show epileptiform activity, later bilateral frontocentral spikes were observed.
Seizures became more complex, usually starting with behavioral arrest “freezing” followed by tonic flexion or extension of the upper limbs and continued as asynchronous clonic jerking of the face, eyelids, and limbs and ended as a series of spasms. As she became older, the tonic phase was less prominent and the jerks were more asynchronous and irregular. Reactivity was disturbed during the seizures.
The seizures were drug resistant: phenobarbital and vigabatrin had partial effect; levetiracetam, clonazepam, valproic acid, topiramate, and phenytoin did not control the seizures, but seizures disappeared for 1 year at the age of 3 years. This happened without any intervention, there was no change in medication with vigabatrin, topiramate, and clobazam. During this period, she improved in walking and acquired some nonverbal communication skills. Seizures reoccurred at the age of 4 years. Therefore, ketogenic diet was implemented with good result: seizure freedom was achieved 3 months after initiation of the diet. The girl remains hypotonic, nonverbal, with severe intellectual disability and motor delay. Repeated brain MR and metabolic screening showed normal results. The clinical phenotype of the patient is summarized in Table 1.
A novel nonsense variant c.2578C>T (p. Gln860*) in hemizygous state in exon 18 of CDKL5 according to the reference sequence NM_003159.2 was found in patient 1. The predicted reading frame is interrupted by a premature STOP codon, leading to a serious effect on the protein and probably nonsense-mediated mRNA decay. The variant was confirmed by Sanger sequencing of exon 18 of the CDKL5 gene (Fig. 1) and was not found in the healthy parents, so we assume it occurred de novo. To our knowledge, this variant has not been previously described, is not listed in HGMD Professional (www.hgmd.cf.ac.uk/ac/index.php), and is absent from global/population databases such as the 1000 Genomes Project (www.1000genomes.org), Exome Aggregation Consortium (http://exac.broadinstitute.org), and Exome Variant Server (http://evs.gs.washington.edu/EVS). According to the ACMG criteria published in Richards et al. (2015), the variant has one very strong (PVS1: null variant in a gene wherein loss of function [LOF] is a known mechanism of disease), one strong (PS2: de novo: both maternity and paternity confirmed in the patient with the disease and no family history), one moderate (PM2: absent from controls in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium), and ≥2 supporting criteria (PP3: multiple lines of computational evidence support a deleterious effect on the gene or gene product; PP4: the patient's phenotype or family history is highly specific for a disease with a single genetic etiology); therefore, we consider it as pathogenic and causal for EIEE2 in this patient.
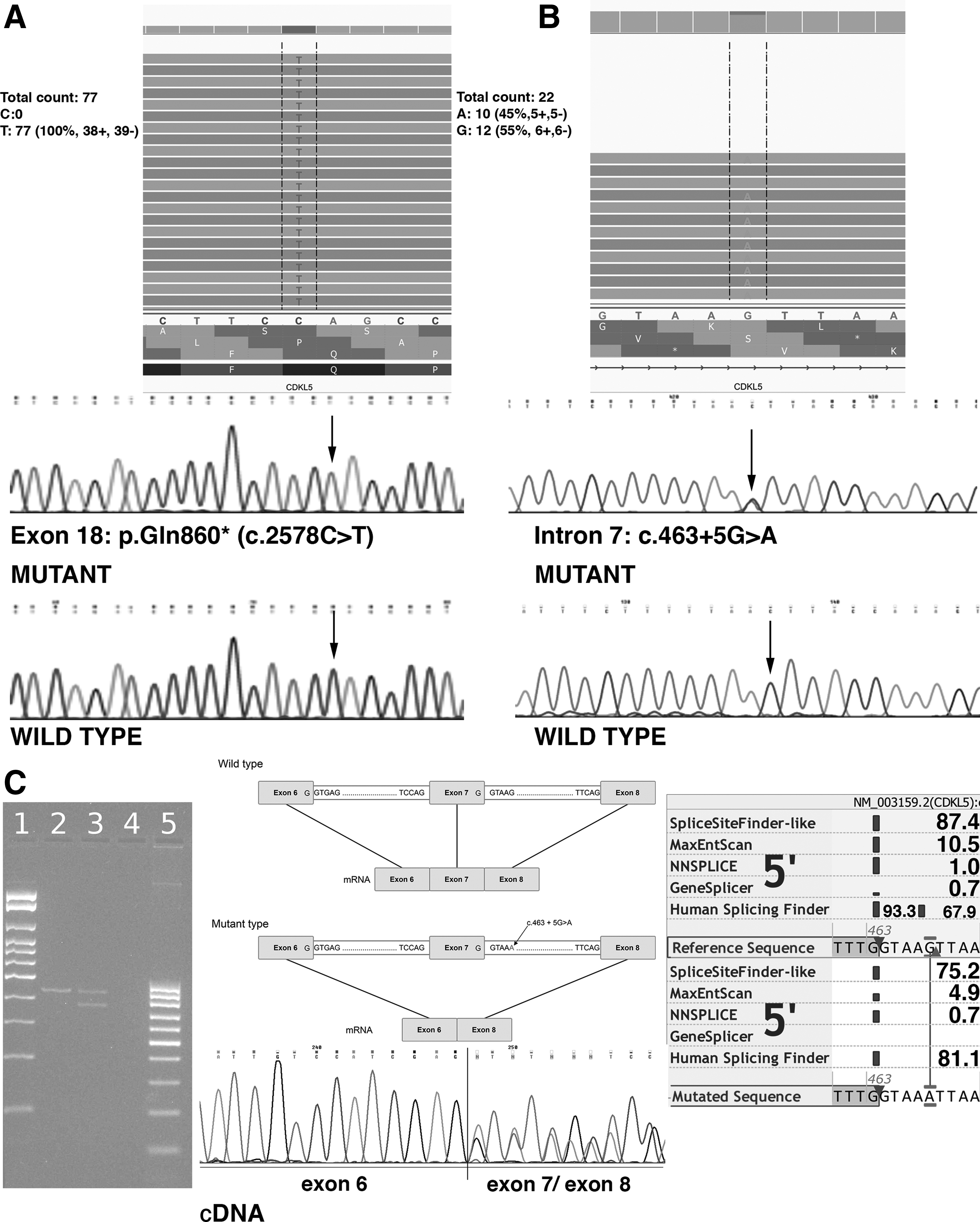
Schematic view of two novel variants in the CDKL5 gene and RNA study.
In patient 2, MPS identified a heterozygous intronic variant close to the donor splice site of exon 7 of CDKL5 gene (NM_003159.2). The variant occurs de novo, because it was not found in the healthy parents (parentity was tested). According to the ACMG rules, the variant has one strong and ≥2 supporting criteria. Therefore, the variant is evaluated as likely pathogenic.
Computer analysis using Alamut v.2.1. (Interactive Biosoftware, Rouen, France) based on the evaluation by a few predictors SpliceSiteFinder-like (www.umd.be/HSF/), MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html), NNSPLICE (www.fruitfly.org/seq_tools/splice.html), GeneSplicer (www.cbcb.umd.edu/software/GeneSplicer/gene_spl.shtml), Human Splicing Finder (www.umd.be/HSF/) was performed. These different algorithms predicted high reduction of the splice site score (Fig. 1C). The difference between the sequences higher than 10%, such as in our case, predicts a strong effect in the splicing process (Shapiro and Senapathy, 1987).
PCR product from cDNA of patient 2 with splice site variant was loaded on 2% agarose gel. In comparison with the control sample, two fragments (differing in 60 nucleotides corresponding to exon skipping of exon 7) were observed due to the formation of an aberrant shorter transcript.
Sanger sequencing demonstrated the presence of exons 4, 5, 6, 7, and 8 on the wild type allele and exons 4, 5, 6, and 8 on the mutant allele.
Discussion
Most of the CDKL5-related encephalopathies were yet described in females and only rarely in males. This may be caused because variants in CDKL5 were known as the cause for the so called atypical Rett syndrome. Therefore, CDKL5 sequencing was frequently the next step after MECP2 testing, which was performed predominantly in girls.
We present two novel variants in the CDKL5 gene, one in a female and one in a male. The overall clinical manifestation of the male patient was similar to the phenotype reported in a few boys with rare point variants in CDKL5 (Elia et al., 2008; Wong and Kwong, 2015). Both children presented with different seizure types, two cases in particular showed very complex seizure types with features of tonic, clonic, and myoclonic convulsions as well as series of spasms. Seizures were drug resistant, but ketogenic diet was at least partially effective in patient 1 and granted seizure freedom in patient 2. Global hypotonia and poor eye contact since early infantile age were present in both children. We did not observe gastrointestinal problems or scoliosis in our patients, but this can be due to their young age. Both patients had transient sleep problems due to myoclonic jerks; however, their epileptic origin has not been proven in either child. We have not observed any stereotyped hand movements typical for Rett syndrome patients. According to our knowledge, no co-occurrence of Fallot tetralogy or any other congenital cardiac anomaly with only CDKL5 variants has been described. The only report describing a boy with severe encephalopathy, congenital cataracts, and tetralogy of Fallot attributes the phenotype to the interstitial deletion of Xp22 comprising also CDKL5 (Van Esch et al., 2007).
There is enough evidence that the variants are pathogenic and, therefore, are the cause of the disease in the presented patients. In patient 1, hemizygous variant p. Gln860* predicting premature stop codon, thus causing a serious effect on the protein, arose de novo. According to ACMG guidelines, we consider the variant as pathogenic, whereas in patient 2, the intronic variant c.463+5G>A affects the splicing and leads to the formation of aberrant transcript.
The splice donor site variant: NM_003159.2: c.463+5G>A in the intron 7 of the CDKL5 gene may cause leaky splicing mechanism and leads to the formation of a 20 amino acids shorter protein without exon 7 (Fig. 1C). We showed that variant in intron 7 c.463+5G>A changes the 5′ donor splice site of intron 7. Aberrant splicing has been already described as a mechanism that could lead to EIEE2 (Weaving et al., 2004; Archer et al., 2006).
It has been suggested that splice site variants effect on the phenotype correlates with the proportion of functional CDKL5 due to mutated alleles (Bahi-Buisson et al., 2008). The precise effect on the severity of the phenotype is not known. It is expected that a milder phenotype correlates with less severe mental retardation and the ability to walk even though gait is atactic. The clinical presentation in patient 2 is milder than in patient 1. This may be caused by leaky splicing. Also, variability of X inactivation in the different brain regions could lead to various phenotype manifestation (Weaving et al., 2004). The other hypothesis is that exon 7 skipping (deletion of 60 nucleotides) causes in-frame deletion, protein is shortened only by 20 amino acids, and this may cause milder phenotype in the patient. Similar clinical phenotype with splice site variant IVS6-1G>T that also results in exon 7 skipping has been reported as pathogenic (Archer et al., 2006). Our patient, similarly to the patient in Archer et al. (2006), had severe mental retardation, hypotonia, epilepsy seizure starting in the first months, and “honeymoon periods” with subsequent return of seizures.
Conclusions
We showed enough evidence that the variants are pathogenic and, therefore, are the cause of the disease in the presented patients. In one patient, stop codon variant causes a serious effect on the protein and the intronic variant in the second patient affects the splicing. Using RNA study, we were able to verify the formation of aberrant transcript in patient 2.
Footnotes
Acknowledgments
This study was supported by the Charles University in Prague, Second Faculty of Medicine, project GA UK No. 438216, and MH-CZ DRO University Hospital Motol Prague, Czech Republic 00064203-6005.
Author Disclosure Statement
No competing financial interests exist.