
Abstract
Lentiviral vectors are promising vaccines because they can transduce and express antigens in dendritic cells in vivo, leading to potent immunization. To improve the safety and efficacy of lentivector vaccination, we sought to target vector transduction to antigen-presenting cells by modifying the viral envelope. To do this we screened a nonimmunized human single-chain antibody phage display library for phage that bound mouse bone marrow-derived dendritic cells (BMDCs) and isolated three single-chain antibodies (scFvs) that bound to more than 20% of cells in the BMDC culture. The three scFvs also bound to dendritic cells, macrophages, monocytes, and B cells from mouse spleen, but not to neutrophils, eosinophils, or T cells. Immunoblotting demonstrated that two unique scFvs, C2 and C7, recognized MHC class II. We constructed chimeric envelope proteins, by fusing these two scFvs to the amino terminus of the amphotropic murine leukemia virus envelope (MLV-A). These chimeric envelopes were expressed on the surface of lentiviral vector particles and enhanced infection (5- to 10-fold) of BMDC cultures, compared with lentiviral vectors with unmodified MLV-A envelope. Similarly, the chimeric envelopes enhanced (10- to 20-fold) the infection of primary lymph node class II-positive cells. One of the envelopes, C2, gave increased interferon-γ production from splenocytes of vaccinated mice compared with MLV-A, achieving a level similar to that obtained with vesicular stomatitis virus glycoprotein G, when used to deliver an ovalbumin model antigen gene. These results demonstrate that surface-targeting lentiviral vector transduction of antigen-presenting cells gives efficient and potentially safer immunization.
Introduction
Adenoviral vectors are potent immunogens and it has proved possible to engineer adenovirus serotype 5 vectors (Ad5) to improve DC specificity and maintain immunogenicity (Cheng et al., 2007). However, clinical use of Ad5 vectors may be problematic in individuals who have previously been exposed to Ad5, as the response to Ad5 may dominate that of the transgene (Watkins et al., 2008). A similar problem with dominance of antivector immune responses has been described for poxvirus vaccines (Smith et al., 2005). Lentivectors have also been proposed as cancer vaccine candidates because they transduce DCs in vivo and induce potent cellular and humoral immunity (Breckpot et al., 2007). As lentivectors do not encode any viral proteins the immune response is focused on the antigen transgene. Many animal studies have used lentivectors with a vesicular stomatitis virus glycoprotein G (VSV-G) envelope and ubiquitously active promoters to drive antigen expression (Esslinger et al., 2003; Palmowski et al., 2004; Kim et al., 2005; Chapatte et al., 2006; Dullaers et al., 2006; He et al., 2006; Iglesias et al., 2006, 2007; Negri et al., 2007; Garcia Casado et al., 2008). When such lentivectors are injected into the skin for vaccination they transduce keratinocytes, skin-derived DCs, and DCs and B cells in the draining lymph nodes (Lopes et al., 2008). The skin-derived DCs are particularly effective in the stimulation of T cells (He et al., 2006).
Similar to the work with Ad5, targeting lentivectors to DCs will be desirable to improve safety and efficacy. One approach is to restrict expression of the antigen to DCs. We have demonstrated that this can be achieved with the Dectin2 promoter, although the level of antigen expression was lower than with a ubiquitously active promoter and a higher vaccine dose was necessary (Lopes et al., 2008). Perhaps a more attractive alternative is to target viral uptake to DCs by modifying or exchanging the viral envelope protein. One report describes efficient immunization with a lentivector carrying a modified Sindbis virus envelope with enhanced tropism for dendritic cells (Yang et al., 2008). Our aim was to develop an alternative approach, using single-chain antibodies (scFvs) that recognize dendritic cells to retarget lentivector vaccines. In this paper we describe the panning of a phage display library to isolate scFvs that recognize mouse DCs. Further characterization of two unique scFvs demonstrated that they recognized mouse MHC class II (MHC-II). We produced lentivectors with these scFvs fused to the N terminus of the envelope from murine leukemia virus amphotropic strain (MLV-A). These lentivectors with chimeric envelopes infected MHC-II-positive cells more efficiently than did those with an MLV-A envelope. One of the chimeric envelopes proved efficient in immunization, generating a T cell response equivalent to that following immunization with a lentivector carrying the VSV-G envelope.
Materials and Methods
Cell culture
293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) plus 10% fetal calf serum (FCS; PAA Laboratories, Pasching, Austria), at 37°C, 10% CO2. NIH 3T3 cells were cultured in RPMI 1640 medium (Invitrogen) plus 10% FCS at 37°C and 5% CO2. Murine DCs were obtained from the bone marrow of C57BL/6 mice by culturing cells in Iscove's modified Dulbecco's medium (IMDM) plus 10% FCS and recombinant mouse granulocyte-macrophage colony-stimulating factor (GM-CSF, 40 ng/ml; PeproTech, Rocky Hill, NJ) as previously described (Lopes et al., 2008).
Phage panning
A 15 billion-member nonimmune human scFv phage display library, constructed from B cells of 57 donors (supplied by A.W. Marasco, Dana-Farber Cancer Institute and Harvard Medical School, Boston, MA), was used as the source of human scFvs. Freshly isolated mouse peritoneal macrophages from C57BL/6 mice (2 × 106 cells) were first incubated with 5 × 1011 plaque-forming units. Nonabsorbed phages were removed by washing with cold phosphate-buffered saline (PBS) and were used to infect Escherichia coli TG1 at log phase for amplification and rescued for the next round of selection. Murine bone marrow-derived DCs cultured for 5 days (2 × 106 per sample) were then incubated for 1 hr at 4°C with phage-scFvs (5 × 1011 plaque-forming units). Nonspecifically absorbed phages were removed by intensive washing; specifically bound phages were eluted with 0.1 M glycine-HCl (pH 2.5) and neutralized with Tris base.
This DC panning was repeated four times. For preparing single-phage clones, individual colonies from the final panning cycle were grown in SB medium containing ampicillin (100 μg/ml) in 96-well plates at 37°C. M13KO7 helper phage (New England BioLabs, Ipswich, MA) was added to the cultures and shaken for another 2 hr. Kanamycin was added to a final concentration of 70 μg/ml. Individual phage clones were rescued and tested for their binding ability.
Sequence analysis
The scFv samples were purified and analyzed by DNA sequencing using the forward primer 5′-CATAATGAAATACCTATT G CCTA-3′ and the reverse primer 5′-CTTATTAGCGTTTGCCAT T-3′. The family assignments were obtained with the program DNAPlot.
Expression and purification of scFvs
The VH and VL gene fragments of selected scFvs were isolated from the pFarber phage display vectors by NcoI and NotI digestion and inserted into pSyn1 vectors cleaved with the same restriction enzymes. The soluble fragments were expressed in E. coli XL1-Blue (Stratagene, La Jolla, CA) and purified from the periplasmic fractions. All scFvs contained a hexahistidine (His6) tag that allows purification by immobilized metal affinity chromatography, and a Myc tag.
Binding activity of scFvs
3T3 cells, murine bone marrow (BM)-derived DCs, and C57BL/6 splenocytes (106 per sample) were harvested and incubated with bacterial culture supernatants containing phage particles or scFv soluble fragments for 1 hr at 4°C on a rocking platform. After intensive washing bound phages were stained with anti-M13 biotin-conjugated antibody (Serotec, Oxford, UK) and detected with streptavidin–fluorescein isothiocyanate (FITC) (Dako, Carpinteria, CA). When soluble fragments were analyzed, bound scFvs were detected with biotin-conjugated His probe (Santa Cruz Biotechnology, Santa Cruz, CA) and streptavidin–FITC (Dako). Cells were costained with phycoerythrin (PE)-conjugated anti-mouse CD11c, PE-conjugated anti-mouse CD19, allophycocyanin (APC)-conjugated anti-mouse CD3, or APC-conjugated anti-mouse MHC class II (eBioscience, San Diego, CA). Samples were analyzed with a FACSCalibur (BD Biosystems, San Jose, CA).
Lentiviral vectors
The plasmid encoding TPO-SU (thrombopoietin [TPO] C-terminal truncation mutant TPO171, fused to the amphotropic MLV envelope at position +1 of the surface [SU] subunit) was described by Verhoeyen and colleagues (2005). We replaced the TPO fragment with C2 or C7 scFv, respectively, using the SfiI–NotI site. Virus was produced by cotransfection of chimeric envelope plasmids with pCMVRΔ8.91 and lentiviral vector plasmids expressing green fluorescent protein (GFP) (pHRSIN) or a dual-promoter plasmid encoding GFP and ovalbumin (OVA) epitopes plus MHC class II-associated invariant chain (IiOVA) (Rowe et al., 2006). In this vector GFP expression is driven by the spleen focus-forming virus (SFFV) promoter and IiOVA expression by the ubiquitin promoter. Virions were collected from the supernatant of producer cells 48 and 72 hr after transfection and pelleted at 4000 rpm overnight. Vectors were suspended in PBS and stored at −80°C. Titers were determined by measuring RT activity (reverse transcriptase assay; Roche Applied Science, Penzberg, Germany) according to the manufacturer's instructions and expressed as nanograms of RT activity per milliliter. Lentiviruses (LVs) pseudotyped with Ampho-SU were also titered by infecting 293T cells and measuring GFP expression by flow cytometry (fluorescence-activated cell sorting [FACS]).
Lentiviral transduction
For lentiviral transduction 293T and NIH 3T3 cells were plated at 105 per well in 24-well plates and infected with LVs overnight. GFP expression was detected by FACS analysis 3 days after infection. Mouse bone marrow-derived DCs were harvested on day 5 of culture, replated at 5 × 105 per well in 24-well plates and infected with 200 ng of RT activity of lentivirus overnight. Six days after infection, DCs were collected and GFP expression was analyzed by FACS. Total lymph node cells were isolated, plated at 106 per well, and infected for 6 hr with LV (expressed as nanograms of RT activity, as indicated in Fig. 3). Three days after infection cells were stained with anti-MHC class II–APC (BD Biosciences) and transduction in the MHC class II population was quantified by detecting GFP expression by FACS.
Immunoblotting
Five microliters of concentrated virus was loaded in a denaturing sodium dodecyl sulfate (SDS)–10% polyacrylamide gel; chimeric envelopes and p24 were detected with goat anti-RLV serum (obtained from the National Cancer Institute, Bethesda, MD) and anti-HIV p24 (ADP365), respectively, and revealed with horseradish peroxidase (HRP)-conjugated antibodies (BD Biosciences). For immunoprecipitation, 5 × 108 splenocytes from C57BL/6 mice were lysed in 0.5 ml of radioimmunoprecipitation assay (RIPA) buffer, and then incubated with 1 μg of IBL-5/22 (Santa Cruz Biotechnology) and protein G–Sepharose (Sigma-Aldrich, St. Louis, MO). Lysate (L, 50 μl), supernatant (S, 50 μl), and pelleted proteins (P, 5 μl of 50 μl) were separated on an SDS–12.5% polyacrylamide gel, and then transferred to nitrocellulose. Membranes were probed with purified scFvs (approximately 1 μg/ml) detected with biotin-conjugated His probe (Santa Cruz Biotechnology) and streptavidin–HRP (BD Biosciences).
Immunization
Adult C57BL/6 mice were injected subcutaneously (footpad and base of tail) with the indicated amounts of lentiviral vectors encoding GFP and OVA epitopes and pseudotyped with wild-type Ampho, C2-SU, and C7-SU; LV pseudotyped with VSV-G was injected as positive control, and PBS as negative control. Vector carrying C2-SU and encoding GFP only was injected as negative control for the scFvs.
Enzyme-linked immunospot assay
Twelve days after immunizations mice were killed and spleens were harvested. Spleens from each group (two mice) were pooled and used for ex vivo enzyme-linked immunospot (EliSpot). EliSpot plates (Millipore, Bedford, MA) were coated overnight with anti-IFN-γ (BD Biosciences) and serial dilutions of splenocytes in RPMI–5% FCS were added ± OVA class I peptide(257–264). After a 24-hr incubation plates were developed according to the manufacturer's instructions. Spots were counted with an EliSpot reader (AID Diagnostika, Strassberg, Germany). Each column shows the mean from three independent experiments together with the standard deviations (indicated with error bars). Statistical comparisons (pairwise analysis of variance [ANOVA]) are shown between indicated samples. Significant (*p < 0.05) and highly significant (**p < 0.01) differences are shown.
Results
Selection of phage antibodies that recognize murine bone marrow-derived DCs
To identify single-chain antibodies which target murine dendritic cells we performed panning of a human non immune phage display library using mouse BMDCs as substrate. The cell panning techniques not only provides multiple targets, it also allows the selection of scFvs against molecules in their native conformation. The panning was monitored by measuring phage titer after each round of selection; after rounds 3 and 4 cell-based ELISA was performed with single phage clones to test binding to BM DCs. After the fourth round 48 clones were also analyzed by flow cytometry (Fig. 1A and B). Ten phage clones showed binding to more than 20% of cells in BMDC cultures measured by flow cytometry, whereas they did not bind to unrelated murine cell line 3T3. Three unique scFvs (C2, C7, and A9) were identified by sequencing; they were encoded by VH1 and Vλ2 or Vλ3. A further seven phage clones, shown to be identical scFvs on sequencing (E1), bound between 10 and 20% of cells in the BMDC cultures. The remaining positive phage identified by ELISA bound less than 10% of cells; two of these were sequenced and were unique (E3 and E4) (Fig. 1C).
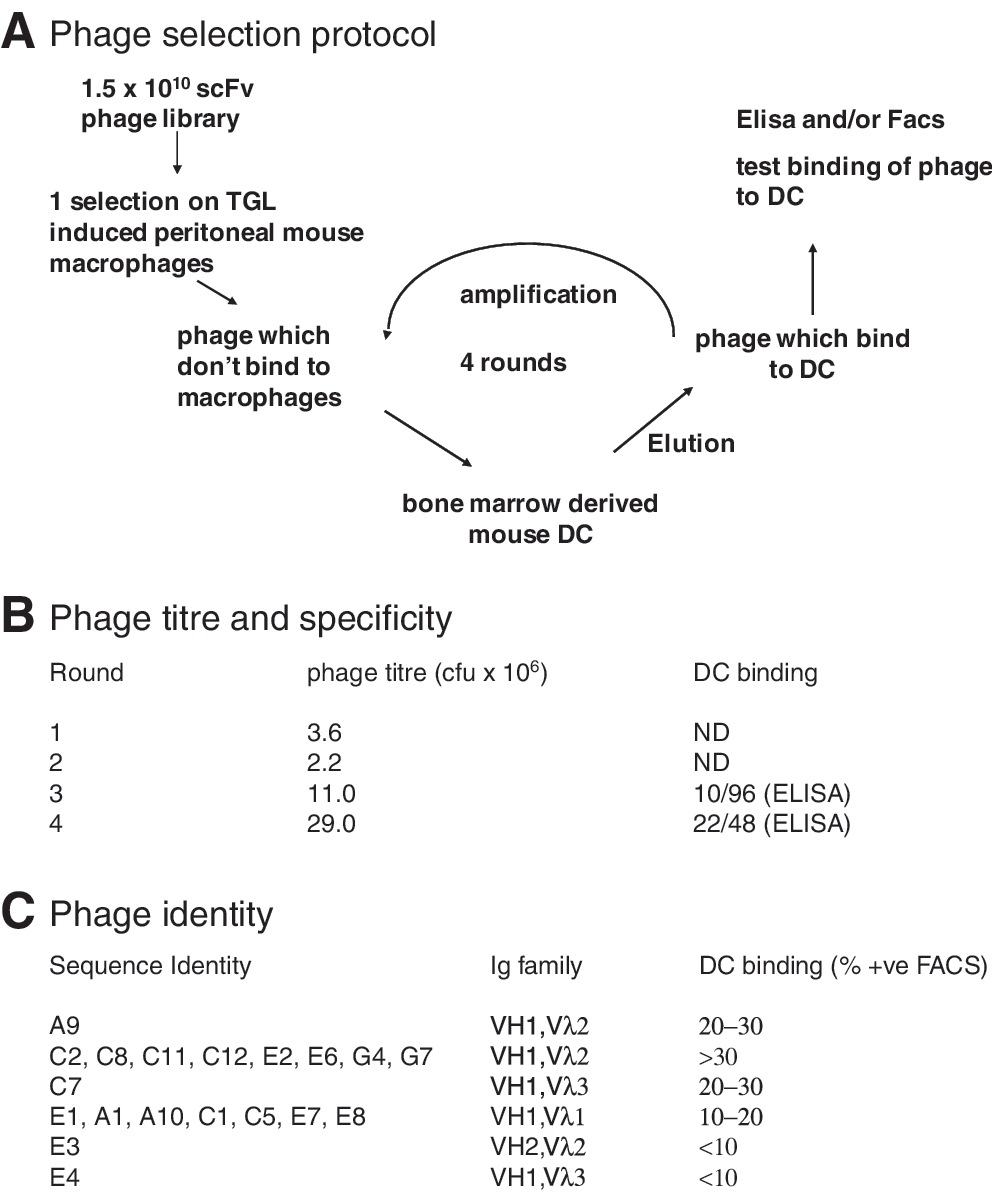
Phage selection. (
scFvs selected from the phage library bind to APCs in vitro
Five unique scFvs were expressed in E. coli tagged with His6 and purified by metal affinity chromatography. The binding activity of the monomeric scFv was similar to that of the phage clones; they stained similar percentages of murine BMDCs as the whole phage (Fig. 2A), but did not bind to 3T3 (Fig. 2B and data not shown). To further investigate the specificity of the scFvs that showed highest binding to BMDCs, we analyzed their binding to fresh spleen cells. Typical flow cytometric profiles for scFv C2 with single antibody costaining is shown in Fig. 2B. CD11c was used to identify DCs, CD19 for B cells, and CD3 for T cells. Further staining used F4/80 for macrophages, resident monocytes were identified as CD11b+GR-1intermediate, inflammatory monocytes as F4/80+CD11b+GR-1intermediate, neutrophils as CD11b+GR-1high, and eosinophils as CD11b+F4/80+. A9, C2, and C7 soluble fragments showed the same pattern of specificity, staining spleen macrophages, DCs, resident monocytes, and B cells. These scFvs did not show binding activity on neutrophils, eosinophils, or inflammatory monocytes and showed little binding to T cells (data not shown). Thus they show specificity for antigen-presenting cells that express MHC class II. To test the hypothesis that these scFvs might recognize MHC class II molecules, we immune-precipitated MHC class II from spleen cell lysate, using the rat monoclonal antibody IBL-5/22. Both C2 and C7 could detect immune-precipitated MHC class II, of a size consistent with the 34-kDa α chain, whereas A9 gave no signal on the spleen cell lysate (Fig. 2C). Thus our selection protocol isolated two different scFvs that recognize MHC class II; A9 may recognize a distinct target, or may fail to detect the same target in an immunoblot. The C2 scFv also binds to the A20 cell line, which is derived from BALB/c mice and expresses IA-d (data not shown) (Walker et al., 1982), suggesting that it has broad specificity for MHC class II α chains.

scFv-binding activity. Purified soluble single-chain antibodies (scFvs) were analyzed by flow cytometry for the ability to bind to (
C2-SU and C7-SU chimeric envelopes
To generate gene delivery systems that specifically target antigen-presenting cells (APCs) we fused the scFvs C2 and C7 to an amphotropic MLV glycoprotein. We chose this envelope for scFv incorporation as it is a well-established technology that has been used for many targeting experiments (Cosset and Russell, 1996). The scFv-coding sequences were inserted in the amino terminus of the amphotropic SU at position +1 followed by a 7-amino acid linker; the position of insertion had been previously shown to allow the functional display of peptides on the virions (Verhoeyen et al., 2005). The resulting chimeras were called C2-SU and C7-SU, respectively, and were used to pseudotype lentiviral vectors encoding either GFP (Demaison et al., 2002), or GFP with an ovalbumin antigen that expresses MHC class I and class II epitopes fused to the invariant chain (Rowe et al., 2006) (Fig. 3A).
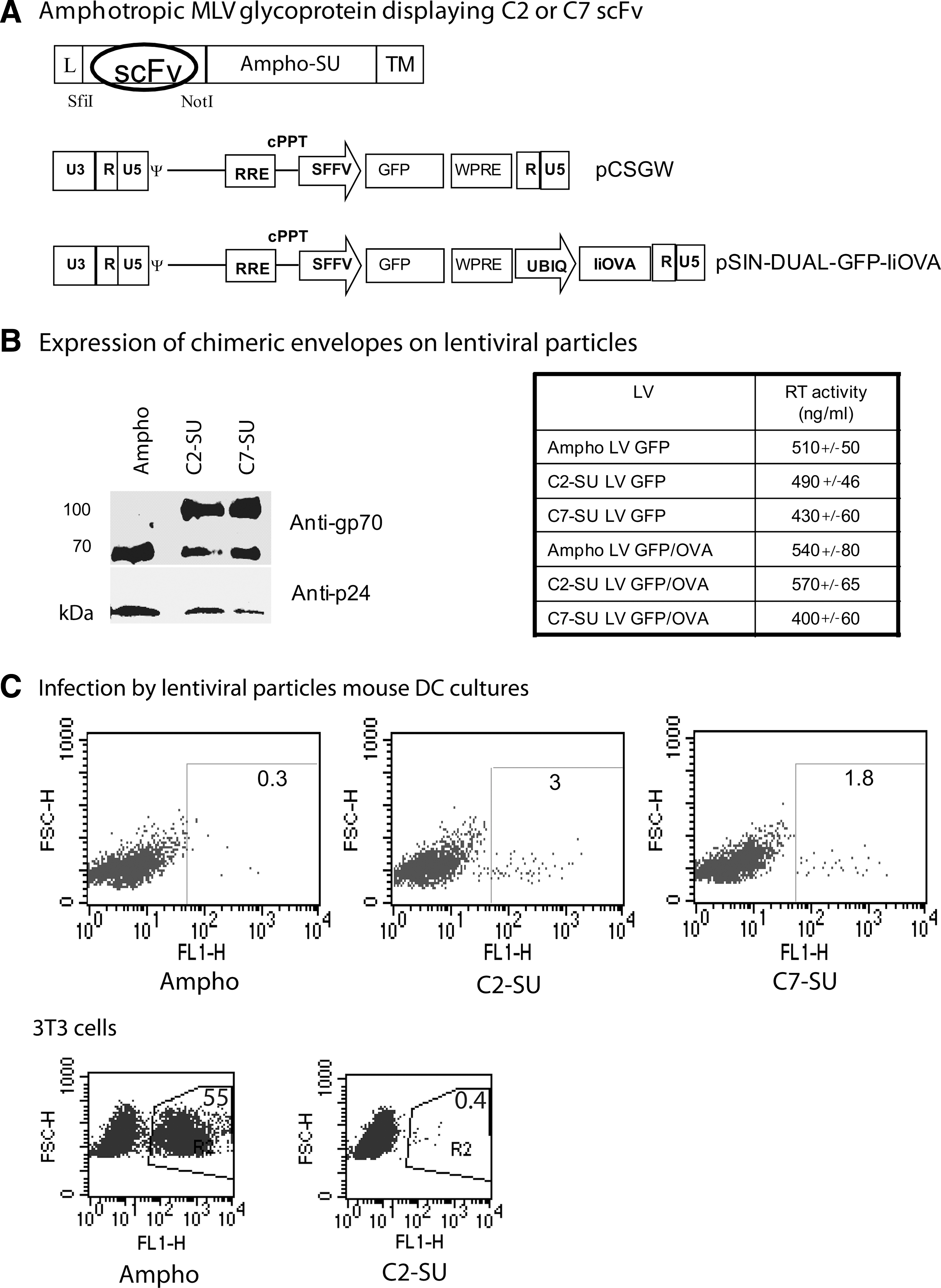
Murine leukemia virus (MLV) chimeric glycoproteins displaying scFv fragments. (
Lentiviral particles were generated by transient transfection of 293T cells with plasmids encoding HIV Gag–Pol, the envelope glycoproteins (either chimeric C2-SU or C7-SU, or wild-type amphotropic MLV) and the LV encoding either GFP (LV-GFP) or GFP and OVA epitopes (LV-GFP/OVA). Viral pellets were analyzed by immunoblotting, to detect envelope incorporation (Fig. 3B). Both chimeric envelopes were incorporated into the viral particles at similar levels to the MLV-A glycoprotein; a smaller glycoprotein of the same size as the native MLV-A was also detected in both chimeric samples, probably due to cleavage of the scFv from the chimeric envelope. No envelope was detected in the pellet when amphotropic, C2-SU, or C7-SU envelopes were transfected without the HIV gag–pol plasmid, demonstrating that the envelopes found in the pellets were associated with viral particles (data not shown). Viral titers were measured by RT activity and expressed as nanograms of RT per milliliter; the titers of all the vectors were comparable (Fig. 3B). Infection of 3T3 or 293T cells with amphotropic LV-GFP showed that 500 ng of RT per milliliter corresponded to approximately 106 IU/ml; however, neither C2-SU nor C7-SU LV-GFP was infectious on these cells, showing that N-terminal expression of these scFvs blocked infection of cells via the amphotropic receptor (Fig. 3C and data not shown). This also shows that the envelope of the same size as MLV-A detected in chimeric samples is nonfunctional, perhaps because of trimerization with the chimera.
Infectivity of chimeric enveloped virions
We then investigated whether the presence of C2 and C7 scFvs on the surface of LV particles influenced infection of BMDCs. Bone marrow-derived dendritic cells were incubated with LV-GFP (200 ng of RT) pseudotyped with wild-type amphotropic envelope, or C2-SU or C7-SU chimeric envelope, and the transduction efficiency was measured by GFP expression (Fig. 3C). As we previously reported (Strang et al., 2004), LVs pseudotyped with wild-type amphotropic envelope were poorly infectious on murine BMDCs. Note that 200 ng of RT represents a multiplicity of infection (MOI) of approximately 1 in this experiment, and MOIs greater than 10 are required for the significant transduction of these cells seen with VSV-G-pseudotyped vectors. However, LVs displaying C2 or C7 gave significant BMDC infection.
We next wished to determine whether LV-GFP pseudotyped with wild-type amphotropic envelope, or with chimeric C2-SU or C7-SU envelope, would infect antigen-presenting cells in vivo. We reported that subcutaneous injection of LV-GFP pseudotyped with VSV-G results in transduction of dendritic cells in the draining lymph node (Lopes et al., 2008). When we subcutaneously injected LV-GFP pseudotyped with wild-type amphotropic envelope, or with chimeric C2-SU or C7-SU envelope, we did not observe any GFP-positive cells in the draining lymph nodes, which we ascribe to the fact that the titers are at least 10 times lower than that of LV-GFP pseudotyped with VSV-G and only a limited volume can be injected subcutaneously (data not shown). As a surrogate assay we therefore infected freshly isolated lymph node (LN) cells with a larger amount of the LVs. We found that LVs pseudotyped with the chimeric C2-SU and C7-SU envelopes were effective in transduction of MHC-II-positive cells from lymph node, whereas LV with amphotropic envelope was unable to transduce these cells (Fig. 4A).
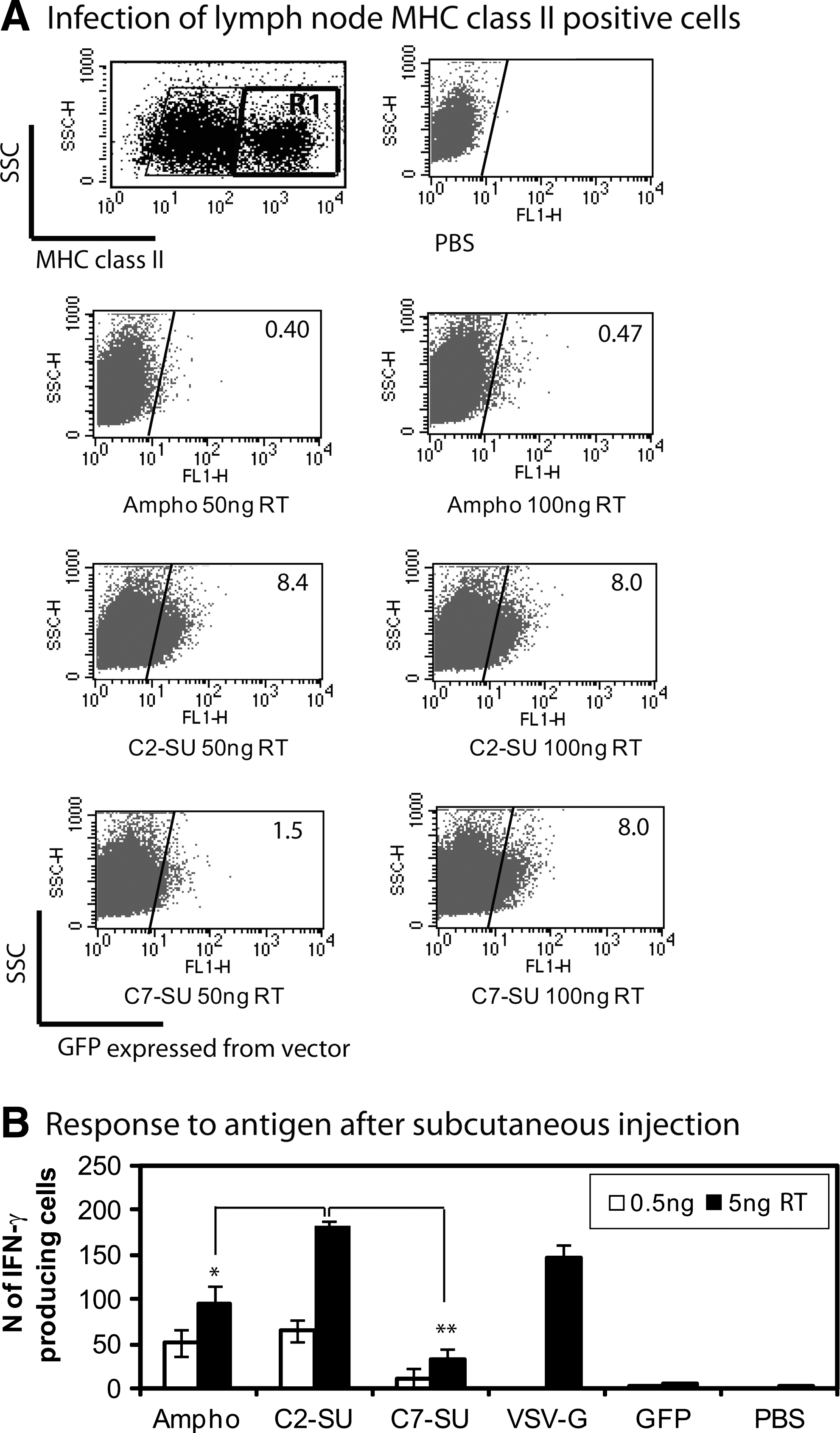
Immunization with targeted lentiviral vectors. (
In vivo injection of LVs displaying scFvs induces IFN-γ release from splenocytes
To investigate whether lentiviral vectors pseudotyped with chimeric envelopes could immunize mice when injected in vivo, we vaccinated mice subcutaneously with the indicated amounts of C2-SU, C7-SU, and amphotropic LVs encoding GFP and OVA epitopes. A group of mice was vaccinated with LVs pseudotyped with VSV-G as positive control and a group with PBS. We also used LVs carrying C2 and encoding GFP only as a negative control for the scFvs. Twelve days after injection we measured IFN-γ release by splenocytes after incubation with OVA peptide(257–264) in an ex vivo EliSpot assay.
Combined data from three separate experiments, immunizing once with two different doses of each LV, are presented in Fig. 4B. We found that LVs pseudotyped with amphotropic envelope, C2-SU, or C7-SU induced CD8+ T cell responses. C2-SU LV stimulated significantly more IFN-γ-producing cells than Ampho-LV at the higher dose whereas C7-SU LV produced significantly lower results than C2-SU LV or Ampho-LV at both doses. The response to C2-SU LV was comparable to the VSV-G LV response. There was no further increase in IFN-γ production when mice were immunized with 50 ng of any LV (data not shown). The display of an scFv on the surface of LV-GFP particles did not induce any response to OVA.
Discussion
From our scFv screen we found antibodies that bound to MHC-II. The initial intention was to remove scFv that bound to macrophages, allowing the isolation of more specific DC-binding antibodies. Our rationale was that negative selection on a highly related cell type would allow us to identify novel DC-specific targets. As MHC-II is expressed on macrophages, we must conclude that the negative selection step was ineffective, although it did reduce the phage titer. It is possible that general pattern recognition of M13 by macrophages gave a nonspecific decrease in library titer, or that we depleted the highest affinity phage that recognized shared antigens such as MHC-II. Despite this problem with negative selection, four rounds of positive selection resulted in identification of four unique phage that bound mouse bone marrow-derived DCs. The scFvs encoded by these phage bound with relatively high affinity, so that monomeric binding to intact cells could be detected by FACS analysis. Immunoblotting demonstrated that two of these scFvs recognized MHC-II α chain. We are continuing to characterize the targets of the other two scFvs; one of them shows target cell specificity similar to those that recognize MHC-II α chain. Previous approaches to generate scFvs that recognize DCs have either constructed them from the heavy and light chains of existing monoclonal antibodies, or selected phage using a recombinant DC surface protein (Demangel et al., 2005; Galibert et al., 2005; Nchinda et al., 2008). Our experience with cell panning suggests that MHC-II may be a dominant target molecule using this approach, at least with the protocol we used. MHC-II molecules are expressed by antigen-presenting cells such as all subsets of dendritic cells, macrophages, and B cells, and MHC-II expression can also be upregulated on other cells by inflammatory cytokines. Targeting to MHC-II-positive cells is a reasonable strategy for vaccination; after injection into the skin, vaccine vectors will contact resident dendritic cells and macrophages and will also traffic to the draining lymph nodes and contact dendritic cells and B cells.
The MHC-II-specific scFvs were incorporated into lentivector particles as N-terminal chimeras with the MLV-A envelope. These chimeric envelopes no longer infected cells via the amphotropic receptor, but showed relatively efficient infection of MHC-II-positive cells such as bone marrow-derived DC cultures and freshly isolated cells from lymph node. MLV-A envelope was relatively inefficient in infection of these cells. Despite recognizing the same target, the C2 chimeric envelope conferred higher infection efficiency, particularly at lower doses of lentivector. Likewise the C2 scFv also bound to a higher percentage of bone marrow-derived DCs. This could be due to a different binding site, different affinity, or a combination of these factors. Lentivectors with the C2 chimeric envelope were also efficient in immunization. In a comparison with the same number of particles with a VSV-G envelope, an equivalent T cell response to a model antigen was observed.
Targeting of enveloped viruses, using single-chain antibodies fused to murine leukemia virus envelopes, often results in relatively inefficient infection because the function of the chimeric envelope protein is compromised to some extent (e.g., Martin et al., 2003). As an alternative solution to this problem, binding-deficient but fusion-competent mutants were isolated from Sindbis and measles viruses and used to retarget binding without affecting the efficiency of fusion (Ohno et al., 1997; Nakamura et al., 2005). In these viruses the binding and fusion functions could be separated because they reside on different envelope glycoproteins. Such envelopes can also be used for retargeting lentiviral vectors (Morizono et al., 2005; Funke et al., 2008). For future scFv targeting of lentivectors to DCs it will likely be more efficient to incorporate C2 and other scFvs into one of these envelopes.
Footnotes
Acknowledgments
This work was supported by an MRC Programme Grant (R. Weiss, PI) and a Cancer Research UK Programme Grant. The authors thank Philip Taylor (Cardiff University) for analysis of scFv binding to splenocytes.
Author Disclosure Statement
No competing financial interests exist.