
Abstract
Abstract
Himadri, P., S. K. S. Sarada, M. Chitharanjan, and S. Dhananjay. Role of oxidative stress and inflammation in hypoxia-induced cerebral edema: a molecular approach. High Alt. Med. Biol. 11:231–244, 2010.—The present study reports the possible role of oxidative stress and inflammation (role of nuclear factor, NFκB) in hypoxia-induced transvascular leakage in brain of rats. The rats were exposed to a simulated altitude of 25,000 ft for different durations: 0, 3, 6, 12, 24, and 48 h. Brain water content, transvascular leakage, oxidative stress, and proinflammatory parameters were studied at different durations of hypoxic exposure. The results revealed that maximum increase in transvascular leakage in brain of rats was observed at 24 h of hypoxic exposure (240.16 ± 1.95 relative fluorescence unit (r.f.u)/g tissue) compared with control (100.58 ± 1.79 r.f.u/g tissue). There was a significant increase in reactive oxygen species (ROS) and lipid peroxidation (MDA), with concomitant reduction in antioxidants. Hypoxic exposure resulted in a significant increase in NFκB protein expression levels and in the DNA binding activity in the 24-h hypoxic exposure (p < 0.001) compared with control. There was a significant increase in proinflammatory cytokines, with concomitant upregulation of cell adhesion molecules. Simultaneously, to rule out the fact that inflammation causes cerebral edema, the rats were pretreated with curcumin (100 mg/kg body weight) 1 h prior to 24-h hypoxia. Curcumin pretreatment significantly attenuated the hypoxia-induced cerebral transvascular leakage (p < 0.05), with concomitant downregulation in the expression of brain NFκB levels (p < 0.001). The present study therefore reveals that inflammation (NFκB) plays a significant role in hypoxia-induced cerebral edema.
Introduction
High altitude cerebral edema (HACE) is a high altitude malady characterized by nonspecific pathophysiological symptoms, such as headache, loss of coordination, weakness, and decreasing levels of consciousness, including disorientation, loss of memory, hallucinations, and psychotic behavior. However, Clarke (1988) reported that, even though less common, HACE also occurs at extreme altitude (over 7000 m) in apparently well acclimatized climbers. If left untreated, HACE may result in death owing to brain herniation (Bartsch and Roach, 2001; Dietz, 2006). It has been observed in one of the human studies that brain tissue oxygen tension falls immediately with exposure to high altitude (Sick et al., 1982). A fall in partial pressure of inspired oxygen results in increased cerebral blood flow, followed by increased permeability of the cerebral endothelium during hypoxia (Roach and Hackett, 2001). Hypoxia-induced increase in regional cerebral blood flow has been reported in a variety of mammalian species (Krasney et al., 1990). It was reported earlier that increased cerebral blood flow results in increased intracranial pressure because of raised capillary hydrostatic pressure, which is thus responsible for many of the clinical manifestations of HACE (Bailey et al., 2006). It was also reported earlier that, cytokine activation, as well as free-radical-mediated damage to barrier function under hypoxia, may lead to fluid accumulation in the brain (Bailey et al., 2006; Dietz, 2006). However, a debate exists over inflammation or the involvement of some other factors in the development of high altitude cerebral edema (Hackett and Roach, 2004). Some reports have revealed that hemodynamic factors, such as sustained vasodilatation (Jensen et al., 1990), elevated cerebral capillary pressure (Krasney, 1994), and impaired cerebral autoregulation (Levine et al., 1999), most likely contribute to the formation of cerebral edema formation. However, Deng et al. (2009) recently reported that blocking of Rho associated kinase (ROK) inhibits the activation of NFκB and production of proinflammatory cytokines, leading to reduced transvascular leakage in brain of rats. But, so far, the exact molecular mechanism of fluid accumulation in the brain has not been explained. Therefore, the molecular mechanism behind cerebral edema formation owing to hypobaric hypoxia is yet to be explored.
It is a well-known phenomenon that reactive oxygen species (ROS) levels increase with exposure to high altitude (Bakonyi and Radak, 2004). Recently, it was shown that ROS are involved and may even play a causative role in AMS, HAPE, and HACE (Bailey et al., 2001; Baumgartner et al., 2002; Baumgartner et al., 2002; Bailey et al., 2004; Purushotham et al., 2008). However, the mechanism by which ROS play a role in HACE is not known. The redox sensitive transcription factor, nuclear factor kappa B (NFκB), is said to be regulated by oxidative conditions, and the activated form of NFκB is a heterodimer, which usually consists of two proteins, a p65 subunit and a p50 subunit (Baeuerle and Baltimore, 1996). It has also been reported that hypoxia-induced vascular injury is associated with the activation of the NFκB pathway (Howard et al., 1998). In turn, NFκB activation leads to upregulation of many genes involved in the recruitment, adhesion, and activation of leucocytes, thus creating a positive feedback loop for enhancing vascular damage (Lum and Roebuck, 2001; Witt et al., 2005), for alterations in the cerebral microvasculature with increased paracellular permeability, and also in the alterations in key tight junction proteins (Brown et al., 2003).
Although research supports the hypothesis that NFκB activation is stimulated by inflammatory or oxidative stress, debate continues as to the exact mechanism(s) that leads to its activation with respect to the type of stress. Further, the mechanism(s) by which ROS or inflammatory cytokines regulate NFκB remain unclear. Moreover, whether inflammation is the cause of HACE or a consequence of HACE is not yet known. Therefore, a time-dependent study of hypoxic exposure was planned using Sprague–Dawley rats to study the molecular mechanism involved in causing vascular leakage in brain of animals exposed to simulated hypobaric hypoxia. Further, we were interested in understanding the role of oxidative stress and the activation of transcription factor NFκB in causing transvascular leakage in brain of rats. We also proposed to determine whether NFκB activation (inflammation) appears before or after fluid accumulation in brain of rats under hypoxia. Our hypothesis in this study is that inflammation (NFκB) certainly plays a significant role in hypoxia-induced cerebral transvascular leakage. Further, if our hypothesis were true, then treatment with a known NFκB blocker (curcumin) during hypoxia would result in reduced transvascular leakage in the brain of rats.
Materials and Methods
Animals
The experiments were conducted using male rats (Sprague–Dawley strain) with average body weights (BWs) of 150 to 200 g. Rats were maintained at 25 ± 1°C with 12:12 hour light to dark cycles and given food and water ad libitum. All animal procedures and experimental protocol were approved by the Institutional Animal Ethics Committee and followed the guidelines of the Universities Federation for Animal Welfare (UFAW) guidelines for animal research.
Exposure to Hypoxia
The experiment was conducted in two phases.
Phase I experiment
A total of 72 rats were used in the phase I experiment; rats were divided into 6 groups of 12 rats each. Group 1 served as control (normoxia or 0 h); group 2 was exposed to hypoxia for a 3-h duration; group 3, for 6 h; group 4, for 12 h; group 5, for 24 h; and group 6, for 48 h.
Phase II experiment
The NFκB blockade study was carried out using curcumin in 24 rats, which were divided into 4 groups of 6 rats each. Group 1, which was exposed to 0-h hypoxia (control), received only vehicle; group 2 (hypoxia) received only vehicle and was exposed to hypoxia for 24 h; group 3 (normoxia + curcumin) was supplemented with curcumin at 100 mg/kg BW; and group 4 (hypoxia +curcumin) was supplemented with curcumin at 100 mg/kg BW and exposed to hypoxia for 24 h.
The temperature of the hypobaric chamber was maintained at a thermoneutral zone of 25 ± 1°C (to nullify the effect of temperature and to see only the hypoxic effect), with an altitude of 7620 m, a barometric pressure of 280 mmHg, and an airflow rate of 4 L/h. The partial pressure of arterial oxygen in control rats was found to be 95 ± 2 mm Hg, and in hypoxia-exposed rats it was 38 ± 2 mmHg, indicating that the rats were exposed to reduced levels of partial pressure of oxygen in the hypobaric chamber. The animals were provided with adequate quantities of food and water during exposure to hypoxia.
Determination of brain water content and vascular permeability
Brain water content
To quantify the brain water content in normoxic (control) and hypoxia-exposed animals, the wet weight of the brain tissue was taken immediately after removal. The samples were dried at 110°C for 24 h and reweighed to give the dry weight. The brain water content was calculated by taking the wet:dry ratio and expressed as w/d ratio (Schoch et al., 2002).
Vascular permeability
To quantify the vascular permeability in brain of rats, 200μL of sodium fluorescein dye (Sigma, St. Louis, MO, USA) at a concentration of 5 mg/kg BW in phosphate buffer saline (PBS) was injected through the tail vein in both control and hypoxia-exposed rats. In brief, 30 min before the predetermined time of hypoxic exposure, the rats were injected with the sodium fluorescein dye. Later, the animals were placed back in the hypoxic chamber up to the determined time. After exposure, the animals were anesthetized and then perfused with PBS (20 mL) through the left heart ventricle to remove the fluorescent tracer from the vascular bed. Subsequently, the whole brain was removed, washed with cold saline (0.9% NaCl), and divided into two equal parts. One part was kept in 3% formamide (Sigma) and left undisturbed for about 18 h at room temperature. The other part was kept in an oven at 55° to 60°C for 48 h. After 18 h, the tissues in formamide were centrifuged for 10 min at 3000 rpm. Then the fluorescence of the supernatant was collected in a dark room, and a reading was taken spectrofluorometrically at 485-nm excitations and 530-nm emissions (Baba et al., 1998). After 48 h, the dried tissues were weighed again. The fluorescence per gram was calculated. Results were presented as r.f.u./g of tissue.
Sample preparation
Whole brain was removed and rinsed in cold saline and made into 10% homogenate (0.154 M/L KCl) at 4°C for estimating various biochemical parameters.
Estimation of oxidative stress
The production of ROS in brain homogenate at different hours of hypoxic exposure (0, 3, 6, 12, 24, and 48 h) was determined by using a nonfluorescent dye [DCFH-DA (2, 7, dichlorofluorescein diacetate)], and the fluorescence was measured by spectrofluorimeter (Varian, USA) with an excitation at 485 nm and emission at 530 nm (Le Bel et al., 1990). Similarly, malondialdehyde (MDA) in brain tissue homogenate at different hours of hypoxic exposure (0, 3, 6, 12, 24, and 48 h) was determined by 2-thiobarbuturic acid assay, and absorbance was measured at 532 nm (Ohkawa et al., 1979). The reduced glutathione (GSH) was determined in the brain tissues by the method of Talt and Tan (1974) using dinitrobenzoic acid (DTNB) reagent, and absorbance was measured at 412 nm. Glutathione peroxidase (GPx) and superoxide dismutase (SOD) levels in the brain tissues were measured using a commercial kit (Randox, Crumlin, UK) as per manufacturer's instructions. Total protein content was estimated using the method of Bradford (1976).
Protein expression studies
Sample preparation
After completion of the determined time of exposure to hypoxia, the animals were sacrificed and the brains were perfused with cold PBS. The whole brain was removed and washed with cold saline and then homogenized in a buffer containing 0.01 M/L tris HCL, pH 7.6, 0.1 M/L NaCl, 0.1 mM/L dithiothreitol, 0.001 M/L EDTA, and 100 μg/mL protease inhibitor cocktail (Sigma). The contents were centrifuged at 3000 rpm for 15 min at 4°C; the supernatant was collected and stored at −80°C.
Nuclear protein extract preparation
Nuclear protein extracts were prepared according to a method reported previously (Hu et al., 2005), with some modifications. Briefly, brain tissues were homogenized with a manual homogenizer (Remi Motors, Mumbai, India) in ice-cold buffer containing 10 mM/L HEPES, pH 7.9, 10 mM/L KCL, 0.1 mM/L EDTA, 1.5 mM/L MgCl2, 1 mM/L dithiothreitol, 0.5 mM/L phenlymethysulfonyl fluoride, and 10 μL/mL of cocktail (protease inhibitors: antipain, chymostatin, pepstatin, and leupeptin). Cytoplasmic fraction was centrifuged at 8000 rpm (Sigma 3/18K, Germany) for 4 min at 4°C, and the nuclear fraction was reconstituted in a buffer containing 20 mM/L HEPES, pH 7.9, 400 mM/L NaCl, 1 mM/L phenylmethylsulfonyl fluoride, and 10 μL/mL of cocktail. The nuclear suspension was centrifuged at 14,000 rpm for 5 min, and the supernatant containing the nuclear extracts was collected and stored at −80°C.
Protein expression study (western blotting)
Proteins (50 μg) were separated on 10% (NFκB, TNF-α, IL-1, and IL-6) and 8% (ICAM-1, VCAM-1, P-selectin, E-selectin) sodium dodecyl sulfate polyacrylamide gel electrophoresis (Bio Rad, Hercules, CA, USA) and electroblotted onto nitrocellulose membranes (Millipore, Billerica, MA, USA). Membranes were blocked in PBST (0.1 M/L phosphate buffered saline, pH 7.4, 0.1% Tween 20) with 5% nonfat milk for 2 h at room temperature and thoroughly washed with PBST. The membranes were then incubated with primary antibodies (NFκB, TNF-α, IL-6, ICAM-1, VCAM-1, P-selectin, and E-selectin) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted in PBST for 2 h at room temperature. The membranes were thoroughly washed with PBST and then probed with their respective secondary antibodies conjugated with horseradish peroxidase for 1 h. Finally, membranes were washed 4 to 5 times with PBST. Membranes were developed using a kit (Chemiluminescence substrate; Sigma), and bands were visualized on x-ray film (Kodak, Rochester, NY, USA). Densitometric analysis was carried out using the Gel Doc system (UVP, BioImaging Systems, Upland, CA, USA).
Transcription factor (NFκB) activation studies: electrophoretic mobility shift assay (EMSA)
The EMSA for NFκB was carried out using a commercial kit (Pierce, Rockford, IL, USA). The binding mixture (25 μL) containing 10 μg of protein of nuclear extract and 1 μg of poly dI-dC were incubated in a tris-EDTA buffer (10 mM/L tris HCL, pH 7.4, 50 mM/L NaCl, 50 mM/L KCl, 1 mM/L MgCl2, 1 mM/L EDTA, 5 mM/L DTT) on ice for 15 min. Later 10 ng of biotinylated, double-stranded NFκB probe (NFκB oligonucleotides probe supplied by Operon, the sequence being NFκB, F 5′-AGT TGA
NFκB Blockade study
To determine the optimum dose of curcumin, the rats were treated with different concentrations of curcumin (50, 100, 200, and 300 mg/kg BW) orally and exposed to 24-h hypoxia. Brain water content and transvascular leakage studies were carried out (data not shown). The optimum dose for curcumin was found to be 100 mg/kg BW, where minimum brain water content and transvascular leakage were obtained. Therefore, curcumin was administered to rats orally at 100 mg curcumin/kg BW 1 h prior to hypoxic exposure (24 h), and we then determined the antioxidant and anti-inflammatory activity (NFkB) of curcumin in brain of rats exposed to hypoxia, as described above.
Statistical analysis
Statistical analysis was performed using SPSS for Windows (15.0) software (SPSS Inc., Chicago, IL, USA). Comparisons among six experimental groups for various time points and a curcumin-treated groups were made by using one-way ANOVA with the Student–Newman–Keuls test for multiple comparisons among groups, whereas comparisons of normoxia (0 h) and hypoxia-exposed (24 h) animals were made using the Student t test. Differences were considered statistically significant for p < 0.05. Results are expressed as mean ± SD.
Results
Determination of cerebral edema
Brain water content
To determine the exact time for initiation of cerebral edema formation as a result of hypoxic exposure, we quantified the brain water content by taking wet–dry weights of brain tissue and expressed this w:d ratio at different time intervals of hypoxic exposure: 0, 3, 6, 12, 24, and 48 h (Fig. 1A). The water content in the brain of rats increased significantly over the time course of hypoxic exposure and reached a peak at 24 h (w/d = 53% increase, p < 0.001), compared with the control (0-h exposure). Although further hypoxic exposure to 48 h led to a decline in brain water content compared with 24-h hypoxic exposure, it was highly significant compared to control.
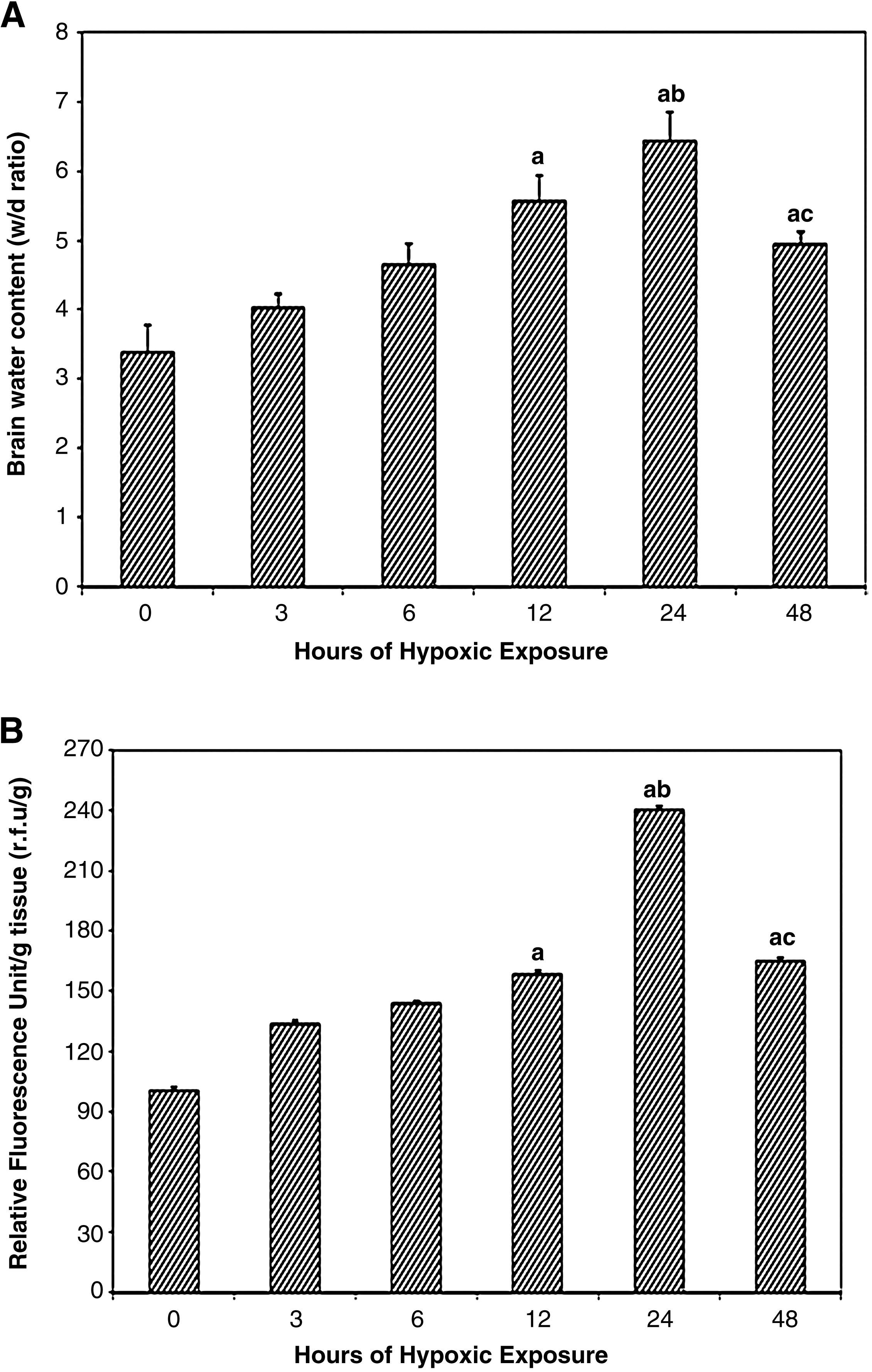
Determination of cerebral edema in rats exposed to different hours of hypobaric hypoxia:
Vascular permeability
Similarly, exposure to hypoxia showed a significant increase in transvascular leakage relative to control animals. There was a gradual increase in brain transvascular leakage up to 24-h hypoxia exposure (42% increase, p < 0.001) compared with the 0-h exposure (control). The maximum leakage in brain was obtained at 24-h hypoxic exposure compared with control. Further exposure to 48 h resulted in a decline in brain transvascular leakage compared with 24-h hypoxic exposure, but was still significant compared with control (p < 0.001) (Fig. 1B).
Estimation of ROS, lipid peroxidation, and antioxidant levels
A gradual increase in ROS generation was observed in the brain of rats exposed to hypoxia at different time points, the maximum being at 24 h (p < 0.01) of hypoxic exposure when compared with control. This was followed by a slight decline in 48-h hypoxic exposure, but was still significant compared with control (p < 0.001) (Table 1). To determine whether increased ROS levels lead to increased membrane lipid peroxidation, we determined the MDA levels in the brain tissue of rats exposed to hypoxia for different durations. The results revealed that, as the time of exposure to hypoxia increased, the MDA levels increased accordingly; that is, a gradual and significant increase (p < 0.001) in brain MDA levels was observed from 6-h hypoxia exposure up to 48-h hypoxic exposure compared with control (Table 1).
Values are mean ± SD (n = 12). Significant differences between groups were determined by one way analysis of variance followed by Student-Newman-Keuls test.
p < 0.001 compared with normoxia (0h); bp < 0.05 compared with hypoxia groups (3, 6 and 12 h); cp < 0.01 compared with all other hypoxia groups (3, 6, 24 and 48 h); dp < 0.01 compared with all other hypoxia groups (3, 6, 12 and 48 h).
Because the maximum increase in cerebral transvascular leakage was observed during 24-h hypoxic exposure, other biochemical parameters (GSH, GPx, and SOD) were determined by exposing the animals for 24 h only.
Antioxidant levels
A nonsignificant reduction in brain GSH levels was observed in the hypoxia-exposed animals compared with control. The brain SOD and GPx levels were reduced significantly (p < 0.05) in the hypoxia-exposed rats compared with control (Table 2).
Values are mean ± SD (n = 12).
p < 0.05 compared with normoxia (0 h).
Brain NFκB p65 protein expression
To determine the role of NFκB in cerebral transvascular leakage, a key transcriptional factor that regulates inflammatory mediators, we determined the relative levels of NFκB by western blotting. Animals exposed to different durations of hypoxia showed a gradual increase in brain NFκB levels from the 3-h exposure up to 12 h. Further exposure to hypoxia resulted in to slight decline in the brain NFκB levels under 24- and 48-h hypoxic exposure, but maintained a significant upregulation of NFκB expression (p < 0.001) compared with control (Fig 2). There were nearly 33-fold and 32-fold increases in the brain NFκB levels, respectively, in the 12- and 24-h hypoxia-exposed rats compared with control (Fig. 2).
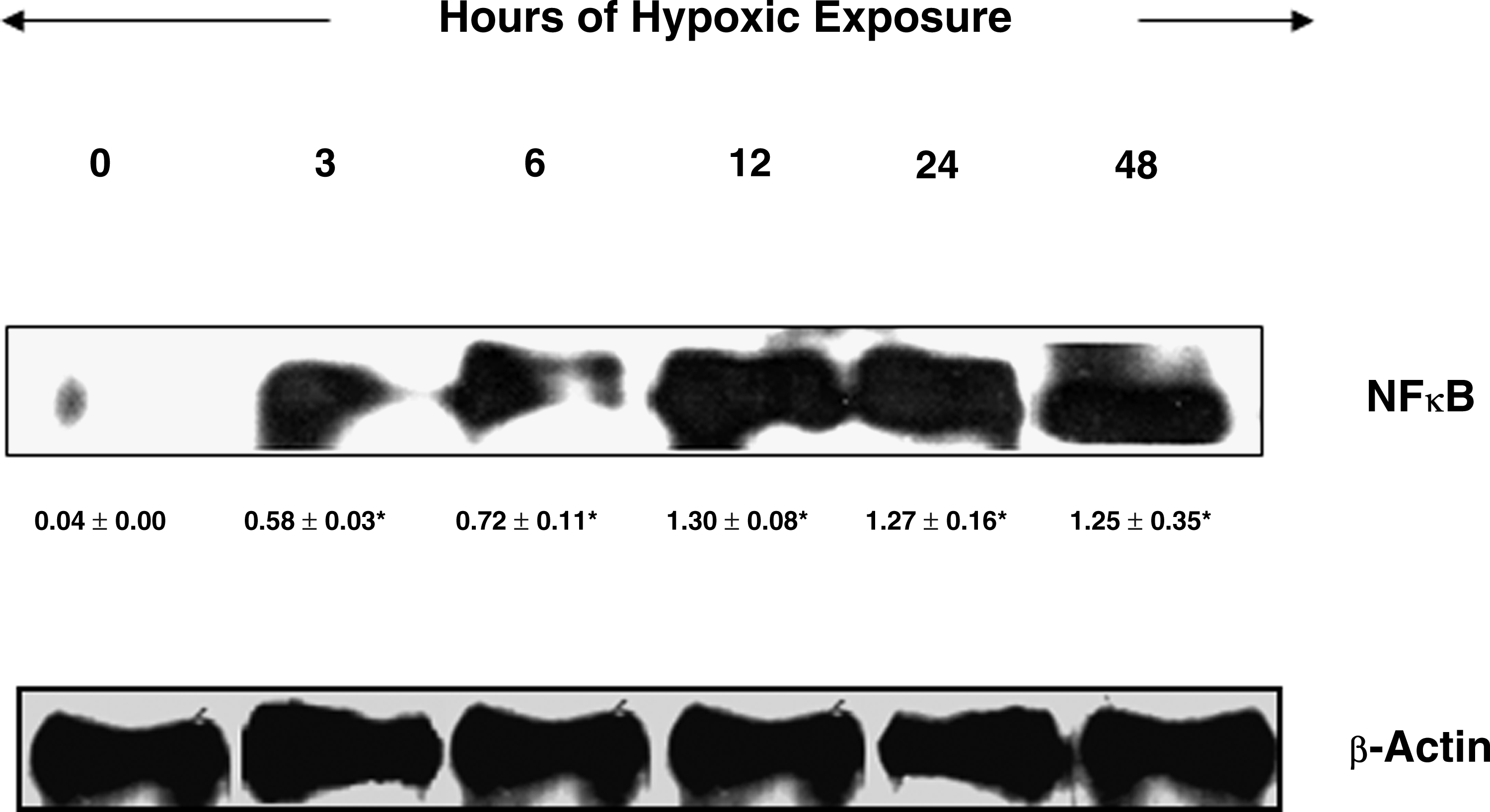
Expression of NF
Brain NFκB–DNA binding assay
To further confirm whether increased upregulation of NFκB also resulted in increased DNA binding activity of the p65 domain of NFκB, an enzyme mobility shift assay (EMSA) was performed in the brain nuclear extract of hypoxia-exposed animals at different time periods. There was an appreciable increase in NFκB–DNA binding activity in the brain homogenate of rats exposed to different hours of hypoxic exposure compared with control. However, a nearly twofold increase was observed under 12-, 24-, and 48-h hypoxic exposure (Fig. 3). The results therefore revealed that, as the hypoxia exposure time increased, the DNA binding activity also progressively increased.
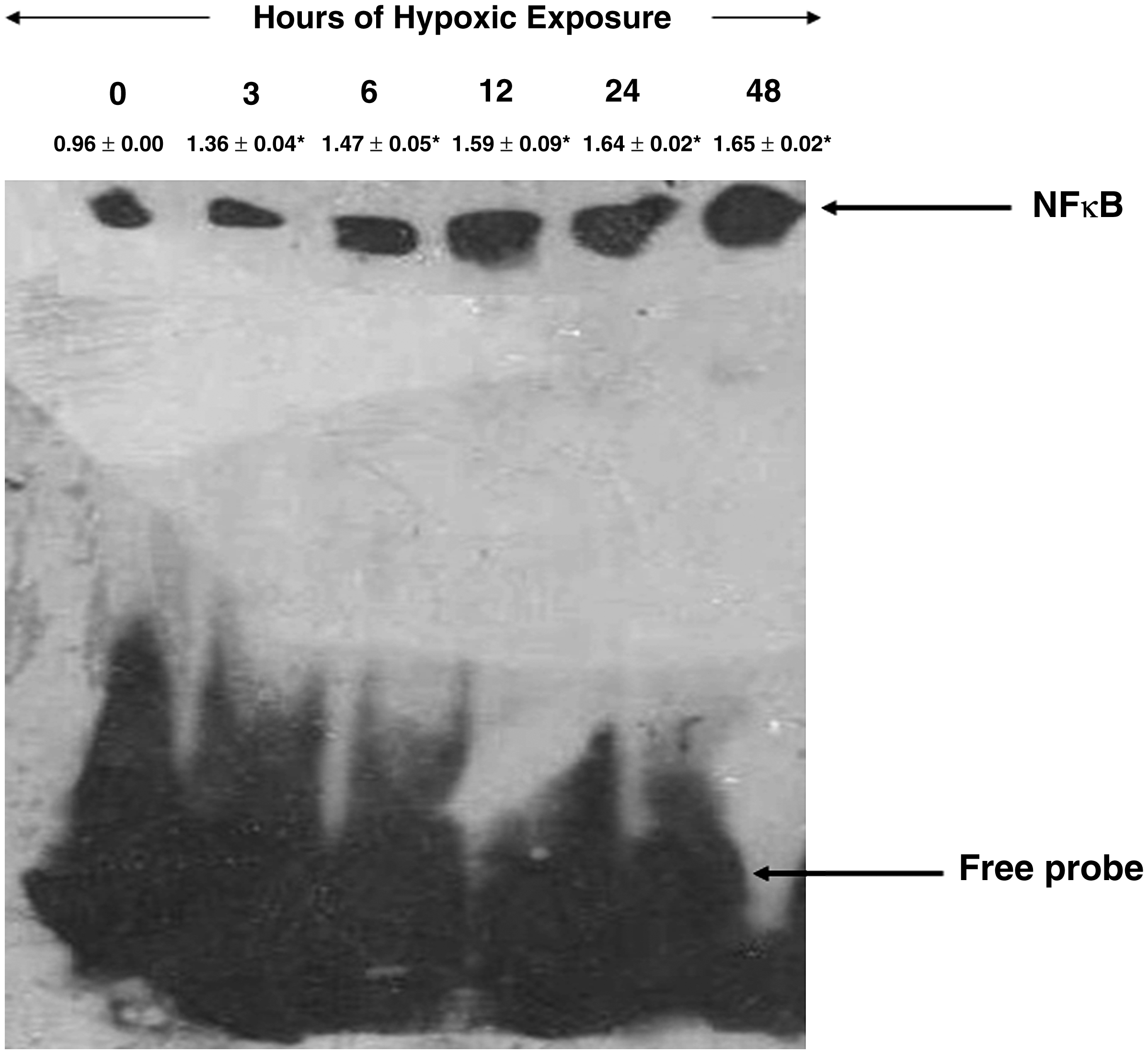
Expression of NFκB-DNA binding activity in the brains of rats exposed to hypoxia (7620 m, 25 ± 1 °C) at different time points. Nuclear extracts were prepared and analyzed for NFκB binding to DNA by EMSA. The arrow indicates position of NFκB and free probe. The densitometry analyses were represented above their respective NFκB expression levels. Hypoxic exposure resulted in a nearly 2-fold increase in NFκB-DNA binding levels in 12-, 24- and 48-h exposure when compared with control (0 h). Values are mean ± SD (n = 12). *p < 0.05 compared with normoxia (0 h).
Expression of proinflammatory cytokines: IL-1, IL-6, and TNF-α; cell adhesion molecules: ICAM-1, VCAM-1; selectins: E-selectin and P-selectin
The proinflammatory cytokines, for example, brain IL-1, expression was upregulated from 3-h hypoxic exposure onwards. However, further increase in hypoxic-exposure duration (6, 12, and 24 h) also showed an increase in the IL-1 levels in brain of rats compared with control (Fig. 4A). A virtually negligible TNF-α expression was observed in control, whereas a gradual increase in TNF-α levels was observed in brain of rats under different durations of hypoxic exposure (Fig. 4B). A similar pattern was also observed in IL-6 expression in brain of rats from 3-h up to 48-h hypoxic exposure (Fig. 4C).

Expression of proinflammatory cytokines (
ICAM-1 expression was virtually not detectable in brain of the control rats and also in 3- and 6-h hypoxia-exposed rats; however, a significant increase in ICAM-1 levels was observed from 12-h hypoxic exposure onward, with a distinct peak level at 24-h hypoxia exposure (Fig. 5A) compared with control, whereas the VCAM-1 levels in brain of rats increased time dependently with increase in time of hypoxic exposure. A nearly ninefold increase in VCAM-1 levels was observed in brain of rats exposed to hypoxia for 24-h duration compared with control (Fig. 5B).
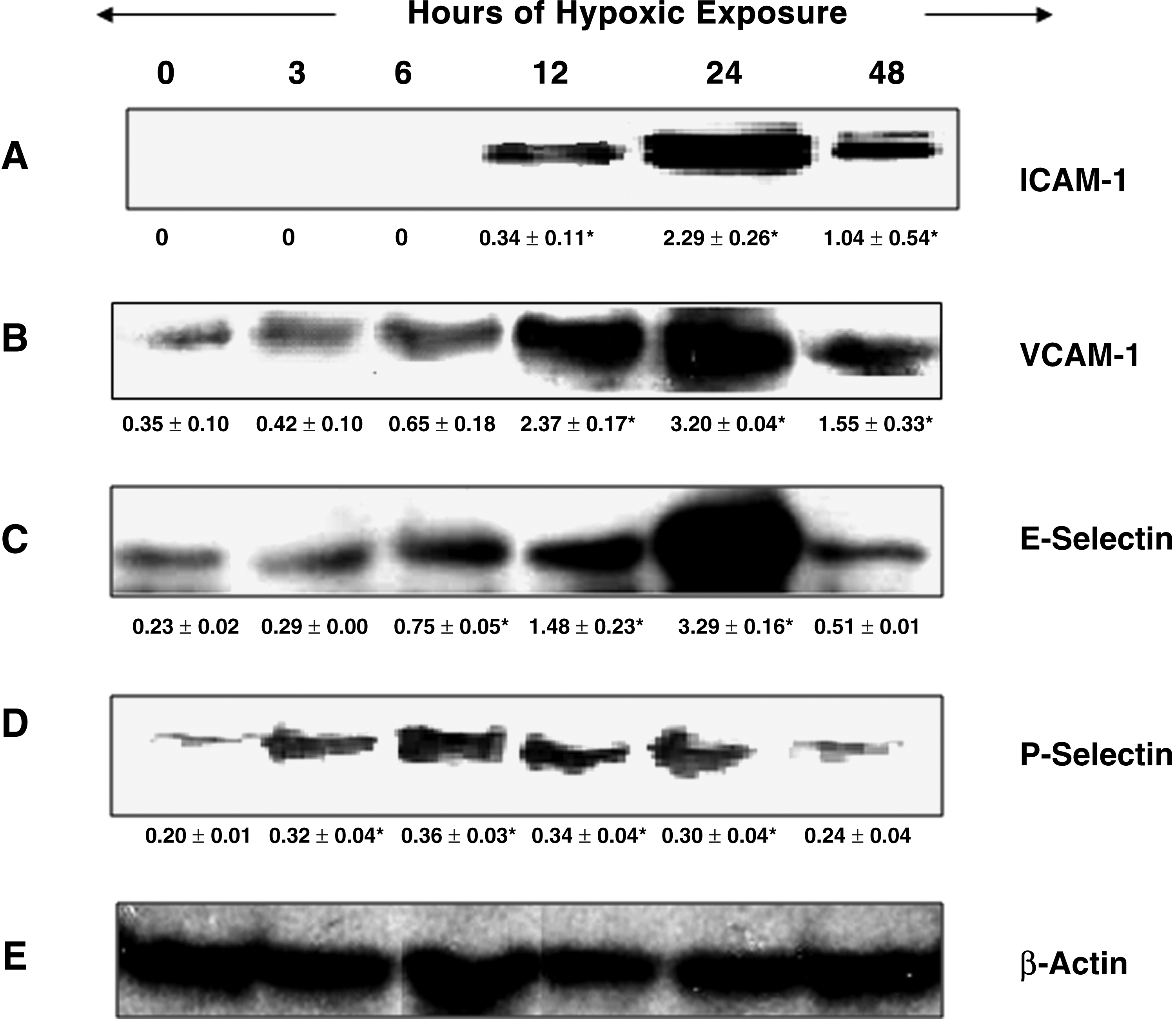
Expression of cell adhesion molecules
Brain E-selectin levels started increasing gradually with time of hypoxic exposure. A significant peak upregulation of E-selectin levels in the brain homogenates was seen in the 24-h hypoxic exposure (14-fold) compared with control (Fig. 5C), whereas, in the case of P-selectin levels in the rat brain homogenate, 6-h exposure exhibited a significant peak level (nearly twofold increase) compared with control. More or less similar significant upregulation was observed in brain P-selectin at 12- and 24-h hypoxic exposure compared with control. Further exposure (48 h) resulted in a nonsignificant upregulation of brain P-selectin levels compared with control (Fig. 5D).
NFκB blockade by curcumin
NFkB protein expression blockade studies were carried out to determine whether blocking the NFκB protein expression leads to reduced fluid accumulation in brain of rats exposed to hypoxia. The results revealed that rats administered curcumin (100 mg/kg BW) and exposed to hypobaric hypoxia for 24 h showed significant reduction in brain water content (63%) compared with control (24-h hypoxia) (Fig. 6A). Similarly, the brain transvascular leakage was also reduced to nearly 50% compared with the 24-h hypoxia-exposed rats (Fig. 6B).
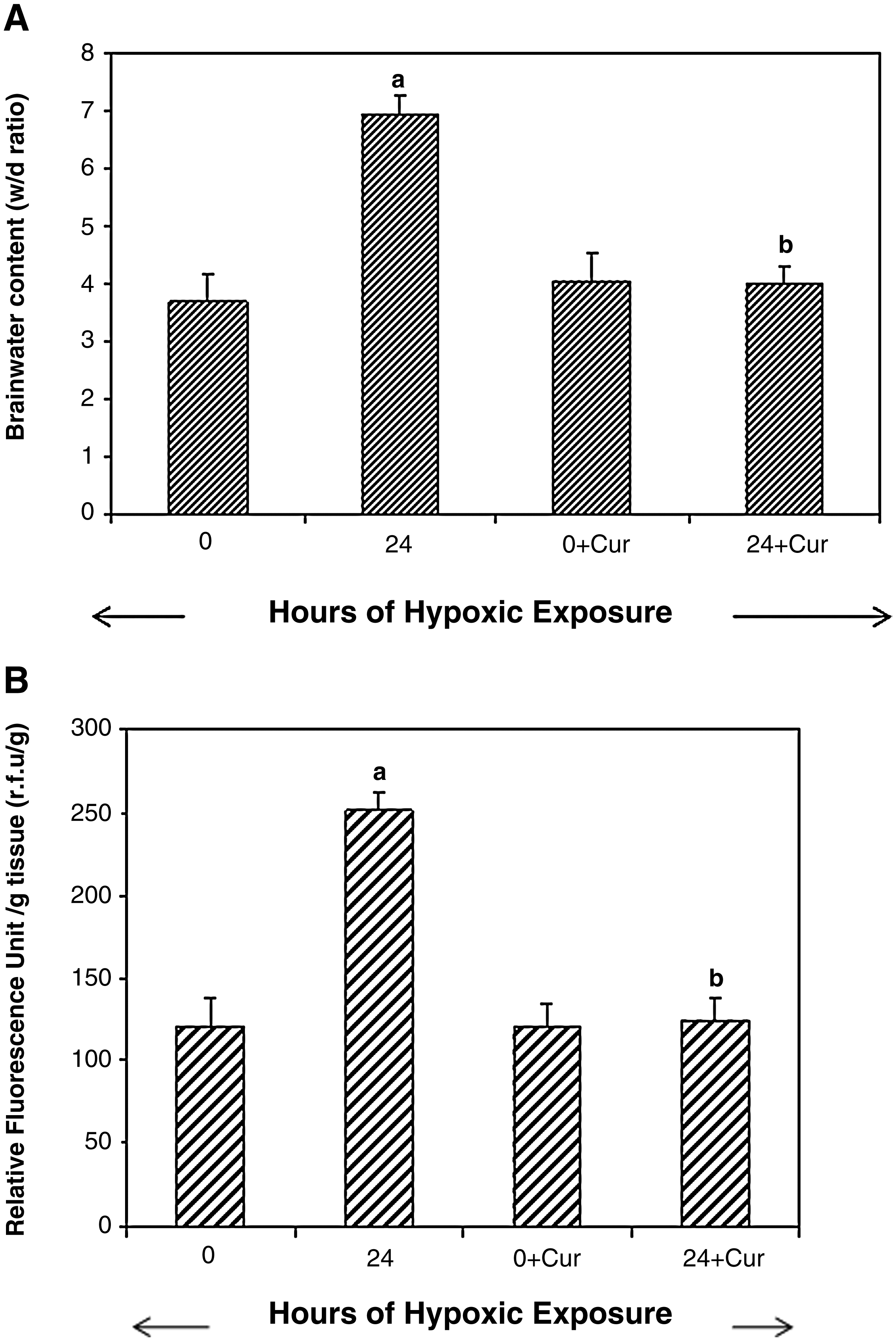
Determination of cerebral edema in rats exposed to 24-h hypobaric hypoxia using curcumin:
To further confirm our results showing that curcumin acts as both an antioxidant and anti-inflammatory agent, biochemical estimation (ROS, MDA, GSH, SOD, and GPx) and also western blotting were carried out using specific antibodies to the p65 domain against NFκB protein in the nuclear extract from brain of rats exposed to hypoxia. The results demonstrated that curcumin administration significantly reduced the levels of ROS and MDA (p < 0.001) in brain of rats exposed to hypoxia compared with control (24-h hypoxia). A nonsignificant increase in brain GSH levels was observed in the curcumin- administered, hypoxia-exposed animals compared with control. The brain SOD and GPx levels increased significantly (p < 0.001) in the curcumin-administered, hypoxia-exposed animals compared with control (Table 3). A further NFκB blocking study revealed that curcumin administration significantly (p < 0.001) downregulated the NFκB levels (40-fold) compared with control (24-h hypoxic exposure) (Fig. 7). To ensure that equal concentrations of protein had been loaded, β-actin protein expression was determined in the brain homogenates by western blotting.
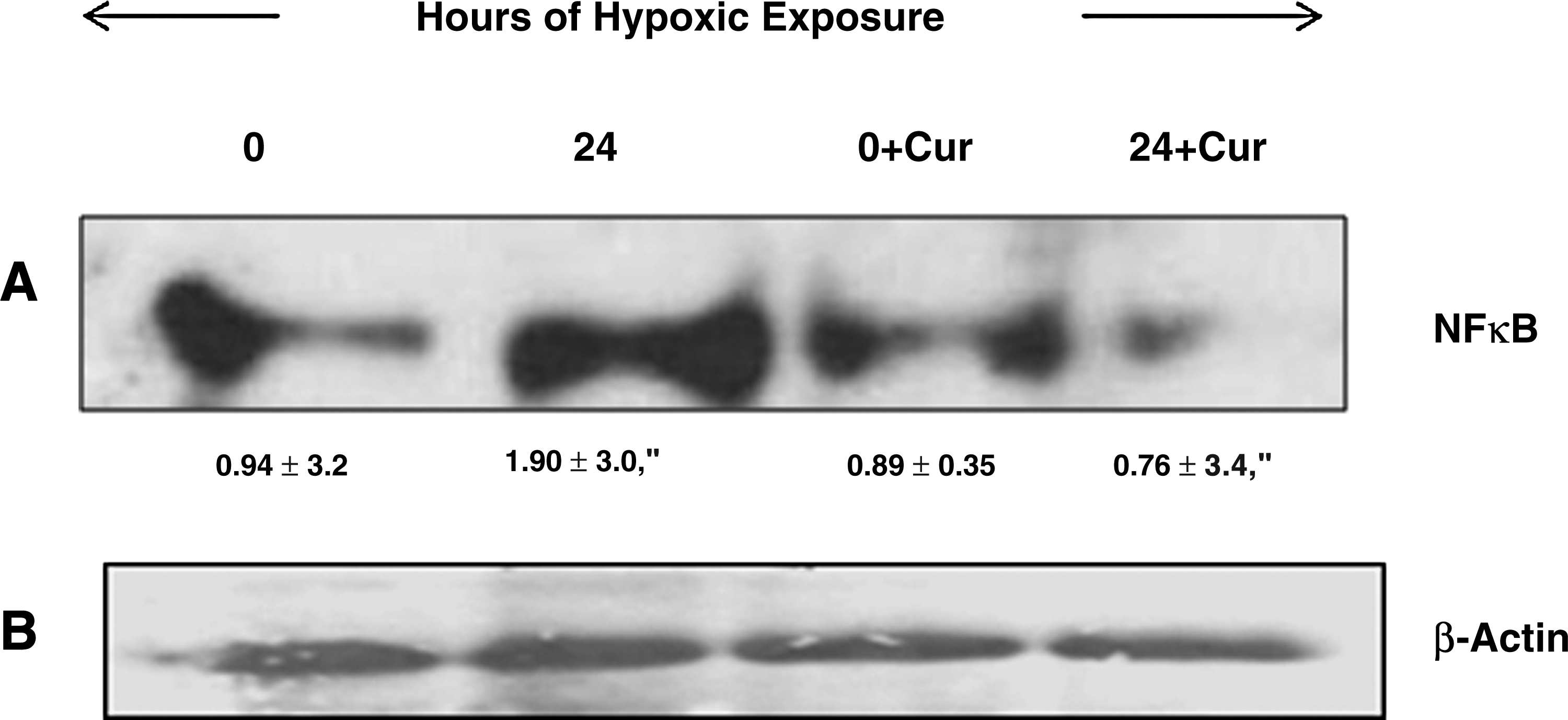
Effect of curcumin on NFκB protein expression in the brain nuclear fraction of rats exposed to hypoxia (7620 m, 25 ± 1°C) for 24 h. The figure represents western blot analysis, followed by its densitometric analysis. Curcumin (100 mg/kg BW) was administered orally to rats 1 h prior to hypoxic exposure (24 h). A nearly 40-fold reduction in NFκB levels in curcumin-administered rat brain was observed compared to control. Values are mean ± SD (n = 6): a, normoxia (0 h) vs. hypoxia (24 h), p < 0.001; b, hypoxia vs. hypoxia + Cur (24 h); p < 0.001; Cur, curcumin.
Curcumin (100 mg/kg BW) was administered orally to rats 1 h prior to exposure (24 h). Values are mean ± SD (n = 6). Significant differences between groups were determined by ANOVA followed by Student-Newman-Keuls test.
normoxia vs. hypoxia, p < 0.001; bhypoxia vs. hypoxia+cur (24 h), p < 0.001.
Cur = curcumin.
Discussion
The present study was undertaken to determine whether NFκB activation, proinflammatory cytokines, and cell adhesion molecules during exposure to hypobaric hypoxia appear before or after the cerebral edema. This study was performed on rats because the physiological and molecular responses of rats appear to be very similar to those of humans adapting to high altitude (Dempsey and Forster, 1982). The redox sensitive transcription factor, NFκB, is known to play a significant role in the pathophysiological consequences of hypoxia. We therefore studied the role of ROS and inflammation (NFκB) in the hypoxia-induced cerebral edema formation. The generation of cerebral transvascular leakage was performed using a fluorescent probe, sodium fluorescein dye that can be measured easily in the brain to quantify brain edema formation. Rats exposed to different durations of hypoxic exposure (0, 3, 6, 12, 24, and 48 h) resulted in a significant increase in transvascular leakage with increase in time of hypoxic exposure, with a peak level at 24-h exposure. This was followed by an increase in ROS production and lipid peroxidation in the brain of hypoxia-exposed animals compared with control. On the other hand, there was a decrease in brain antioxidant enzyme levels (GPx and SOD). Further, we observed a significant increase in brain NFκB levels and proinflammatory genes regulated by it (IL-1, IL-6, TNF-α, VCAM-1, ICAM-1, P-selectin and E-selectin) in brain of rats exposed to different durations of hypoxia. Interestingly, it was observed that curcumin administration 1 h prior to hypoxic exposure (24 h) significantly (p < 0.001) reduced the transvascular leakage in brain of rats by attenuating the upregulation of NFκB compared with hypoxia-exposed rats.
In the present study, the maximum brain water content was obtained in rats exposed to 24-h hypoxia duration compared with control. Further exposure to hypoxia resulted in a marginal decrease in its edema index. This could be attributed to considerable water loss caused by severe hyperventilation during exposure to a prolonged period of hypoxia. To confirm further, we measured the transvascular leakage by a direct method using a fluorescent probe, sodium fluorescein. We found that fluid accumulation, as revealed by an increase in fluorescein content in brain of rats, began at 12-h hypoxic exposure (p < 0.001) and reached a peak at 24-h exposure.
It is well known that, paradoxically, cells under hypoxia increase their production of ROS, resulting in increased oxidative stress (Kumar et al., 1999; Sarada et al., 2002; Sarada et al., 2008; Ruchko et al., 2009). Although the availability of oxygen to the body is reduced during hypoxic exposure, ROS are overproduced in hypoxia. For this, several reports have provided much evidence that has been incriminated in hypoxia-induced ROS generation, which include (1) the primary source of free-radical generation in the cell during hypoxia is due to a decrease in the mitochondria redox potential causing ROS production from the electron transportation chain (ETC) mainly at the level of cytochrome III (Guzy and Schumacker, 2006; Pialoux et al., 2009), and (2) the increase of oxygen radical release through the activation of xanthine oxidase (Sohn et al., 2003) at phospholipase A2 (Neidlinger et al., 2005) and at Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Jones et al., 2000). Therefore, it is speculated that under hypoxic conditions not enough oxygen molecules are available to accept the electrons from oxidative phosphorylation, resulting in electron accumulation in the mitochondria (Kanter, 1994) and ultimately leading to enhanced ROS generation. Since in our present study hypoxia resulted in increased transvascular leakage, we investigated the role of oxidative stress, that is, ROS, in the development of HACE. To determine the exact time for induction of oxidative stress, animals were exposed to different hours of hypoxia. It was found that a gradual increase in ROS generation from 3-h hypoxia exposure onward occurred, with a significant peak level at 24 h (p < 0.001) of hypoxic exposure, as evidenced by an increase in Dichlorofuorescein diacetate (DCFH-DA) in the brain tissue. The increased ROS production led to increased brain MDA levels under hypoxia. Hypoxia is known to increase plasma and tissue MDA levels at high altitude (Kumar et al., 1999; Sarada et al., 2002; Sarada et al., 2008). During an extensive study in humans at high altitude during Operation Everest III, the levels of lipid peroxidation increased by 23% at 6000 m and by 79% at an altitude of 8848 m, indicating that the level of oxidative stress increases with increase in altitude (Joanny et al., 2001). Our results are therefore in accordance with these earlier findings.
Since exposure of animals to hypobaric hypoxia for 24 h showed a maximum ROS generation, we studied the levels of antioxidant enzymes in brain of rats exposed to hypoxia for 24 h (GSH, SOD, and GPx). There was a significant reduction in the activity of both brain SOD and GPx (p < 0.05) levels and a nonsignificant reduction in levels of GSH compared with control. Some studies have compared the activity levels of serum GPx of native highlanders (4000 m) to subjects from sea level and found that people from high altitude had lower levels of GPx activity (Imai et al., 1995). The gradual and continuous rise in the levels of brain MDA up to 48-h hypoxic exposure in the present study explains the reduction in all three (GSH, GPx, and SOD levels) antioxidant enzymes in order to cope with oxidative stress under hypoxic conditions. At 5500-m simulated altitude, the levels of liver and lung SOD and GPx decrease, which indicates that the capacity of antioxidant systems somewhat decreases on exposure to high altitude (Nakanishi et al., 1995). Several human and animal studies have collectively reported that high altitude-associated hypoxia causes oxidative damage to lipids and proteins. This damage may be owing to either increased levels of ROS production and/or decreased levels of antioxidant capacity. Because there was a continuous increase in the levels of free-radical production in the brain tissue of hypoxia-exposed rats, the body's balance between free-radical production and the antioxidant system was lost, indicating the initial involvement of oxidative stress in causing cerebral transvascular leakage. Earlier studies revealed free-radical-mediated neuronal damage, altered blood–brain barrier permeability, and inflammatory response when exposed to hypoxia (Bailey et al., 2001; Baumgartner et al, 2002; Bailey et al., 2004; Bailey, 2005).
It is well known that increased physical activity at high altitude increases the vulnerability of the body to oxidative stress and can lead to oxidative damage (Bakonyi and Radak, 2004). However, Bigard et al. (2000) reported that rats exposed to moderate hypoxia showed increased spontaneous physical activity (SPA), but this SPA was reduced in rats under severe hypoxic conditions. In relation to this context, Dapp et al. (2006) reported that mice exposure to 10.5% hypoxia resulted into an initial drop in SPA. Perhaps this could partially explain the decrease in ROS production in the present study in rats between 24- and 48- h hypoxia.
The increase in oxidative stress, in turn, upregulates the redox-sensitive transcription factor NFκB, which will further transcribe a number of genes involved in causing inflammation. The nuclear extract of rat brain at different hours of hypoxic exposure in the present study resulted in a gradual and significant (p < 0.001) upregulation of NFκB levels from 3-h hypoxic exposure to 48 h, showing peak levels at 24-h hypoxic exposure compared with control (normoxia). NFκB is constitutively expressed in the central nervous system at low levels (Kaltschmidt et al., 1994), which is necessary for basic cell activity (Birbach et al., 2002), but was found to be stimulated above basal levels by stimuli associated with hypoxic stress (Hu et al., 2005; Zhang et al., 2006). Our results are therefore in consistence with these studies. The phosphorylation of the NFκB inhibitory protein IκBα at position Ser-32 and Ser-36 catalyzed by IκB kinase leads to activation of NFkB (Traenckner et al., 1995). Translocation of NFκB into the nucleus regulates transcription of the genes involved in inflammatory responses. The DNA binding studies in the present study have shown enhanced NFκB binding (nearly twofold) activity in brain of rats exposed to hypoxia for 24 h compared with control. Moreover, it was proved earlier, as well as our recently published report indicates, that hypoxia activates NFκB–DNA binding activity (Koong et al., 1994; Sarada et al., 2008). A much lower expression of brain NFκB–DNA binding activity was observed in the control rats. However, the most important and interesting observation in the present study is that a significant and distinct increase in NFκB expression appeared much before the onset of transvascular leakage in brain of rats, indicating that NFκB certainly contributes to causing transvascular leakage in the brain of rats by upregulating the proinflammatory cytokines, as well as cell adhesion molecules. Therefore, to the best of our knowledge, we report for the first time the involvement of inflammation (NFκB) in the development of cerebral transvascular leakage.
Our earlier and recently published data reported that altered NFκB can mediate an inflammatory cytokine response and also induce gene transcription of proinflammatory and cell adhesion molecules (Hang et al., 2005; Sarada et al., 2008; Srivastava et al., 2008). We therefore, in the present study, measured inflammatory mediators regulated by NFκB in the brain homogenate of rats exposed to hypoxia. There was a significant increase in the expression of proinflammatory cytokines (IL-1, IL-6, and TNF-α) in brain of rats exposed to different hours of hypoxia compared with control. Several studies have demonstrated that upregulation of proinflammatory cytokines (IL-1, IL-6, and TNF-α) in the body in turn stimulates NFκB expression, resulting in enhanced inflammatory responses (Baeuerle and Baltimore, 1996; Grilli and Memo, 1999; Witt et al., 2005). In the present study, there was a consistent and significant (p < 0.05) increase in IL-1 protein expression from 3-h hypoxic exposure onward, and a decrease was observed after 24-h hypoxic exposure, but it was still significant compared with control. It has also been reported that early response to inflammation is owing to the inflammatory cytokine IL-1 (Szaflarski et al., 1995; Dunn et al., 2002). TNF-α is a polypeptide that influences endothelial cell function by promoting neutrophil adherence to vascular endothelium. Maurus and colleagues (2006) reported that TNF-α significantly upregulated cell surface expression of ICAM-1, VCAM-1, E-selectin, and P-selectin in the in vitro hypoxic exposure of human aortic endothelial cells. This is attributed to the fact that these inflammatory cytokines not only help in regulating diapedesis during inflammation, but also in turn activate the NFkB expression and thereby contribute to fluid leakage in brain of rats under hypoxia.
In the present study, we observed that the cell adhesion molecules (VCAM-1, E-selectin, and P-selectin) that are responsible for the adhesion and migration of leukocytes to the inflamed site were enhanced appreciably in brain of rats exposed to hypoxia compared with control, while the ICAM-1 levels were upregulated from 12-h hypoxic exposure onward and were virtually undetected in the control (0 h) and 3- and 6-h hypoxic exposures. Several studies have revealed that the positive regulatory domains required for inflammatory cytokine gene induction are present in the promoter regions of endothelial adhesion molecules (E-selectin, P-selectin, ICAM-1, and VCAM-1) (Collins et al., 1995; Schimmer et al., 2001). This indicates that the increase in cell adhesion molecules in the present experiment might contribute to the adhesion of leukocytes to the vascular wall, thereby leading to transvascular leakage in the brain. Earlier reports showed the presence of E-selectins, ICAM-1, and VCAM-1 receptors at sites of leukocyte recruitment and inflammation in the brain (Zhang et al., 2006). We therefore hypothesize that the observed increase in proinflammatory cytokines and cell adhesion molecules could be attributed to enhanced NFκB levels in the brain of hypoxia-exposed rats. This hypothesis is schematically depicted in Fig. 8.
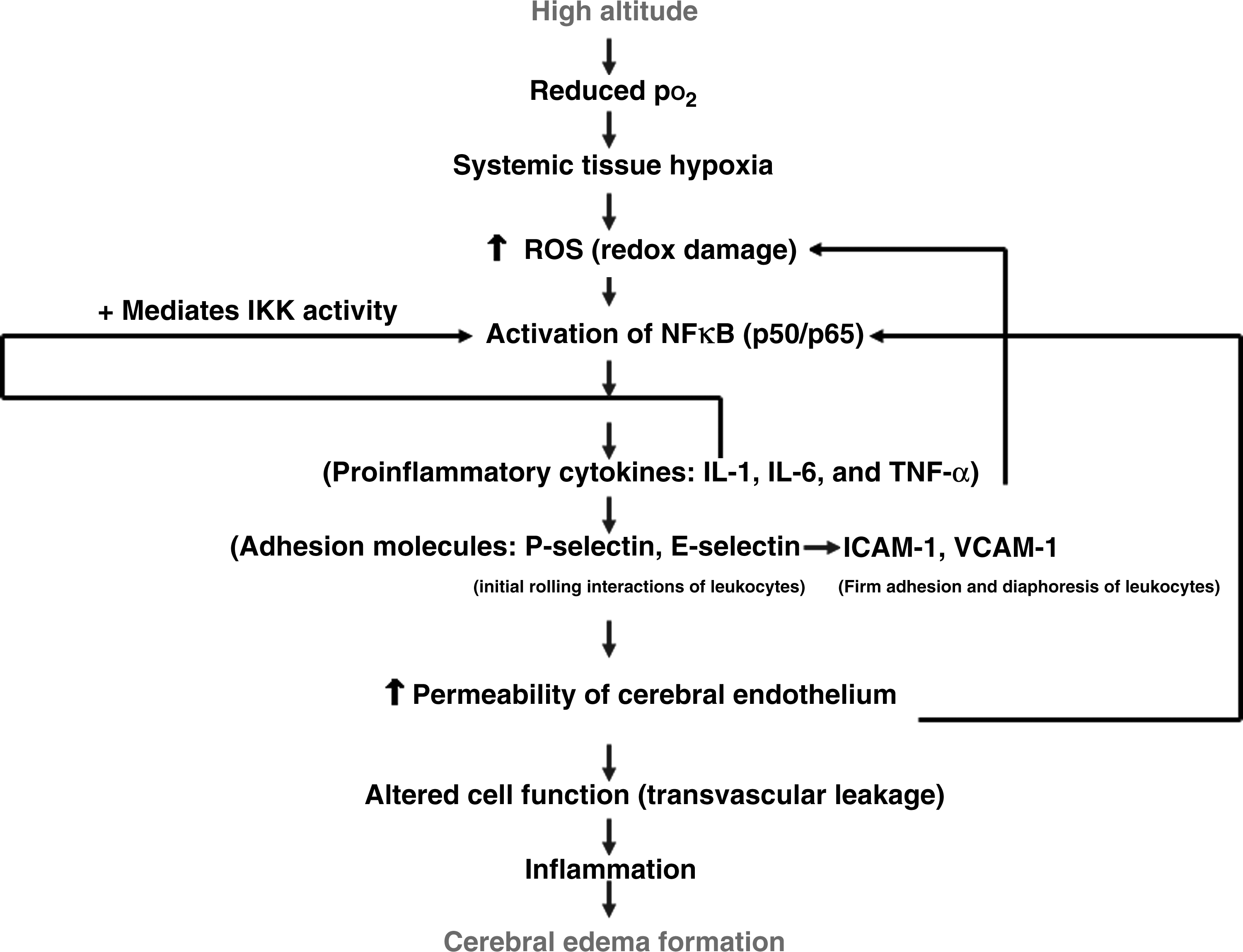
Schematic representation of the possible involvement of inflammatory components in the occurrence of cerebral edema. Increased oxidative stress under hypoxia is a well-known phenomenon. The increased oxidative stress leads to activation of NFκB. The activated NFκB further upregulates the proinflammatory cytokines (IL-1, IL-6, and TNF-α) and adhesion molecules (ICAM-I, VCAM-I, P-selectin, and E-selectin) which in turn leads to transvascular leakage in the brain of rats exposed to hypoxia. pO2, partial pressure of oxygen; ROS, reactive oxygen species; IKK–IκB kinase complex; IL-1, interleukin-1; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; ICAM-1, intracellular cell adhesion molecule; VCAM-1, vascular cell adhesion molecule; E-selectin and P-selectin, selectins in vascular endothelium.
Curcumin is the major constituent of turmeric powder extracted from the rhizomes of the plant Curcura longa L. It has the ability to downregulate NFκB activation, which has been linked to a number of inflammatory diseases (Cho et al., 2007). Moreover, turmeric has also been used for centuries as a traditional medicine to treat inflammatory disorders (Chopra et al., 1958; Nandkarni, 1976). Therefore, NFκB blockade studies were carried out using curcumin to determine whether this strategy would reduce hypoxia-induced fluid accumulation in the brain. Interestingly, we found a significant reduction in brain water content and transvascular leakage in brain of rats, followed by downregulation of brain NFκB expression compared with control. Further, curcumin significantly reduced ROS and MDA levels and maintained the brain antioxidant levels similar to that of control. A study by Jobin and colleagues (1999) reported that the curcumin inhibits IL-1-induced IKK activity. As curcumin is known to be both a natural antioxidant and an anti-inflammatory factor, the use of the natural product therefore provides an attractive and safe alternative to modulate inflammatory disorders. Further, the pharmacological safety of curcumin has been shown by the nontoxic consumption of up to 100 mg/day in humans and 5 g/day in rats (Ammon and Wahl, 1991; Commander and Vermeulen, 1996). In the present study, there was a significant increase in ROS generation and NFκB expression from 3-h exposure onward, but the vascular leakage started with a significant increase from 12-h exposure onward. On the other hand, NFκB expression started declining slightly from 12-h exposure onward, but was still highly significant compared with control. But the ROS generation and lipid peroxidation continued to increase even after 12-h exposure. The lipid peroxidation increased even after 24 h, while maximum brain water content was observed at 24-h hypoxic exposure, indicaing that increased NFκB activity was directly associated with increased cerebral edema. However, in the present study the ability of curcumin to prevent hypoxia-induced brain transvacular leakage is not linked to its antioxidant activity, as administration of vitamin C or vitamin E has shown very little effect on hypoxia-induced vascular permeability (data not shown). This also possibly explains the inability of antioxidants to prevent hypoxia-induced cerebral vascular leakage. This indicates that ROS are not solely responsible for the occurrence of cerebral transvascular leakage, although they might be partially contributing to the occurrence of vascular leakage, probably through upregulation of NFκB. Therefore, in the present study the observed reduction in fluid efflux in brain of rats might be owing to the anti-inflammatory activity of curcumin. We therefore hypothesize that exposure of animals to hypoxia results in increased oxidative stress and enhanced NFκB expression, which in turn results in upregulation of inflammatory mediators, finally leading to cerebral fluid accumulation.
Conclusion
Our study shows that hypobaric hypoxia increased brain ROS and MDA levels with concomitant reduction in antioxidant enzyme levels in brain of rats exposed to different hours of hypoxia compared with control. Further, hypoxia exposure resulted in a significant upregulation of brain NFκB levels and its target genes, the proinflammatory cytokines (IL-1, IL-6, and TNF-α) and cell adhesion molecules (ICAM-1, VCAM-1, E-selectin, and P-selectin). The present study also revealed that blocking of NFκB expression led to reduced transvascular leakage, with significant downregulation of NFκB expression levels, followed by significant downregulation of proinflammatory cytokines and cell adhesion molecules in brain of rats under hypoxia. Our data reported here provide ample evidence that inflammation certainly contributes to hypoxia-induced transvascular leakage in brain of rats. Further knockout studies using transgenic mice and microarray analysis of brain tissue will provide much more information and also reveal the exact molecular mechanism involved in hypoxia-induced cerebral edema. The present study therefore opens a new era for developing better therapeutic drugs for the prevention of HACE.
Footnotes
Disclosures
The authors have no conflicts of interest or financial ties to disclose.