
Abstract
Background:
The role of deep intronic variants in hereditary cancer susceptibility has been largely understudied. Previously, the BRCA2 c.6937 + 594T>G variant has been shown to preferentially promote the inclusion of a 95 nucleotide cryptic exon and to introduce a premature termination codon. Our objective was to further assess the pathogenicity of the BRCA2 c.6937 + 594T>G deep intronic variant.
Patients and Methods:
We examined the association between BRCA2 c.6937 + 594T>G and breast cancer (BC) risk in 464 BC cases and 497 noncancer controls from Puerto Rico.
Results:
The overall frequency of the G allele was 2.1% in this population. There was no association between the TG/GG genotypes and BC risk in the uncorrected model and after correcting for confounders. There was only one carrier of the GG genotype. This individual did not have personal or family history of cancer and did not meet the National Comprehensive Cancer Network criteria for hereditary cancer genetic testing.
Conclusions:
Although previous work has demonstrated that the BRCA2 c.6937 + 594T>G variant affects splicing, this association study does not support a pathogenic role for the BRCA2 c.6937 + 594T>G intronic variant in breast and ovarian cancer syndrome susceptibility. Furthermore, it emphasizes the need to take into account multiple diverse populations in association studies for the assessment of variant pathogenicity.
Introduction
T
It has been shown that for a subset of high-risk families, BRCA1 and BRCA2 testing for hereditary breast and ovarian cancer syndrome (HBOC) does not identify a causative variant (Caminsky et al., 2016). Traditionally, diagnostic screening for cancer risk assessment was performed on genomic DNA and limited to coding exons and intron/exon junctions (Frank et al., 2002). As a result, ∼79% of the BRCA variants reported in the Breast Cancer Information Core database are frameshifts, resulting in the insertion of a premature termination codon, missense or nonsense changes, leaving changes that may alter splicing largely unexplored (National Institutes of Health, 2017). Previous studies have proposed that splicing variants may account for a subset of HBOC cases (Sanz et al. 2010; Gambino et al., 2015).
Recently, transcript analysis for individuals with strong familial cancer history that does not present pathogenic variants in coding sequences or at exon/intron junctions led to the identification of a deep intronic mutation in BRCA2 (c.6937 + 594T>G; rs191253965), promoting the inclusion of a 95 nucleotide cryptic exon and introducing a premature termination codon (Anczuków et al., 2012). Based on molecular analyses showing a preference for the cryptic splice site compared with the canonical site, the authors proposed that this variant should be considered pathogenic (Anczuków et al., 2012).
Due to an observed frequency of 3.8% for the rare allele in the Puerto Rico 1000 Genomes project sample, this population represents a unique opportunity to explore the role of this deep intronic mutation in conferring high risk for HBOC. The present study assesses the association of the BRCA2 c.6937 + 594T>G variant with BC in Puerto Rican women, using an incident case-control study design. We present evidence against a role of this variant in conferring high risk to BC.
Patients and Methods
Study population
This study utilized an incident case-control design. The study was approved by the Ponce Health Sciences University-Ponce Research Institute Institutional Review Board. Participants (464 BC cases and 497 noncancer controls) were selected from our larger BC study, which has been previously described (Matta et al., 2012; Morales et al., 2013). Women 21 years or older, whose three out of four grandparents were born in Puerto Rico, are included in the study. Inclusion criteria for cases were patients who (1) were recently diagnosed histopathologically with primary BC and (2) were treatment-naive (had not received chemotherapy, blood transfusions, or radiotherapy). Cases were not selected for family history or age of onset. All individuals recruited as controls had a negative mammography and breast examination within the last 6 months. Participants completed a standard questionnaire regarding their demographic information, hormonal and pregnancy history, and family history of cancer. In addition, for BC patients, a tumor pathology report was obtained. Personal and family history information was used to estimate pretest likelihood of mutation using the BRCAPRO (Parmigiani et al., 1998) and Myriad II (Frank et al., 2002) models. The carrier risk was calculated independently for maternal and paternal sides of each family.
Genotyping
Genomic DNA from lymphocytes of 464 BC patients and 497 controls was extracted using QIAamp DNA Mini Kit (Qiagen, Valencia, CA), following the manufacturer's protocol. Genotyping of BRCA2 (c.6937 + 594T>G) (rs191253965) was conducted using a TaqMan Custom Allelic Discrimination Assay (Applied Biosystems, Foster City, CA). This assay consists of two primers for amplifying the sequence of interest and two TaqMan minor groove binding probes for detecting allele 1(T) and allele 2(G), using the fluorophores FAM and VIC, respectively. PCR was performed in 5 μL reaction volumes under the following conditions: initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. Genotypes were confirmed by Sanger sequencing in a subset of 29 individuals. Primers for PCR amplification and sequencing were as follows: 5′-TGATGCCATTACCGATCAGA-3′ and 5′-GGGAATCCAAGCTGTCAAAA-3′ (Anczuków et al., 2012).
Statistical analyses
All statistical analyses were conducted with R version 3.2.1 implemented in R Studio (RStudio Team, 2015). Differences in frequency distributions were calculated by Pearson's chi-square test. The association between BRCA2 intron 12 c.6937 + 594T>G variant and BC risk was analyzed by multiple logistic regression analysis using confounding factors as covariates. The impact of genotype on BC risk was quantified by an odds ratio (OR) and by calculating the 95% confidence interval (CI).
Results
The demographic, pregnancy, and hormonal history of the study population are summarized in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/gtmb). Overall, controls were younger (p < 0.0001), more likely to be married and less likely to be widows (p < 0.0001) than BC cases. When comparing reproductive risk factors, a significant difference was observed in the number of children (p < 0.001). For cases, menopause occurred later in life (p = 0.004), and they were less likely to have used estrogen replacement therapy (p = 0.008). Both groups were similar for all other variables measured.
Out of a total of 961 BC cases and controls, 18 nonaffected controls and 21 BC cases were heterozygous for the BRCA2 c.6937 + 594T>G variant. In addition, one control was found to be homozygous for the G allele. The genotype frequencies for BRCA2 c.6937 + 594T>G did not significantly deviate from the Hardy-Weinberg equilibrium in cases (χ2 = 0.3021, p = 0.8) or in noncancer controls (χ2 = 0.587, p = 0.7). There was no significant association when comparing the frequency of carriers (TG+GG) between BC cases and controls in the unadjusted model (OR = 1.19, 95% CI = 0.63-2.27, p = 0.6), after adjusting for age, education level, and civil status (OR = 1.17, 95% CI = 0.58-2.38, p = 0.5) or after adjusting for all confounders (OR = 0.98, 95% CI = 0.34-2.76, p = 0.3) (Table 1).
Adjusted for age, education level, and civil status (n missing = 185).
Adjusted for age, education level, civil status, age at menopause, use of estrogen replacement therapy, number of children, and breast feeding duration (n missing = 598).
CI, confidence interval, OR, odds ratio; Ref., referent.
To further assess the potential clinical relevance of the BRCA2 c.6937 + 594T>G variant, the prior carrier risks of homozygous and heterozygous carriers were estimated using the Myriad tables and BRCAPRO tool (Fig. 1). The distribution and ranges of the pretest likelihood of carrying a BRCA2 mutation, as predicted by BRCAPRO and the Myriad tables, were not different in carriers of the c.6937 + 594 [G] allele versus controls. Overall, only a minority of the carriers (13%) met the National Comprehensive Cancer Network (NCCN) criteria (version 2.2016) for HBOC genetic testing (NCCN, 2016). It is also noteworthy that a 51-year-old participant was found to be homozygous for this variant. She had no personal history of cancer, did not meet the NCCN criteria for genetic testing, and her estimated prior risk of carrying a BRCA pathogenic variant was estimated at 1.5% and 0.1% with the Myriad tables and BRCAPRO model, respectively. This participant reported family history of endometrial cancer, but no family history of breast or ovarian cancer at any age.
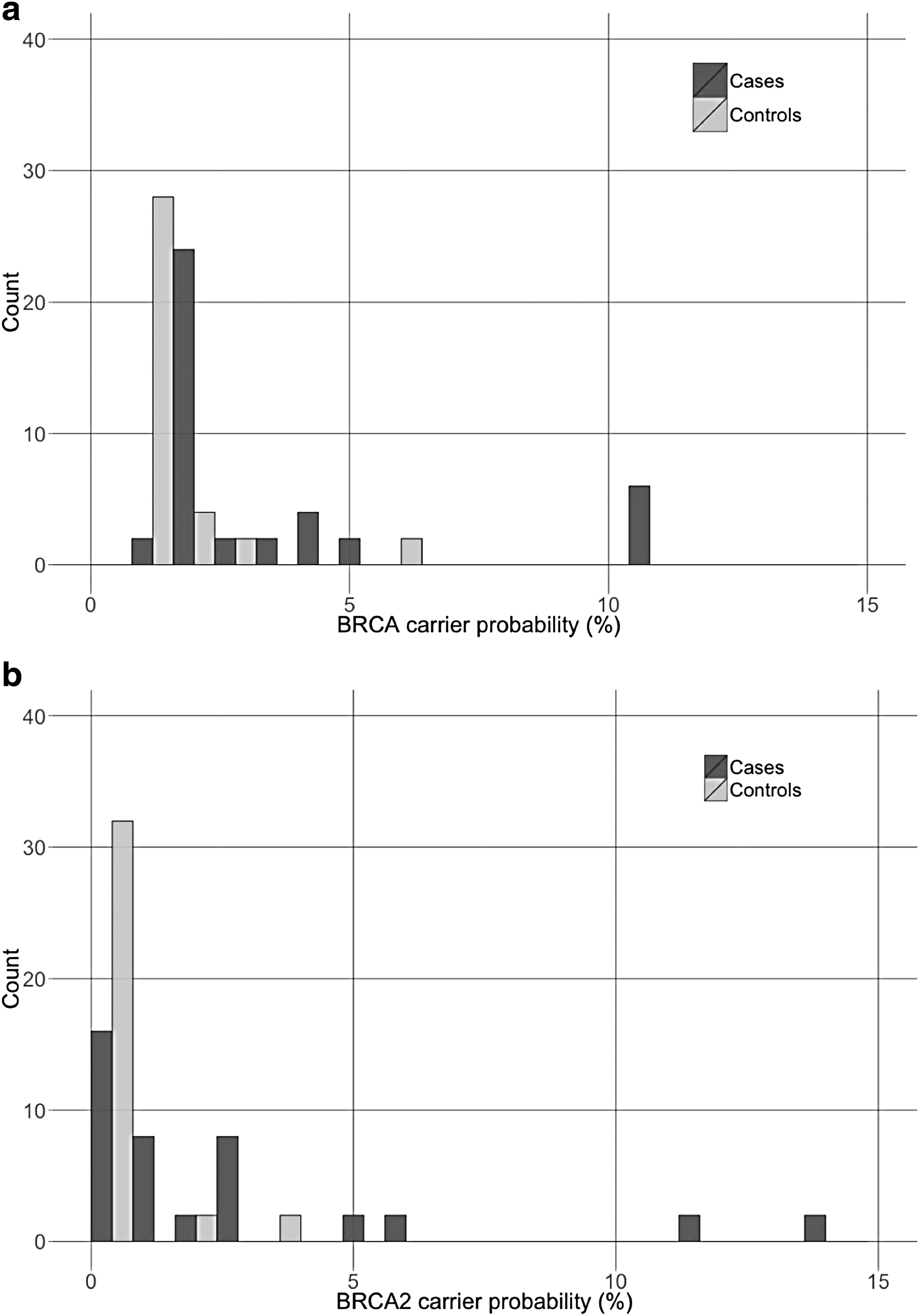
Distribution of the prior carrier probability in carriers of the c.6937 + 594T>G alternate allele grouped by diseases status. BRCA carrier probability as estimated by the myriad tables
Discussion
In the current study, we have assessed the potential pathogenicity of a BRCA2 deep intronic variant, c.6937 + 594T>G, using genetic association and family history studies. Previous molecular studies have indicated that this variant preferentially activate a cryptic splice site within intron 12, leading to the inclusion of a 95 nucleotide cryptic exon in the BRCA2 transcript (Anczuków et al., 2012). We provide evidence that this variant is unlikely to represent a pathogenic variant in the context of high risk of BC. In the current study, we did not observe an association between the genotype at BRCA2 c.6937 + 594T>G and BC risk.
Several hundreds of pathogenic variants have been identified in the BRCA genes (National Institutes of Health, 2017). Combined with the low prevalence of hereditary cancers, each of these variants is expected to be rare in an unselected population. Due to a well-characterized founder effect, the Ashkenazi Jewish population presents higher prevalence of pathogenic variants in the BRCA genes, explained mostly by three founder mutations: BRCA1 185delAG, 5382insC, and BRCA2 6174delT (Neuhausen et al., 1996; Tonin et al., 1996). It is noteworthy that the combined frequency for these three variants in the Ashkenazi Jewish population adds up to 2.6% (Roa et al., 1996; Frank et al., 2002; Phelan et al., 2002). In Hispanic populations, the BRCA1 185delAG variant is also the most frequent, reported in seven countries, including U.S. Hispanics (Dutil et al., 2015). Yet, the reported frequency for this variant in the Exome Aggregation Consortium (ExAC) Database is <0.03% (ExAC, 2016). In the 1000 Genomes Project Phase 3 dataset, the [G] allele at BRCA2 c.6937 + 594T>G has been observed in 10 individuals, which corresponds to an overall frequency of 0.2% (1000 Genomes Consortium, 2016). Since most of the carriers were observed in Puerto Ricans from Puerto Rico (PUR), the frequency of the rare allele was estimated to be 3.8% in that population (1000 Genomes Consortium, 2016). Although variant prevalence can vary significantly across race and ethnicity, it would be unlikely for a single pathogenic BRCA variant to reach such a high population frequency.
The classification of BRCA variants using epidemiological studies is usually complicated by the limited power associated with low population frequencies of the rare allele. Given that the observed allelic frequency for the [G] allele at BRCA2 c.6937 + 594T>G is of 2.1% in this study and an estimated hereditary cancer prevalence of 0.2% (Janavičius, 2010), power calculations indicate that the current sample would be adequate for detecting a risk variant that is associated with an OR of 2.15-fold or higher with a power of 80.7%. Although BRCA pathogenic variants have been associated with ORs in the order of 3.2- to 6.8-fold (Ford et al., 1998; Pruthi et al., 2010), we cannot rule out that this variant have a more modest effect on risk as described for the polymorphic stop codon K3326X (Meeks et al., 2015). However, for the purposes of clinical risk assessment, these variants should not be considered pathogenic (Spurdle et al., 2016).
In BRCA1 and BRCA2, functional in vitro assays revealed that 44-49% of the splicing variants predicted functional by bioinformatics approaches result in aberrant splicing (Sanz et al., 2010; Acedo et al., 2012). In a minigene assay, the BRCA2 c.6937 + 549G>T variant was shown to systematically promote splicing from the cryptic splice site (Anczuków et al., 2012). However, splicing of patients RNA is not always concordant with minigene assay results, which are single allele and lack the genomic context of the patient (Sanz et al., 2010). In the neurofibromatosis type 1 gene (NF1), a splicing variant in exon 37 reproduces the exon skipping patterns observed in patients RNA only if the minigene construct spans exon 31-38, emphasizing the importance of the genomic context of neighboring exons in studying splicing mutations (Buratti et al., 2006). The role of the genomic context and the splicing regulatory factors influencing the splicing outcomes in carriers of the BRCA2 c.6937 + 549G>T variant remain to be determined. Nevertheless, although in vitro functional assays can provide valuable functional data for the clinical classification of splicing alterations, this information should be evaluated in the context of additional evidence, such as family and epidemiological data (Thomassen et al., 2012).
One of the limitations of this study is the younger age of controls when compared with cases. This is especially a concern since BRCA2 mutations carriers tend to develop BC at later ages when compared with BRCA1 carriers (Antoniou et al., 2003). After correcting for age, there was no association between BRCA2 c.6937 + 594T>G and BC risk. This observation is further supported by the family history characteristics, as carriers were not more likely to present with family history of the diseases. In fact, one individual (a control) was identified as a homozygous carrier of the [G] allele at this locus. Biallelic BRCA2 carriers present with autosomal recessive Fanconi anemia, a genomic instability syndrome associated with developmental abnormalities, bone marrow failure, and a high predisposition to cancer (OMIM No. 605724). The nonpathogenicity of BRCA variants can therefore be assessed by the co-occurrence of a known pathogenic mutation in trans and the absence of the Fanconi anemia or other developmental delay phenotypes, to guard against the co-occurrence of hypomorphic variants (Domchek et al., 2013; Eggington et al., 2014). This individual did not present any indication of Fanconi anemia-related phenotypes or cancer.
Several groups have reported that splicing mutations may cause hereditary diseases (López-Bigas et al., 2005), including breast and ovarian cancer syndrome (Sanz et al., 2010; Thomassen et al., 2012; Wappenschmidt et al., 2012; Ahlborn et al., 2015). As more comprehensive genetic analysis assays are incorporated in routine clinical testing, more splicing and variants are likely to be identified. Assessing the pathogenicity of these variants will be important to prevent the complications associated with clinical management of individuals found to carry a variant of unknown significance (Murray et al., 2011). Our results also emphasize the utility of including diverse population in family history and epidemiologic studies aimed at classifying variants underlying genetic diseases.
Footnotes
Acknowledgments
The authors are grateful to all the women who participated in this study. We also want to thank the clinic teams at Auxilio Mutuo and Wanda Vargas at Ponce Research Institute. This work was supported by grant of the National Institute of Health (NIH), National Cancer Institute (NCI) (U54 CA163071, CA163068, and U01 CA116167) with the technical support of the Ponce Research Institute Molecular, and Genomics Core (M.A.G.I.C) (NIH-National Institute on Minority Health and Health Disparities (NIMHD) MD007579). Recruitment of participants was supported by grants from the NIH-National Institute of General Medical Sciences (NIGMS) (S06-GM008239 and 9SC1CA182846). R Rivera-Lugo received a fellowship from NIH-NIGMS (R25GM096955).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.