
Abstract
Background:
Autosomal recessive congenital ichthyoses (ARCI) are a group of rare nonsyndromic genodermatoses characterized by generalized scaly appearance of the epidermis with markedly impaired cutaneous barriers owing to defects in keratinization related genes. In this study, we ascertained a consanguineous Pakistani family affected with ARCI.
Aims:
To investigate genetic defect underlying disease phenotype in the affected family.
Methods:
All available members of the family (affected and unaffected) were sampled. Whole exome sequencing (WES) was performed on DNA of the proband and the data were analyzed for probable pathogenic variants. Segregation of the identified variant was validated by Sanger sequencing.
Results:
Analysis of the WES data identified a novel nonsense mutation, c.762C>G, in the PNPLA1 (patatin-like phospholipase domain containing 1) gene. The protein product of of this gene is involved in lipid organization during cornified cell envelope formation. The variant is predicted to result in the generation of a premature truncation site at amino acid position 254 (p.Tyr254*). This would result in the loss of a large C-terminal portion of the protein suggesting it to be rendered nonfunctional. In silico protein structure modeling confirmed a detrimental effect of the variation on protein structure.
Conclusions:
The study supports the evidence for the prevalence of PNPLA1 mutations in distant ethnic groups. Despite the significant number of reported ARCI cases with PNPLA1 variants, a straightforward genotype-phenotype correlation cannot be established.
Introduction
Autosomal recessive congenital ichthyosis (ARCI) is a genetically and phenotypically heterogeneous group of nonsyndromic cornification disorders broadly classified as harlequin ichthyosis, lamellar ichthyosis (LI), and congenital ichthyosiform erythroderma (CIE); the latter two form extreme ends of a spectrum of overlapping phenotypes with a combined prevalence of 1:200,000-300,000. Thus far, 14 genes, namely, TGM1, ALOXE3, ALOX12B, ABCA12, CYP4F22, PNPLA1, NIPAL4, LIPN, CERS3, SDR9C7, SLC27A4, ST14, SULT2B1, and CASP14 have been associated with ARCI phenotypes (Karim et al., 2017; Richard, 2017). Pathologically, defects in these genes and consequently their protein products interfere with the normal differentiation and desquamation of keratinocytes, leading to the formation of an abnormal stratum corneum with compromised permeability barrier functions. To reestablish the barrier functions, epidermis triggers homeostatic responses in the form of hypermetabolism. Genetic nature of the defect in corneocytes would result in incessant triggering of the hypermetabolism resulting in epidermal hyperplasia, therefore a thickened stratum corneum, characteristic of ARCI (Elias et al., 2012; Akiyama, 2017). In this study, whole exome sequencing (WES) identified a novel nonsense sequence variant in PNPLA1 (patatin-like phospholipase domain containing 1) gene underlying LI-CIE intermediate phenotype in a Pakistani family.
Materials and Methods
Samples collection and study approval
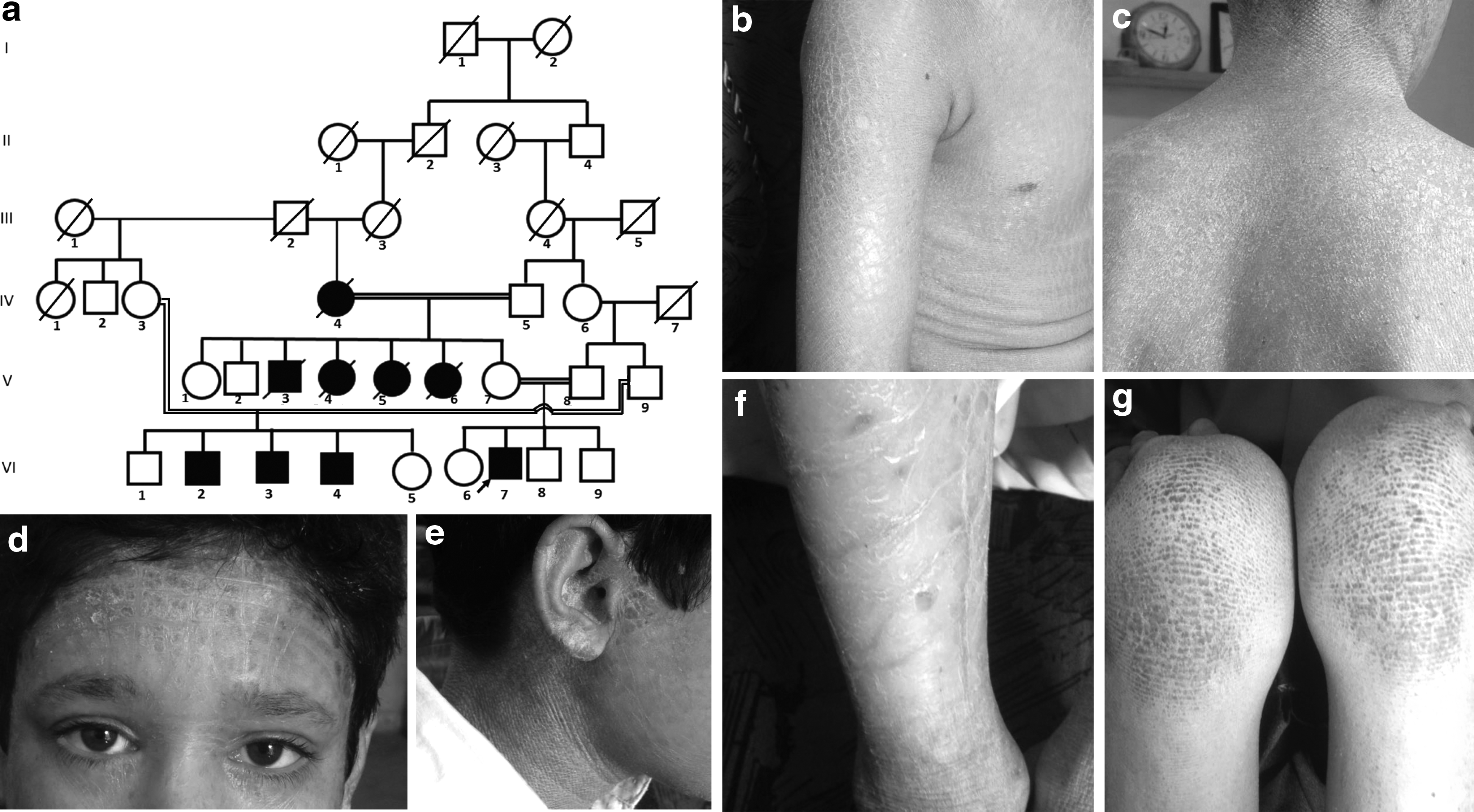
A consanguineous Pakistani family affected with ARCI (Fig. 1a-g) was recruited with informed consent. The pedigree was constructed from the information provided by elders of the family and venous blood samples were collected from four affected (VI-2, VI-3, VI-4, and VI-7) and eight normal individuals (IV-3, V-7, V-8, V-9, VI-1, VI-5, VI-6, and VI-8) of the family (Fig. 1a). The study was approved by the Institutional Review Board of the University.
Whole exome sequencing
To look for the causal genetic defect, WES of the proband (VI-7) was performed. All the exonic regions and flanking exon-intron boundaries were captured and enriched with the help of Agilent SureSelect Human All Exon V5 and loaded on an Illumina HiSeq4000 sequencer. Extensive cycles of extension and imaging through HiSeq4000 generated a total of 98,571,884 reads with an average size of 101 bp that were subsequently mapped to reference sequence by BWA (bwa-0.7.10) and duplicates flagged and removed through Picard (picard-tools-1.118). On the aligned reads, variants (single nucleotide polymorphisms and indels) calling and annotation through GATK (GATK3.v4) and SnpEff (SnpEff_v4.1) gave a total of 88,574 variants.
Shortlisting of variants
To shortlist the potential causative variants in the exome data nonsense, missense, frameshift, indels, and splice site variants in the above-mentioned 14 genes already associated with ARCI were selected. They were further sorted based on their consistency with recessive mode of inheritance that is homozygous or compound heterozygous. The selected variants were checked for their minor allele frequency (MAF) in South Asian (SAS) populations from Ensemble and ExAC database and those with unknown allele frequency or MAF <0.01 were shortlisted (Fig. 2) and checked for segregation in the family with disease phenotype by Sanger sequencing.
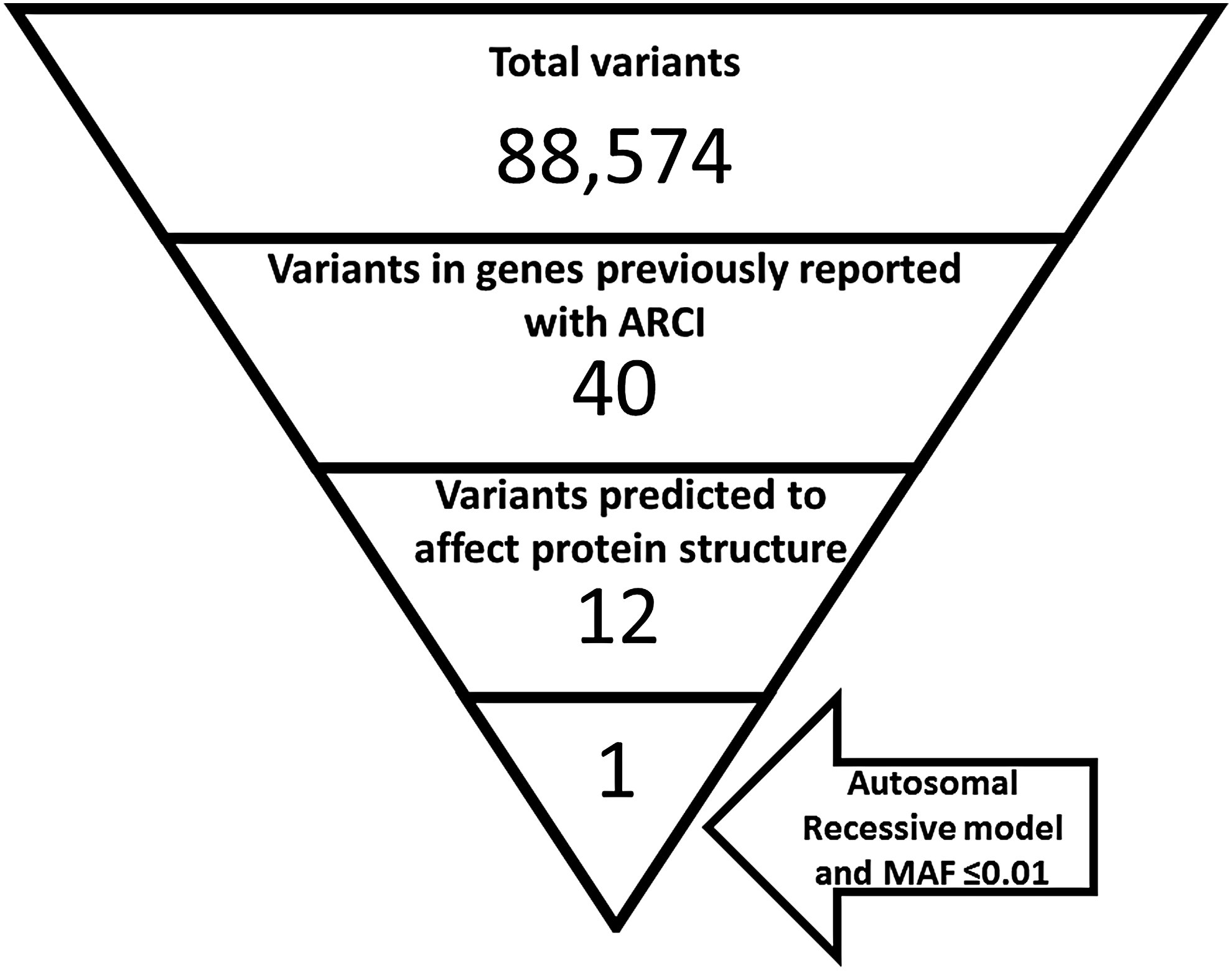
Number of variants at the different stages of filtration in the exome sequencing data of the proband (VI-7). SureSelect V5 kit was used to capture 359,555 target regions (exons and exon-intron boundaries) from 21,522 genes. The captured regions were sequenced using Illumina HiSeq 4000 sequencer that provided a total of 98,571,884 reads (9,955,760,284 bp) covering 50,390,601 bp target regions. On alignment with GrCh37 assembly, 79.8% of the reads were identified as on-target reads, whereas 72.8% of total were nonredundant. The mean depth of the target regions (on-target yield/target regions) was 99.3. The percentage of bases in the target regions with a depth of coverage of 10 × or greater was 97.7, at 20 × or greater 93.3, and at 30 × or greater 87.5. The minimum Phred-scaled quality score used in the analysis was 100.
Sanger sequencing
Segregation of the shortlisted PNPLA1 variant (NM_001145717.1; ENST00000394571.3; c.762C>G) with the disease phenotype in the family was tested through Sanger sequencing. Polymerase chain reaction (PCR) amplification of the genomic region flanking the potential candidate variant was performed with the primers 5′-GAGGCTCCACCATGAATGGTG-3′ (forward primer) and 5′-CTTTCTGGACCCAGCGGTCT-3′ (reverse primer). The PCR products were purified through Thermo Scientific GeneJET PCR Purification Kit (No. K0702) and subsequently subjected to sequencing in Applied Biosystems automatic sequencer 3730XL, using Big Dye terminator cycle sequencing kit V3.1 (PE Applied Biosystems, Foster City, CA). The sequences generated were analyzed in comparison with the reference sequence using BioEdit Sequence Alignment Editor (version 7.0.5.3). Effect of mutation on protein structure was evaluated in silico by generating three-dimensional (3D) protein models for normal and mutant protein using Phyre2. The models were visualized by RasMol Software.
Results
We identified a large inbred family affected with an LI-CIE intermediate phenotype. Five of the nine affected individuals had died before commencement of the study (Fig. 1a). The affected individuals had a history of congenital collodion membrane and presented with fine and white nonerythrodermic scales on trunk, neck, arms, thighs, scalp, ears, and face and larger transparent and plate-like scales on lower limbs. They had normal scalp hair, eye brows, and eye lashes but sparse hair on rest of the body. However, the severity of scales on different bodily sites varied among the patients as is common in ARCI. Other features frequently found with ARCI such as ectropion, eclabium, palmoplantar hyperkeratosis, and nail disorders were absent; yet, they had mild palmoplantar hyperlinearity (Fig. 1b-g).
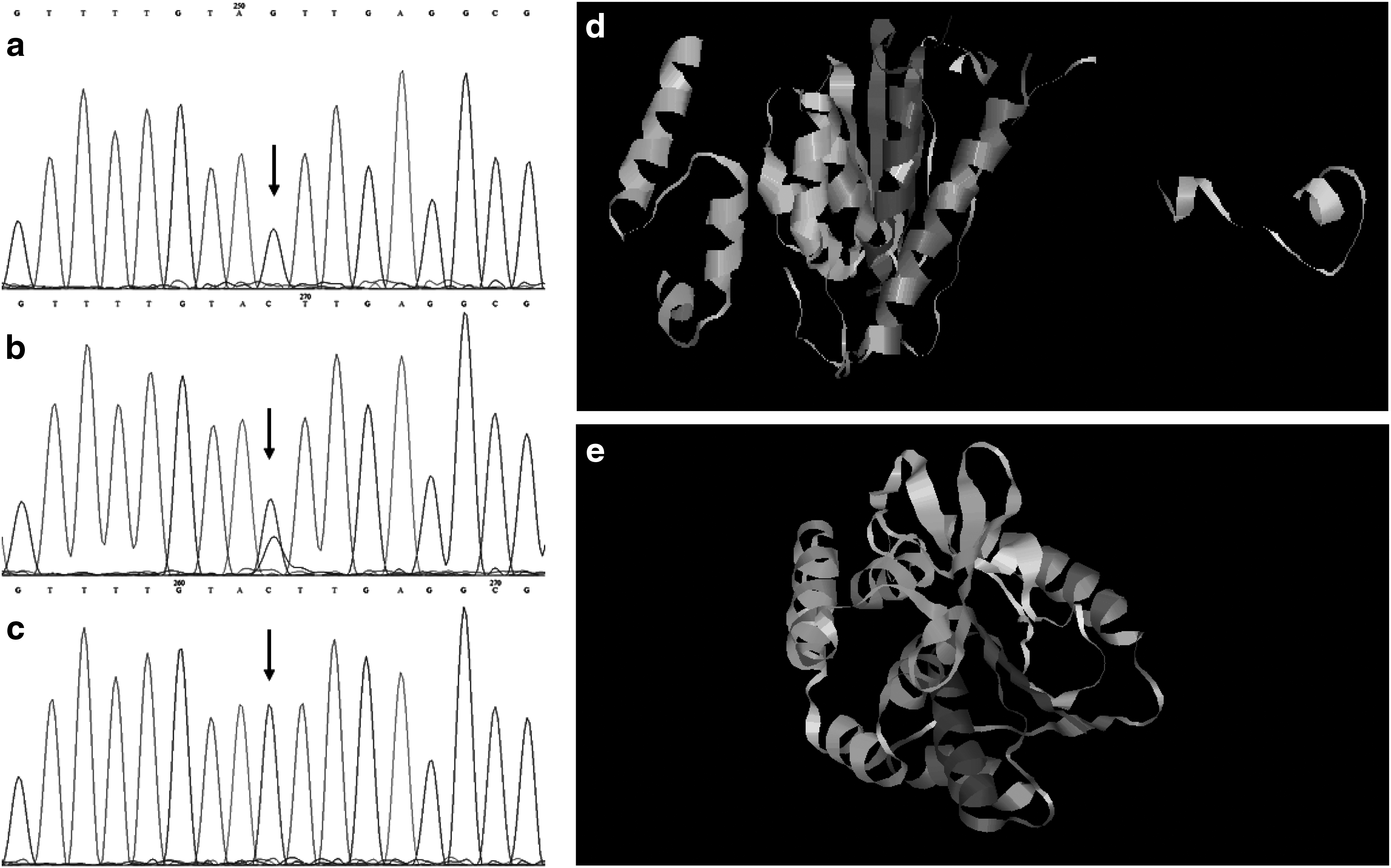
WES of the proband (VI-7) followed by variants filtration by the criteria mentioned in Materials and Methods section revealed a nonsense transversion mutation, c.762C>G in exon 5 of PNPLA1 gene. It was found in homozygous state (Fig. 3a) in the affected individuals (VI-2 VI-3, VI-4, and VI-7) and heterozygous (Fig. 3b) in normal individuals (IV-3, V-7, V-8, V-9, VI-1, and VI-6) of the family. The normal sibling VI-5 was homozygous for the wild-type allele C (Fig. 3c). The variation predicts premature truncation of the protein at amino acid position 254 (p.Tyr254*). The messenger RNA (mRNA) transcribed from the mutant allele would be highly probable to undergo nonsense-mediated mRNA decay (Nagy and Maquat, 1998) that was also predicted by MutationTaster. However, if the protein is produced by the mutant allele, its 3D structure would be inordinately diverted from that of normal as predicted by the Phyre2 (Fig. 3d, e).
Discussion
PNPLA1 is a gene located on the short arm of chromosome 6 (6p21.31) in a positive orientation (ncbi.nlm.nih.gov) coding for a member of the adiponutrin family, a subgroup of patatin-like phospholipases family of proteins (involved in lipid metabolism). Expressed primarily in the granular layer of the epidermis (Chang et al., 2013), various studies have associated defects in the gene with ARCI in humans and dogs (Grall et al., 2012; Tamamoto-Mochizuki et al., 2016). Intact PNPLA1 protein is found to catalyze the esterification of ω-hydroxyceramide with linoleic acid to form ω-O-acylceramides, essential for cutaneous barrier formation (Ohno et al., 2017). PNPLA1 knockout mice with impaired ω-O-acylCer displayed the formation of impaired cornified lipid envelope severely affecting its water barrier functions (Pichery et al., 2017). This compromised cutaneous barrier would elicit a torrent of homeostatic epidermal responses leading to several folds of thickened stratum corneum, hence the quintessential hyperkeratotic scaly appearance.
A tremendous increase in PNPLA1 mutations associated with ARCI in patients with different origins and ethnic groups around the world is seen in years 2017 and 2018 with 43 novel mutations. To date, a total of 46 mutations (36 nonsense/missense, 6 small deletions, 3 splice site, and 1 indel) in PNPLA1 underlying ARCI, have been reported. Of all the reported cases, eight mutations are such that they would predictably lead to complete loss of the protein (nonsense and frameshifts leading to generation of stop codons), six of which were found in a compound heterozygous state with missense mutations. Although considerable variability can be seen in the phenotypic features presented by the patients, a common ground can be seen in the features of generalized fine white to brownish scales accompanying mild erythroderma and presence of collodion membranes in most of the cases. However, the patients with mutations leading to complete loss of the proteins cannot be distinguished from those with missense mutations by the degree of severity of the phenotype (Grall et al., 2012; Boyden et al., 2017; Vahidnezhad et al., 2017; Zimmer et al., 2017).
Most of the reported mutations lie in the highly conserved core patatin domain of the protein (aa residues 16-185); however, recently mutations downstream the core domain has shown the requirement of C-terminal of protein for the patatin domain to perform its enzymatic activity (Boyden et al., 2017; Dökmeci-Emre et al., 2017; Pichery et al., 2017; Vahidnezhad et al., 2017; Zimmer et al., 2017; Diociaiuti et al., 2018). This study identified a novel nonsense mutation, p.Tyr254*, that predicts the loss of a large portion of C-terminal region including proline-rich domain (aa residues 326-451). Loss of the C-terminal domain in two members of adiponutrin family, PNPLA1 and PNPLA2, is found to compromise their catalytic activity and lipid droplets binding ability, thus indicating the importance of C-terminal domain for protein activities (Schweiger et al., 2008; Chang et al., 2013).
In summary, we report third nonsense mutation in PNPLA1 that lies outside the patatin domain underlying ARCI in a Pakistani family thereby supporting the hypothesis of C-terminal domain to be equally important for the protein function. Although a significant number of ARCI cases have been reported with underlying pathogenic variants in PNPLA1, a straightforward genotype-to-phenotype correlation cannot be established because of rampant phenotypic variability.
Footnotes
Acknowledgments
The authors acknowledge all members of the family, who participated in the study.
Author Disclosure Statement
No competing financial interests exist.