
Abstract
To achieve effective gene therapy, it is necessary to selectively and efficiently transfect therapeutic gene into targeted cells. In this study, we developed a combination method using mannosylated lipoplexes, which show selectivity to antigen-presenting cells such as macrophages and dendritic cells, and bubble liposomes (BLs), which are known to enhance transfection efficiency on exposure to ultrasound (US). In cultured mouse macrophages, known for the expression of mannose receptors, the transfection efficiency of this combination method using mannosylated lipoplexes and BLs with US was higher than that of naked pDNA or unmodified lipoplexes and BLs. In the liver and spleen, the in vivo transfection efficiency of this combination method was significantly higher than that of naked pDNA or unmodified lipoplexes and BLs with US. Transfection in hepatic nonparenchymal cells using this combination method was about 12 times higher than that in hepatic parenchymal cells. As far as splenic transfection activities were concerned, the transfection efficiency of this combination method in CD11c+ cells was significantly higher than that in CD11c− cells. In conclusion, we demonstrated that the gene transfection efficiency in macrophages and dendritic cells was significantly increased by this combination method using mannosylated lipoplexes and BLs with US exposure.
Introduction
The antigen-presenting cells (APCs), which are the major target cells for cancer immunotherapy or antiinflammatory therapy, express large number of mannose receptors on the cell membrane (Taylor et al., 2005). We have developed mannosylated liposomes for delivery to the APCs by mannose receptor-mediated endocytosis (Kawakami et al., 2000; Yamada et al., 2004; Wijagkanalan et al., 2008). Recently, we have demonstrated that mannosylated liposomes targeted to APCs, such as macrophages and dendritic cells, can be applied to treatment of inflammatory diseases or cancer (Hattori et al., 2006a,b; Higuchi et al., 2006; Huang et al., 2009). However, for achievement of better therapeutic effects which can be applied to actual medical treatment, higher transfection efficiency in APCs is necessary.
Recently, the uses of external physical stimulation, such as electrical, magnetic field, and water pressure, have been reported to improve gene transfection (Scherer et al., 2002; Gersting et al., 2004; Kobayashi et al., 2004; Kim et al., 2008). Among them, ultrasound (US) exposure method using microbubbles, used as contrast agents for US imaging, is one of the most promising approaches for enhanced gene transfection (Iwanaga et al., 2007; Negishi et al., 2008; Shen et al., 2008; Suzuki et al., 2008). It was reported that direct injection of naked pDNA and bubble liposomes (BLs) following US exposure showed high gene transfection efficiency (Negishi et al., 2008; Suzuki et al., 2008). However, the effectiveness of direct injection of naked pDNA is limited in the injection site. Therefore, systemic administration is necessary to transfect the gene widely in the organ. In addition, the delivery of pDNA to the targeted cells is also necessary to transfect the gene into them. As our mannosylated lipoplex can deliver pDNA efficiently into mannose receptor-expressing cells via intravenous route (Kawakami et al., 2000; Yamada et al., 2004), we considered that the combination of mannosylated lipoplexes and BLs is expected to enhance the transfection efficiency in those cells.
In this study, we developed the combination method using mannosylated lipoplexes and BLs with US to enhance gene transfection in mannose receptor-expressing cells. We investigated gene expression and cellular toxic properties in in vitro and in vivo by the combination method using mannosylated lipoplexes and BLs with US. In addition, the gene expression characteristics in nonparenchymal cells (NPCs) in the liver and dendritic cells in the spleen were also evaluated. The luciferase gene (pCMV-Luc) was used as reporter gene because it is simple to handle and can be detected with high sensitivity.
Materials and Methods
Materials
Dioleoylphosphatidylethanolamine (DOPE) was purchased from Avanti Polar Lipids (Alabaster, AL). 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC) and collagenase type I were obtained from Sigma Chemicals (St. Louis, MO). Anti-CD11c monoclonal antibody (N418)-labeled magnetic beads were purchased from Miltenyi Biotec (Auburn, CA). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG(2K)) was obtained from NOF (Tokyo, Japan) and fetal bovine serum (FBS) was purchased from Equitech-Bio (Kerrville, TX). RPMI-1640, thioglycolate medium, and Hank's medium were purchased from Nissui Pharmaceutical (Tokyo, Japan). Opti-MEM I was obtained from Gibco BRL (Grand Island, NY). All other chemicals were of the highest purity available.
Mice
Female ICR mice (4–5 weeks, 18–22 g) were purchased from the Shizuoka Agricultural Cooperative Association for Laboratory Animals (Shizuoka, Japan). All animal experiments were carried out in accordance with the Principles of Laboratory Animal Care as adopted and propagated by the U.S. National Institutes of Health and the Guidelines for Animal Experiments of Kyoto University.
Construction and preparation of pDNA (pCMV-Luc)
pCMV-Luc was constructed by subcloning the HindIII/Xba I firefly luciferase cDNA fragment from pGL3-control vector (Promega, Madison, WI) into the polylinker of pcDNA3 vector (Invitrogen, Carlsbad, CA). pDNA was amplified in the Escherichia coli strain DH5α and was isolated and purified using a JETSTAR2.0 Plasmid Giga Kit (Genomed, Lohne, Germany).
Construction of lipoplexes and BLs
Man-C4-Chol or DC-Chol and DOPE were mixed in chloroform at a molar ratio of 3:2 to produce lipoplexes. Man-C4-Chol was synthesized by the method described previously (Kawakami et al., 2000). For construction of BLs, DSPC and DSPE-PEG(2K) were mixed in chloroform at a molar ratio of 94:6. The mixture for the construction of each type of liposome was dried by evaporation, and then it was vacuum-desiccated and the resultant lipid film was resuspended in sterile 5% dextrose or saline. After hydration for 30 min at room temperature, the dispersion was sonicated for 10 min in a bath sonicator and for 3 min in a tip sonicator to produce liposomes. The resultant liposomes were sterilized by passage through a 0.45-μm filter (Nihon-Millipore, Tokyo, Japan). The particle sizes and zeta potentials of liposomes were determined using a Zetasizer Nano ZS instrument (Malvern Instruments, Worcestershire, UK). Lipoplexes were prepared at a charge ratio of 1.0:3.1 (−:+) as described in our previous report (Hattori et al., 2004). BLs were prepared according to a previous report (Suzuki et al., 2008). Briefly, 2 ml of liposomes was added to 5-ml sterilized vials, filled with perfluoropropane gas (Takachiho Chemical Ind., Tokyo, Japan), capped, and then pressured with 7.5 ml of perfluoropropane gas. To produce BLs, the vial was sonicated using a bath-type sonicator (As One Corporation, Osaka, Japan) for 5 min.
Harvesting and culture of macrophages
Mouse peritoneal macrophages were collected and cultured according to the previous report of our group (Hattori et al., 2006b). Briefly, macrophages were harvested from ICR mice for 4 days after intraperitoneal injection of 2.9% thioglycolate medium. The washed cells were suspended in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, penicillin G (100 U/ml), and streptomycin (100 μg/ml). Then, macrophages were plated on 24-well culture plates at a density of 2 × 105 cells/1.88 cm2. After incubation for 2 hr at 37°C in 5% CO2–95% air, nonadherent cells were washed off with culture medium and cells were incubated for another 72 hr.
In vitro transfection experiments in macrophages
After 72 hr incubation of macrophages, the culture medium was replaced with Opti-MEM-I-containing lipoplexes (5 μg pCMV-Luc/well). After 30 min incubation, BLs (60 μg/well) were added and macrophages were immediately exposed to US (frequency, 2.062 MHz; duty, 50%; burst rate, 10 Hz; intensity, 4.0 W/cm2) for 20 sec using a 6-mm-diameter probe placed in the well. US was generated using a Sonopore-4000 sonicator (Nepa Gene, Chiba, Japan). Thirty minutes later, the incubation medium was replaced with RPMI-1640 again and incubated for an additional 23 hr. Lipofectamine® 2000 (LF2000; Invitrogen) was used according to the recommended procedures and the exposure time of LF2000 was the same as that for the mannosylated lipoplexes in in vitro. The cells were scraped from the plates and suspended in lysis buffer (0.05% Triton X-100, 2 mM ethylenediaminetetraacetic acid (EDTA), 0.1 M Tris, pH 7.8). Then, the cell suspension was shaken and centrifuged at 10,000 × g, 4°C for 10 min. The supernatant was mixed with luciferase assay buffer (Picagene; Toyo Ink, Tokyo, Japan) and luciferase activity was measured in a luminometer (Lumat LB 9507; EG&G Berthold, Bad Wildbad, Germany). The luciferase activity was normalized with respect to the protein content of cells. The protein concentration was determined with a Protein Quantification Kit (Dojindo Molecular Technologies, Tokyo, Japan).
In vitro imaging of pDNA and lipoplexes in macrophages
At 72 hr incubation after collecting macrophages, the culture medium was replaced with Opti-MEM-I-containing nitrobenzoxadiazole (NBD)-labeled liposomes/rhodamine-labeled pDNA complexes (1 μg pCMV-Luc/well). Rhodamine-labeled pDNA was prepared using the Label IT Nucleic Acid Labeling Kit (Mirus, Madison, WI). NBD-labeled liposomes consisted of Man-C4-Chol, DOPE, and NBD-DOPE in a molar ratio of 60:35:5. After preparation of NBD-labeled liposomes, free NBD-DOPE was removed by gel chromatography using a Sephadex® G-25 M PD-10 column (GE Healthcare, Uppsala, Sweden). Thirty minutes later, BLs (60 μg/well) were added and macrophages were exposed to US. After 30 min incubation, the incubation medium was replaced with RPMI-1640 and macrophages were incubated for an additional 5 hr and examined by fluorescence microscopy (Biozero BZ-8000; Keyence, Osaka, Japan).
In vivo gene expression experiments in mice
Four-week-old ICR female mice were given an intravenous injection of 300 μl lipoplexes via tail vein, using a 26-gauge syringe needle at a dose of 50 μg pDNA. Then, at 5 min after injection, BLs (500 μg/200 μl) were administered intravenously via tail vein. At 5 min after the injection of BLs, US (frequency, 1.045 MHz; duty, 50%; burst rate, 10 Hz; intensity, 1.0 W/cm2; time, 2 min) was applied transdermally to the abdominal area using a Sonopore-4000 sonicator with a probe of diameter 20 mm. The organs were directly exposed to US, using a probe of diameter 6 mm. At predetermined times after injection, mice were sacrificed and their organs collected for each experiment. The organs were washed twice with cold saline and homogenized with lysis buffer (0.05% Triton X-100, 2 mM EDTA, 0.1 M Tris, pH 7.8). The lysis buffer was added at a weight ratio of 5 ml/g for liver or 4 ml/g for other organs. After three cycles of freezing and thawing, the homogenates were centrifuged at 10,000 × g, 4°C for 10 min and the resultant supernatant was used for luciferase assay.
Cellular localization of luciferase activity in liver
At 6 hr after transfection, each mouse was anesthetized with pentobarbital sodium (40–60 mg/kg) and the liver was perfused with perfusion buffer (Ca2+, Mg2+-free HEPES solution, pH 7.2) for 10 min. Then, the liver was perfused with collagenase buffer (HEPES solution, pH 7.5, containing 5 mM CaCl2 and 0.05% (w/v) collagenase type I) for 5 min. Immediately after the start of perfusion, the vena cava and aorta were cut and the perfusion rate was maintained at 5 ml/min. At the end of perfusion, the liver was excised. The cells were dispersed in ice-cold Hank's HEPES buffer by gentle stirring and then filtered through cotton mesh sieves, followed by centrifugation at 50 × g for 1 min. The pellets containing parenchymal cells (PCs) were washed five times with Hank's HEPES buffer by centrifuging at 50 × g for 1 min. The supernatant containing NPCs was similarly centrifuged five times. The resulting supernatant was centrifuged twice at 300 × g for 10 min. PCs and NPCs were resuspended separately in ice-cold Hank's HEPES buffer (2 ml for PCs and 500 μl for NPCs) and luciferase activity of supernatant was measured. The cell numbers and viability were determined by the trypan blue exclusion method and the luciferase activity was normalized with respect to the number of cells.
Measurement of transaminase activity in serum
After transfection, the serum was collected from the anesthetized mice. Alanine aminotransferase (ALT/GPT) and aspartate aminotransferase (AST/GOT) activities in the serum were determined using Transaminase CII-Test Wako kit (Wako Pure Chemical Industries, Tokyo, Japan) according to manufacturer's instructions.
Quantification of luciferase mRNA in splenic CD11c+ cells
At 6 hr after transfection, spleens were harvested and spleen cells were suspended in ice-cold RPMI-1640 medium on ice. Then, red blood cells were removed by incubation with hemolytic reagent (0.83% NH4Cl solution) for 3 min at room temperature. CD11c+ and CD11c− cells were separated by magnetic cell sorting with auto magnetic cell sorting (MACS) system (Miltenyi Biotec) following the manufacturer's instructions. Luciferase activity of each fraction was determined by luciferase assay. Total RNA was isolated from separated cells using GenElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich, St. Louis, MO). Reverse transcription of mRNA was carried out using PrimeScript® RT reagent Kit (Takara Bio, Shiga, Japan). Real-time polymerase chain reaction was performed using SYBR® Premix Ex Taq (Takara Bio) and Lightcycler Quick System 350S (Roche Diagnostics, Indianapolis, IN) with primers. The primers for luciferase and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA were constructed as follows: primer for luciferase cDNA: 5′-TTCTTCGCCAAAAGCACTC-3′ (forward) and 5′-CCCTCGGGTGTAATCAGAAT-3′ (reverse); primer for GAPDH: 5′-TCTCCTGCGACTTCAACA-3′ (forward) and 5′-GCTGTAGCCGTATTCATTGT-3′ (reverse) (Sigma-Aldrich). The mRNA copy numbers were calculated for each sample from the standard curve using an instrument software (“Arithmetic Fit Point Analysis” for the Lightcycler). The results were expressed as relative copy numbers calculated relative to GAPDH mRNA (copy numbers of luciferase mRNA/copy numbers of GAPDH mRNA).
Statistical analysis
The results were expressed as the mean ± standard deviation of more than three experiments. Statistical analysis was performed using analysis of variance and the Turkey–Kramer test for multiple comparisons between groups or Student's t-test for two-group comparisons; p < 0.05 was considered statistically significant.
Results
Physicochemical properties of mannosylated lipoplexes and BLs
The physicochemical properties of DC-Chol:DOPE lipoplexes (bare lipoplexes), Man-C4-Chol:DOPE lipoplexes (mannosylated lipoplexes), and BLs used for all experiments were evaluated by measuring particle sizes and zeta potentials. The mean particle sizes of bare lipoplexes, mannosylated lipoplexes, and BLs were 123 ± 6.0, 128 ± 9.5, and 498 ± 18 nm (n = 3), respectively. Moreover, zeta potentials of bare lipoplexes, mannosylated lipoplexes, and BLs were 47.5 ± 1.4, 46.7 ± 3.8, and 0.08 ± 0.2 mV (n = 3), respectively. These values of mannosylated lipoplexes were the same as in our previous report (Hattori et al., 2006a) and the particle size of BLs was almost identical to that in a previous report (Suzuki et al., 2008).
Transfection efficiency in cultured mouse peritoneal macrophages
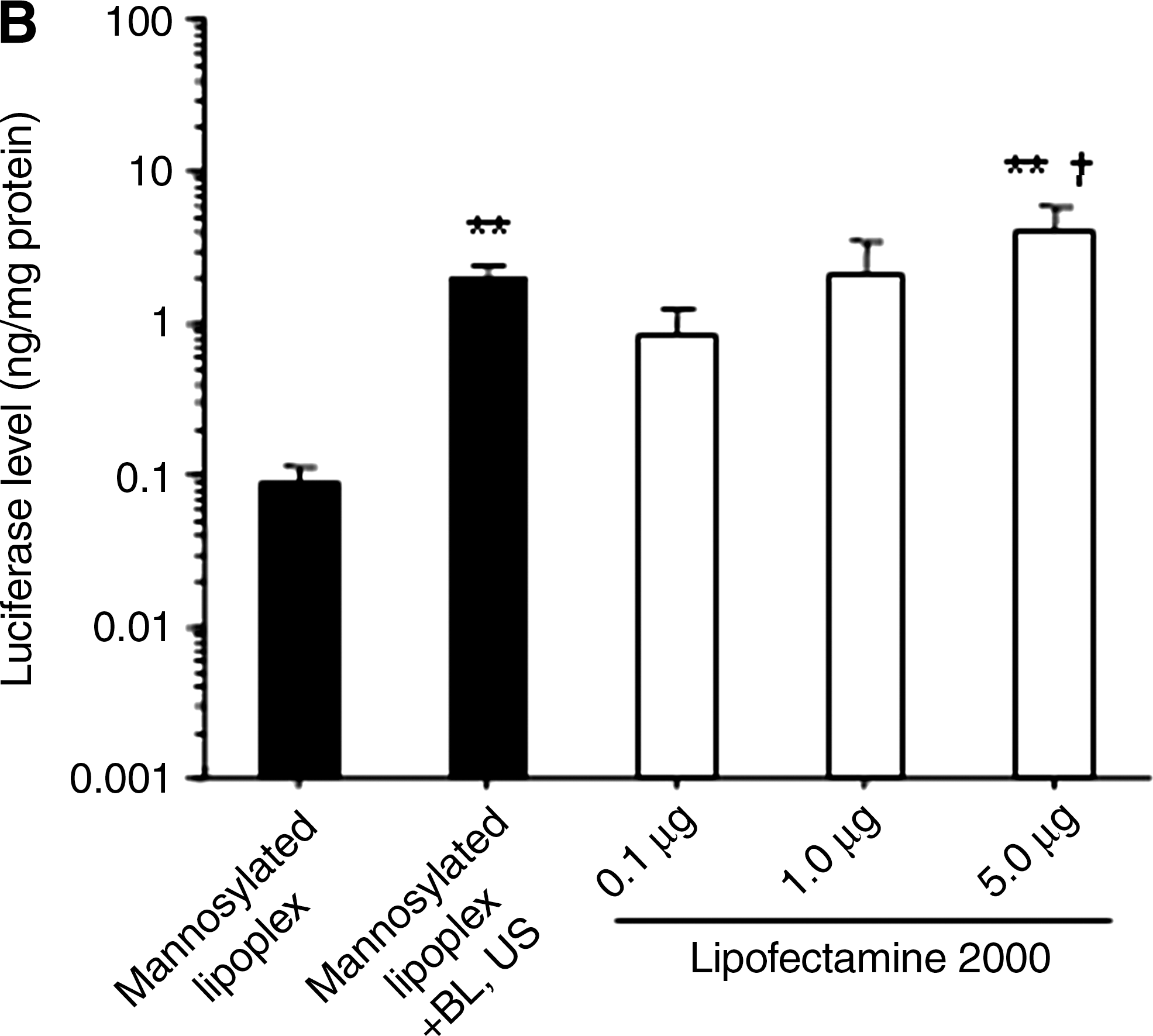
The transfection efficiency of mannosylated lipoplexes and BLs with US was significantly higher than that of naked pDNA or bare lipoplexes and BLs with US in cultured macrophages (Fig. 1A). The transfection efficiency of the combination of mannosylated lipoplexes and BLs was comparable to LF2000 (Fig. 1B). Then, we examined the cytotoxicity of this transfection method by mannosylated lipoplexes and BLs with US. As shown in Fig. 1C, this combination method did not exhibit any cytotoxic effect. However, LF2000 exhibited severe cytotoxic effects.
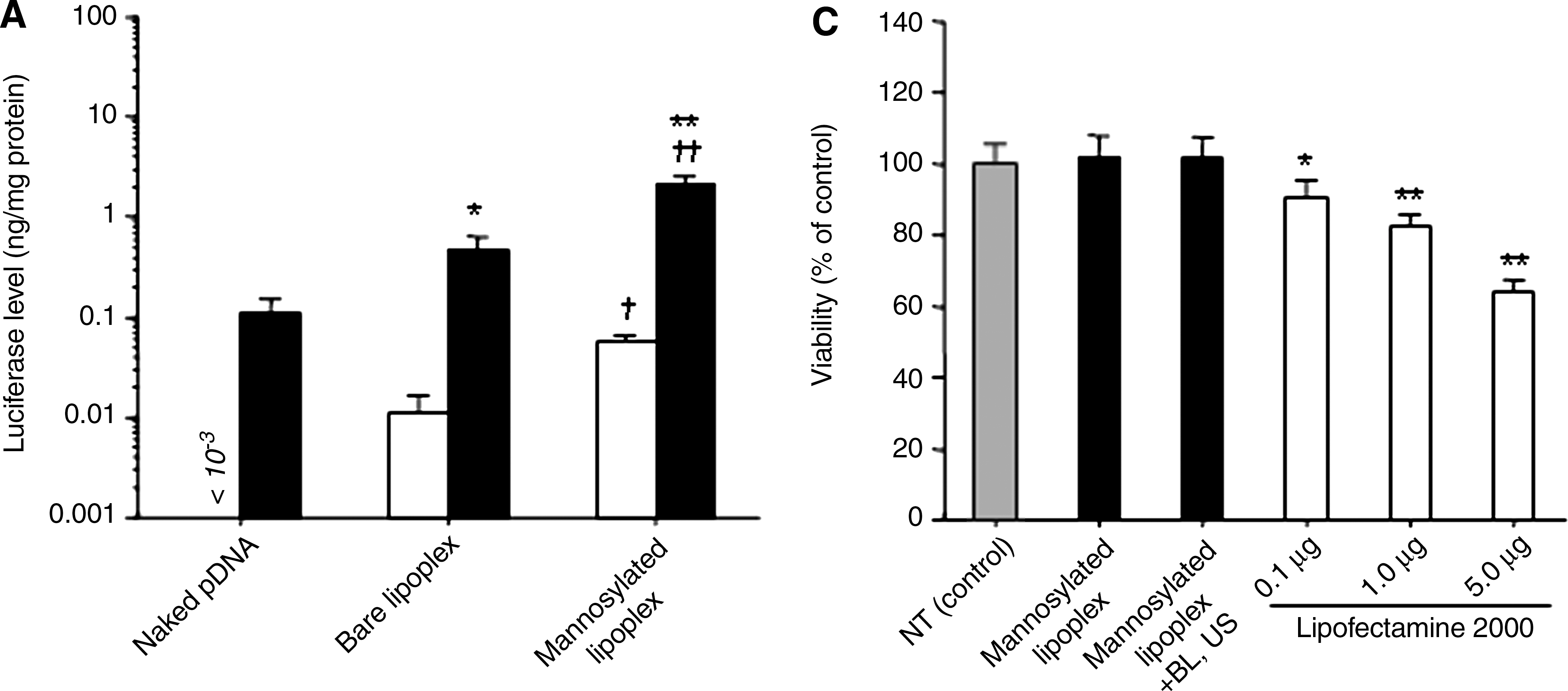
In vitro transfection activity in cultured macrophages by pDNA, bare lipoplexes, and mannosylated lipoplexes, with or without BLs and US. Naked pDNA (5.0 μg/well) was complexed with mannosylated liposomes at a charge ratio of 3.1. (
Intracellular uptake of pDNA in cultured mouse peritoneal macrophages
As shown in Fig. 2, the intracellular uptake of rhodamine-labeled pDNA was significantly increased by the combination method using mannosylated lipoplexes and BLs, compared with transfection by mannosylated lipoplexes. In contrast, the intracellular uptake of mannosylated liposomes by this combination method was smaller than that by transfection using mannosylated lipoplexes. This result means that extracellular mannosylated liposomes are disrupted by the cavitation energy generated by the collapse of BLs following US exposure, and thus, only pDNA carried by mannosylated liposomes is introduced into cells.
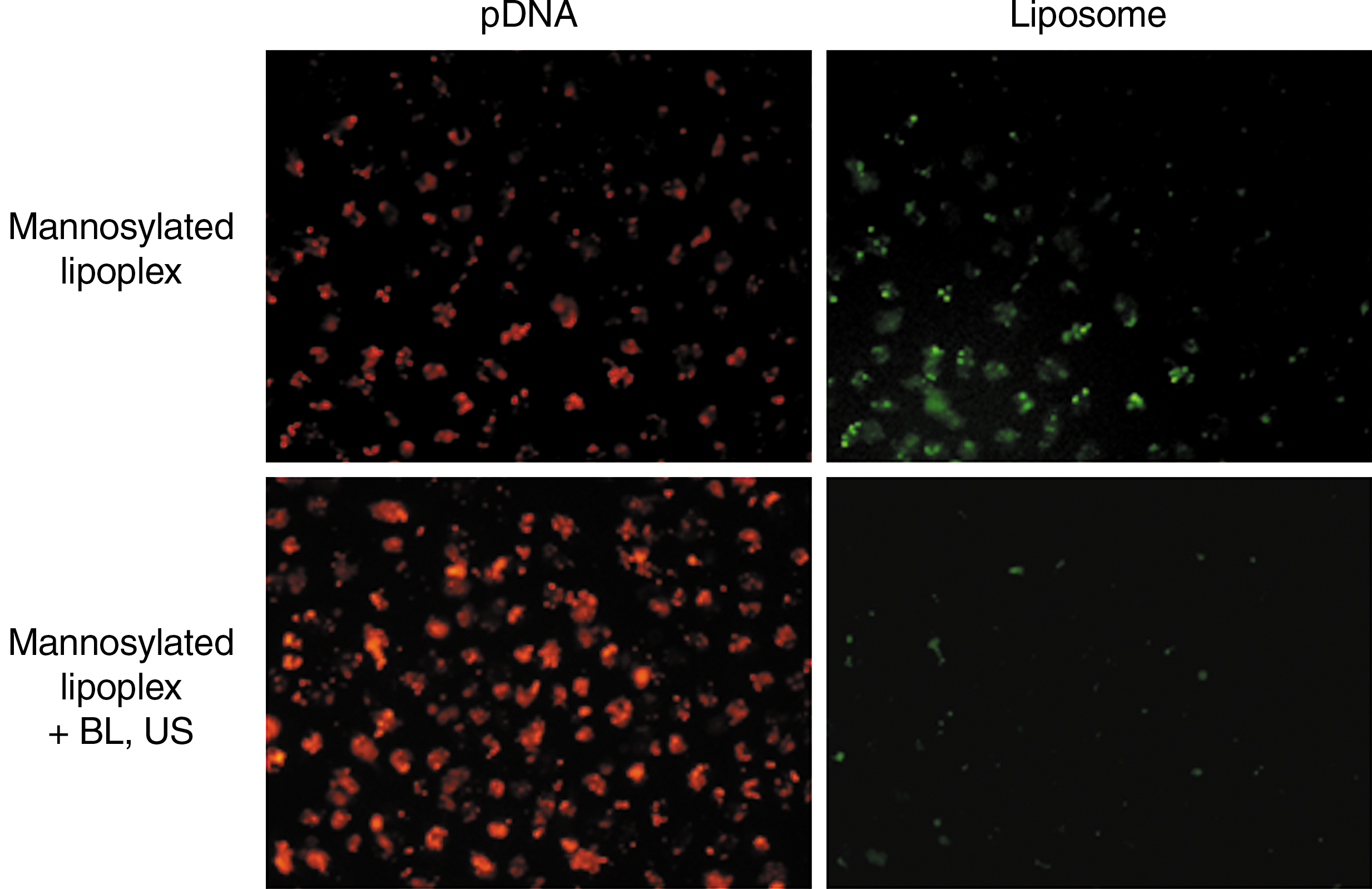
Amount of intracellular uptake of naked pDNA and mannosylated liposomes in cultured macrophages. pDNA (red): TM-rhodamine-labeled pCMV-Luc; liposome (green): mannosylated liposome containing NBD-labeled phospholipids. Microscopy images were recorded 6 hr after the transfection using mannosylated lipoplexes, with or without BLs and US.
Transfection activities after intravenous administration in mice
The transfection efficiency by this combination method was higher than that by naked pDNA or bare lipoplexes and BLs using US in the liver and spleen (Fig. 3). The transfection efficiency by this combination method remained higher than that by mannosylated lipoplexes for 48 hr in the liver and spleen (Fig. 4). Moreover, as shown in Fig. 5, the increase of transfection efficiency by this combination method was not observed in the lung, kidney, and heart.
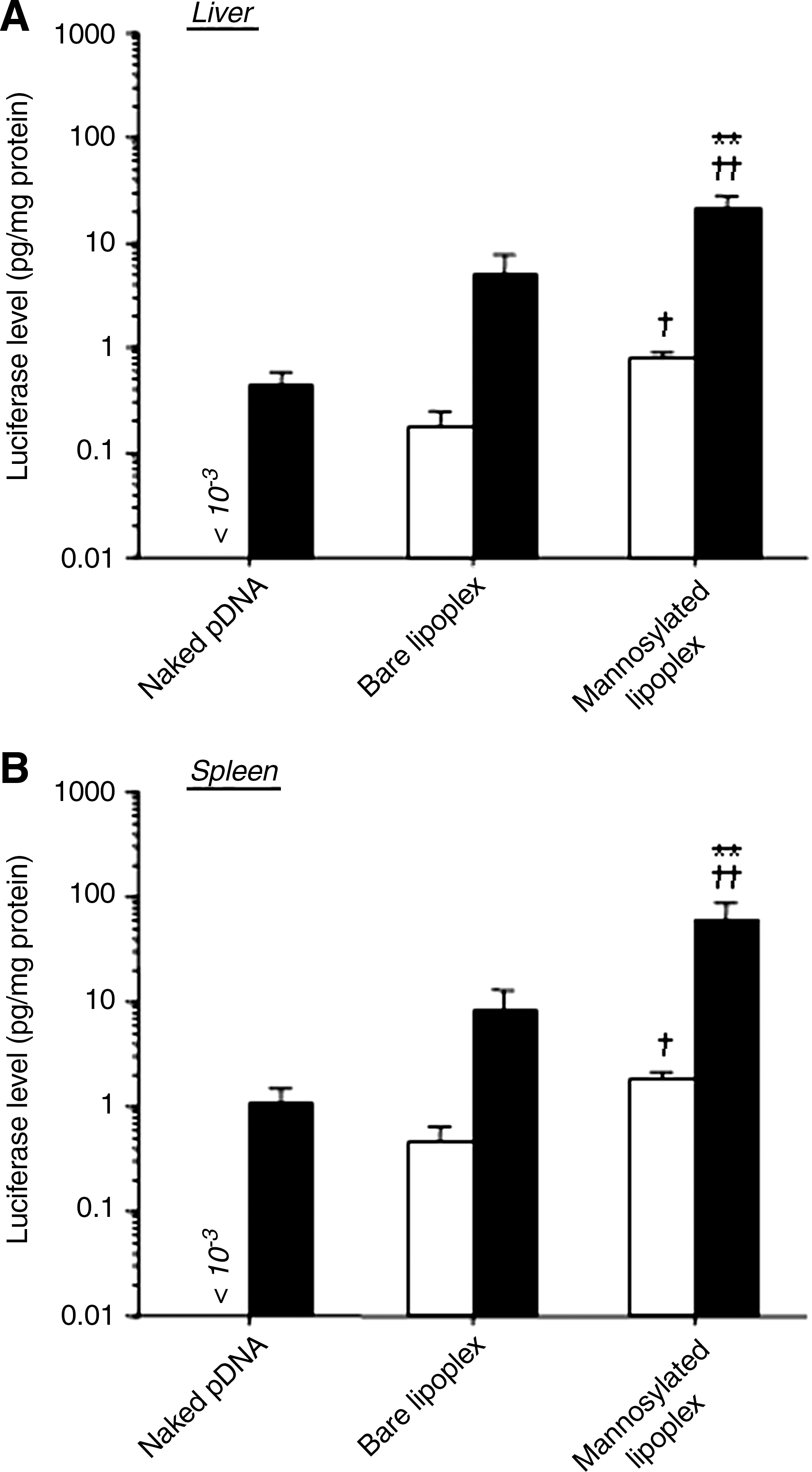
Luciferase expression in the liver (
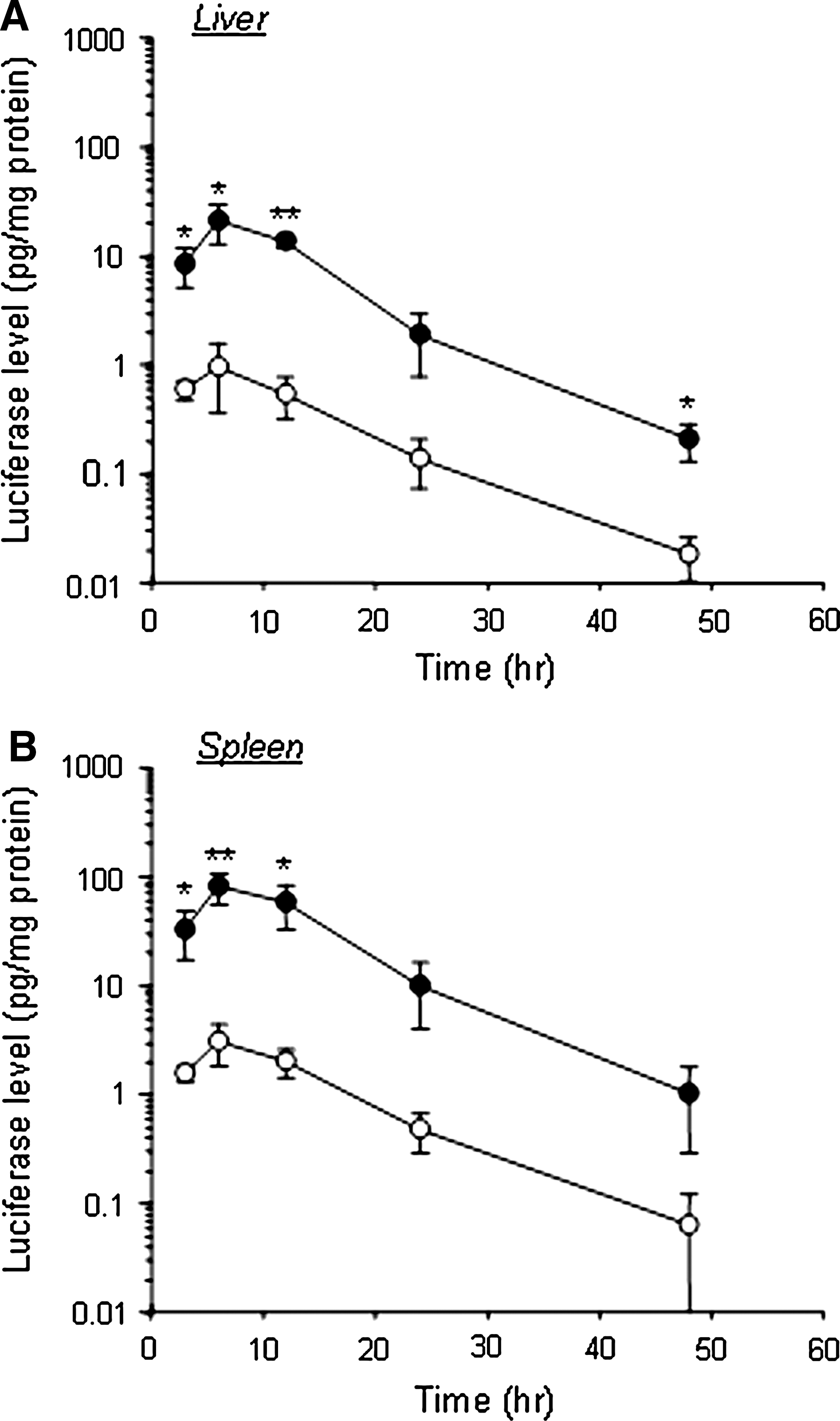
Time-course of luciferase expression in the liver (
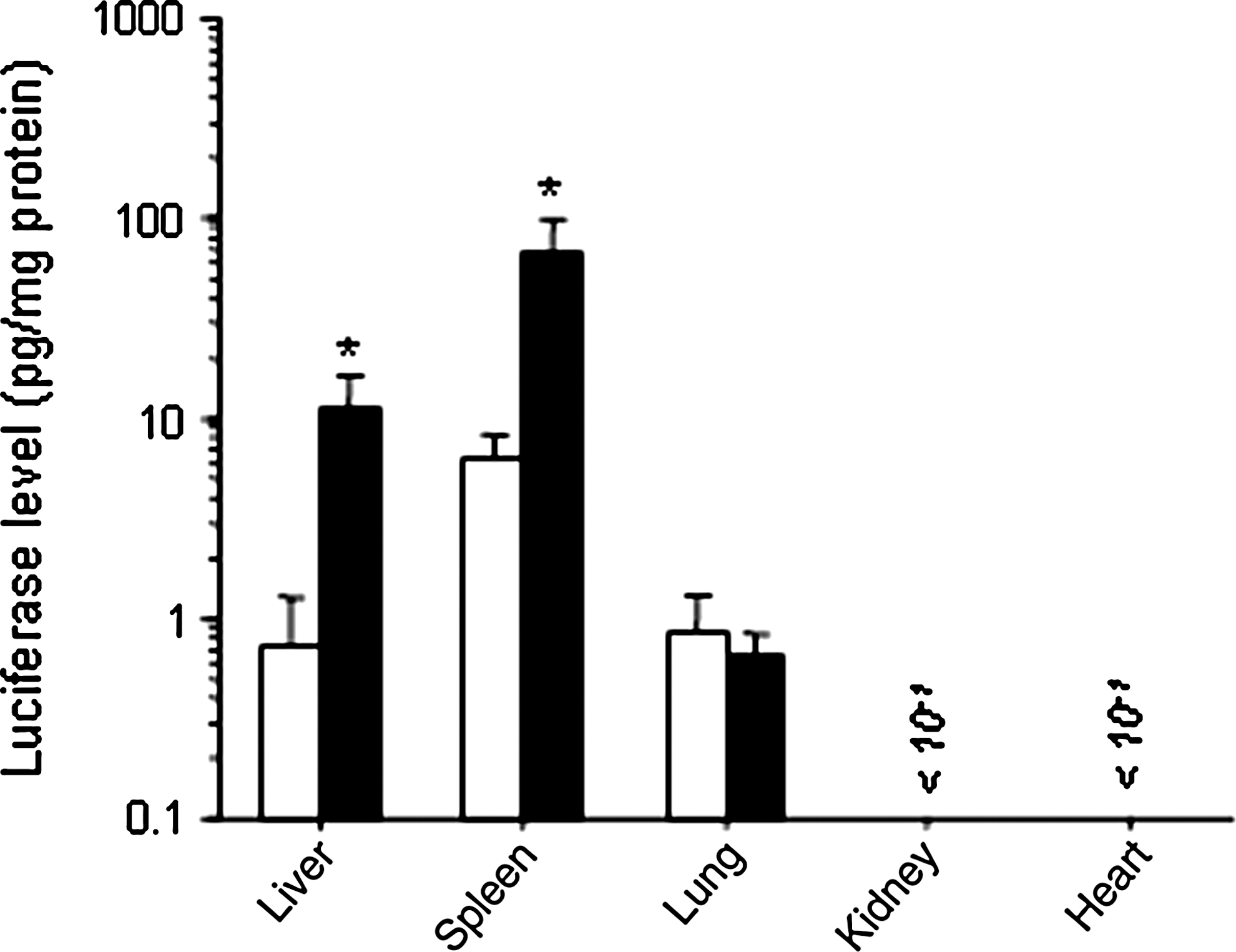
Luciferase expression in each organ after transfection by intravenous administration of mannosylated lipoplexes, with or without BLs and US in mice. Open bars, without; solid bars, with BLs and US. Each value represents the mean ± SD (n = 4). *p < 0.05, compared with or without BLs and US.
Intrahepatic transfection characteristics and hepatic toxicity after treatment
After transfection by this combination method using mannosylated lipoplexes and BLs with US, the gene expression in NPCs was significantly higher than that in PCs (Fig. 6A). This difference in gene expression between NPCs and PCs was similar to that obtained by treatment with mannosylated lipoplexes, but the gene expression in NPCs and PCs was higher than that obtained by treatment with mannosylated lipoplexes. On the other hand, the gene expression in NPCs and PCs was almost the same after treatment by naked pDNA and BLs with US. Further, we examined ALT and AST activities in the serum to investigate the hepatic toxicity after treatment by mannosylated lipoplexes and BLs using US. The ALT and AST activities following transfection by the combination method using mannosylated lipoplexes and BLs with US were almost the same as those following transfection using mannosylated lipoplexes (Fig. 6B).
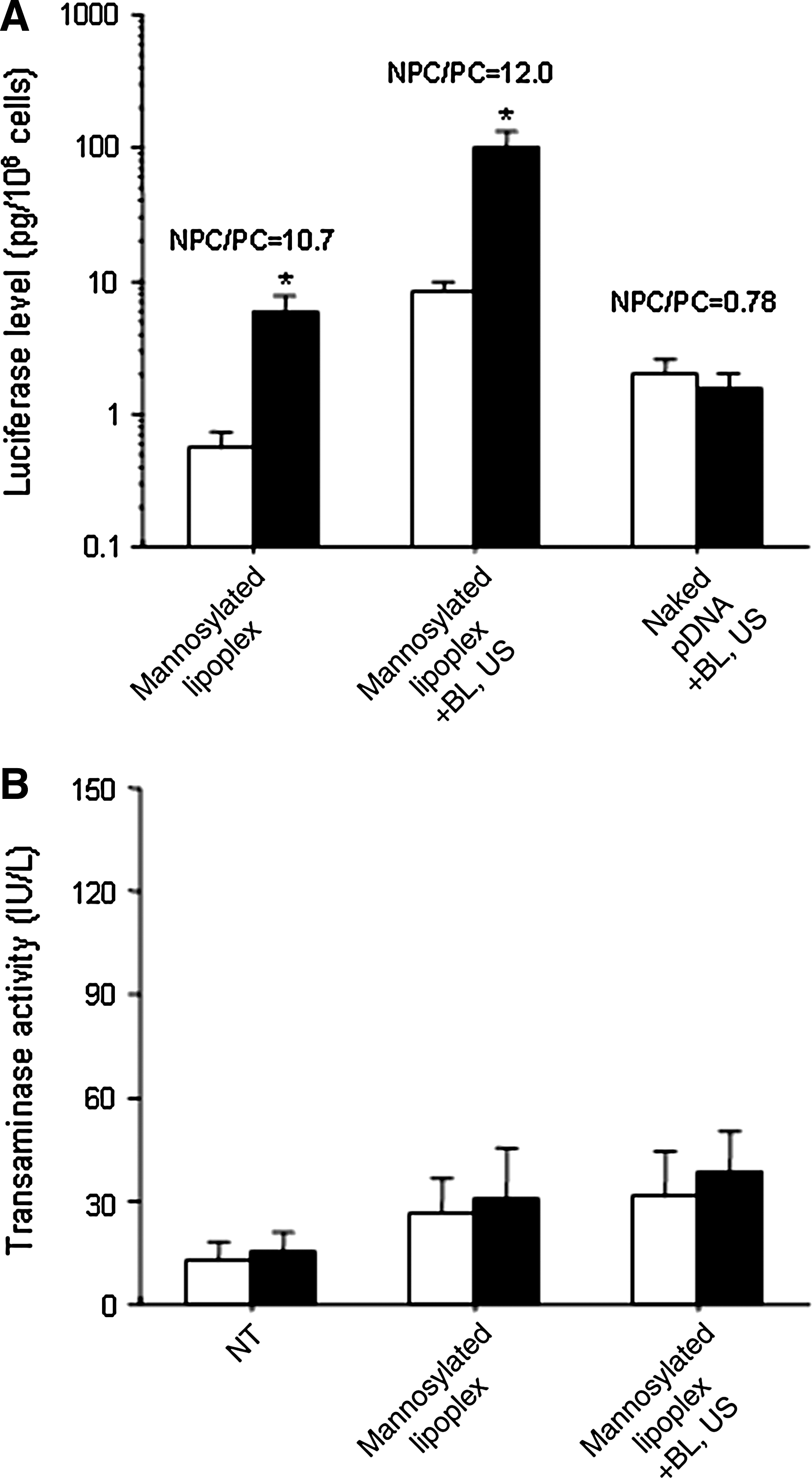
(
Intrasplenic transfection characteristics after treatment
As shown in Fig. 7A, no luciferase activity in the splenic CD11c+ and CD11c− cells at 107 cells was detected using mannosylated lipoplexes. However, with the combination method using mannosylated lipoplexes and BLs, the luciferase activity in the splenic CD11c+ and CD11c− cells at 107 cells was significantly increased, and that in CD11c+ cells, known as dendritic cells, was significantly higher than that in CD11c− cells. Moreover, comparable results were obtained from the experiment involving luciferase mRNA expression (Fig. 7B).
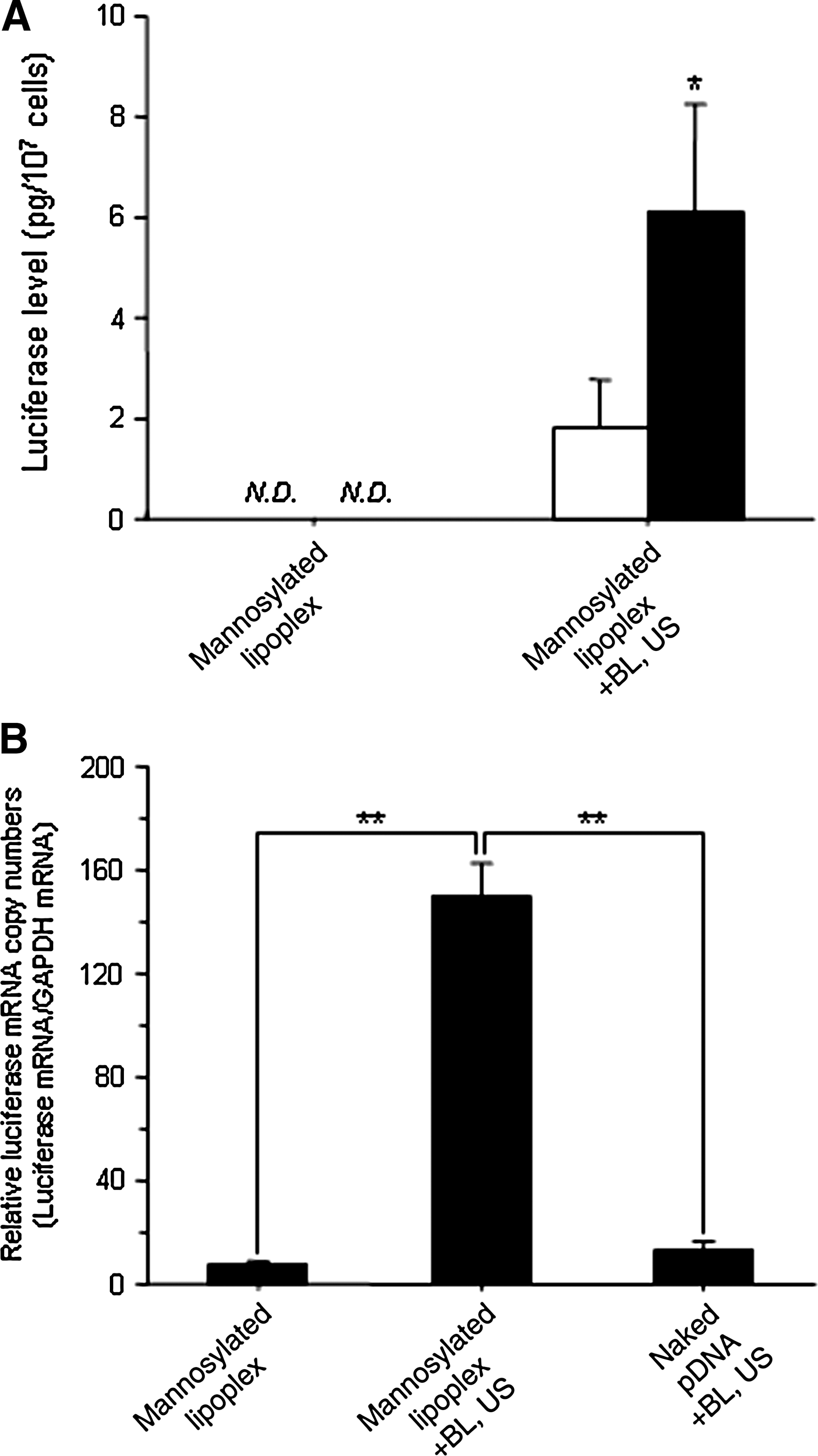
Luciferase expression and luciferase mRNA expression in mouse splenic CD11c+ cells after intravenous administration of naked pDNA or mannosylated lipoplexes, with or without BLs and US at 6 hr after transfection. (
Transfection activities after direct exposure of liver or spleen to US
We investigated gene expression by directly exposing liver or spleen to US after intravenous administration of mannosylated lipoplexes and BLs. As shown in Fig. 8, direct exposure of liver or spleen to US after intravenous administration of BLs results in transfection of the US-exposed organ selectively.
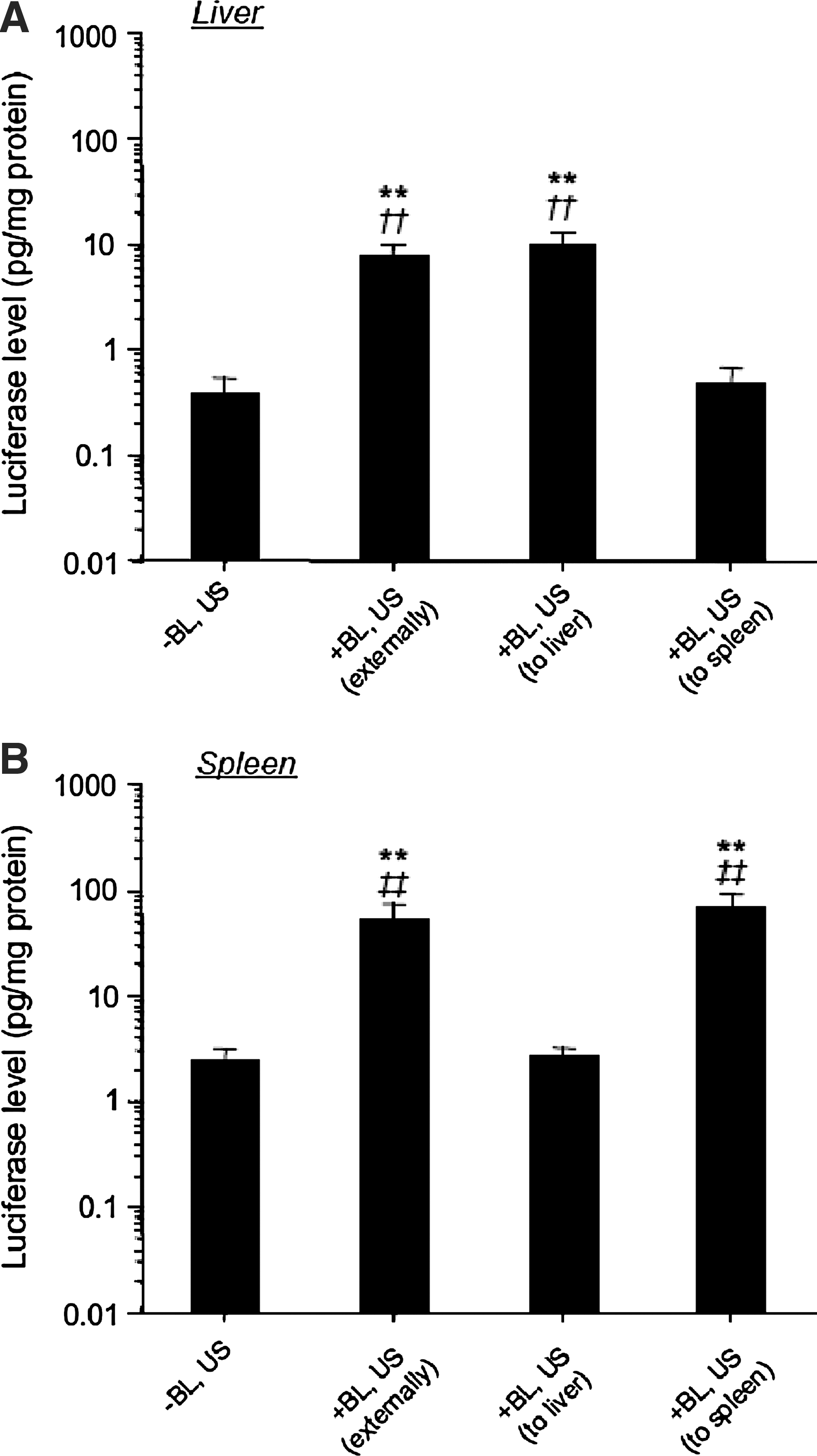
Luciferase expression by mannosylated lipoplexes and BLs with US exposure externally or directly. Luciferase expression in the liver (
Discussion
In this study, using mannosylated liposomes as a carrier of pDNA for controlling the kinetics of pDNA and optimizing the targeting properties, we tried to increase selectively the gene transfection efficiency in the targeted organ or the targeted cell by combining with BLs. Initially, we considered that the in vitro gene transfection efficiency in cultured macrophages, which are known for the expression of mannose receptors, was increased by using BLs and mannosylated lipoplexes as a carrier of pDNA, compared with the transfection using naked pDNA which is the conventional method, or the transfection using bare lipoplexes, which are nontargeted liposomes, as a carrier of pDNA. As shown in Figs. 1A and 2, significantly higher transfection efficiency in cultured macrophages was observed by the transfection using mannosylated lipoplexes and BLs, compared with the transfection using naked pDNA or bare lipoplexes and BLs with US. As this result shows that gene transfection efficiency was increased not only by the improvement of interaction with cell membrane by cationic carriers, but also by the improvement of interaction with mannose receptors by mannose modification, targeted cell-selective and efficient gene transfection is possible by this transfection method using mannosylated lipoplexes and BLs with US. Then we found that the transfection efficiency of this combination method using US was the same as that of LF2000, which is used widely as an efficient transfection reagent (Fig. 1B). Moreover, the cytotoxicity of this combination method using US was significantly lower than that of LF2000 (Fig. 1C). In addition, as shown in Fig. 2, although intracellular uptake of pDNA into macrophages was increased by this combination method, intracellular uptake of mannosylated liposomes was low; therefore, different transfection mechanisms based on intracellular uptake of pDNA via endocytosis were observed by the transfection using lipoplexes. This result may indicate that BLs and US are operated on mannosylated lipoplexes existing on the cell surface, and then pDNA is introduced directly into the cytoplasm. It is generally considered that gene transfection into APCs such as macrophages is difficult (Erbacher et al., 1996; Sakurai et al., 2000). However, in this study, we demonstrated that this combination method using mannosylated lipoplexes and BLs with US could achieve significantly high transfection efficiency in primary cultured peritoneal macrophages.
To achieve effective cancer vaccine therapy, it is necessary to transfect the antigen-presenting gene efficiently into the APCs, such as macrophages and dendritic cells distributed in the liver and spleen (Goerdt et al., 1999; Melief, 2008). Especially, dendritic cells are major target cells for DNA vaccine therapy. As the number of APCs in the spleen is extremely low, it is difficult to transfect the pDNA selectively into macrophages and dendritic cells. We showed that the in vivo selective gene transfection efficiency into mannose receptor-expressing cells in the liver and spleen could be increased by this combination method using US. In this study, US was applied externally using a large probe with diameter 20 mm (see Materials and Methods section). As shown in Figs. 3 and 4, the gene transfection efficiency in the liver and spleen was significantly increased by using mannosylated lipoplexes, compared with transfection using naked pDNA or bare lipoplexes and BLs with US. We have already reported that mannosylated lipoplexes used in this study were accumulated in the liver and spleen, compared with bare lipoplexes (Yamada et al., 2004). Therefore, these results show that in vivo distribution characteristics of carriers are important for enhancement of gene transfection efficiency in the targeted organ by using BLs. Moreover, to investigate the cellular localization of gene expression after transfection by the combination of mannosylated lipoplexes and BLs with US, the gene expression levels of NPCs in the liver and dendritic cells in the spleen were determined. As a result, we demonstrated that this combination method could transfect the gene selectively into hepatic NPCs and splenic dendritic cells (Figs. 6A and 7). The gene transfection efficiency into splenic dendritic cells could be monitored by the luciferase protein in this combination method using US (Fig. 7), whereas a previous study by our group was only able to detect the luciferase mRNA in the transfection method using mannosylated lipoplexes (Hattori et al., 2006b). Therefore, this combination method using US results in high gene transfection efficiency in splenic dendritic cells. Moreover, there was very little in vitro cytotoxicity and hepatic toxicity in mice, using this method (Figs. 1C and 6B). Therefore, this transfection method may be more suitable for clinical application. These results indicate that the combination of mannosylated lipoplexes and BLs with US is a novel method for markedly increasing gene expression in hepatic NPCs and splenic dendritic cells following intravenous injection in mice.
In this study, although US was applied externally for the experiment involving noninvasive conditions, the increase of gene expression by the combination of mannosylated lipoplexes and BLs with US was observed in the liver and spleen, but not in the lung, kidney, and heart (Fig. 5). Although the uptake into the reticuloendothelial system (RES) can be avoided by PEGylation of the liposomes, large amounts of PEGylated liposomes are distributed in the liver and spleen (Elbayoumi and Torchilin, 2006). Moreover, it is known that PEGylated liposomes with a particle size over 500 nm are easily taken up into the RES, compared with smaller conventional liposomes (Ishida et al., 1999). Harashima et al. (1994) also reported a similar effect of particle size on the distribution of liposomes. The BLs used in this study possesses a particle size of approximately 500 nm, which would be preferentially taken up by the RES as suggested by these reports (Harashima et al., 1994; Ishida et al., 1999). Considering that the RES comprises Kupffer cells and splenic macrophages/dendritic cells which express mannose receptors, the treatment of mannosylated lipoplexes is quite reasonable (Fig. 5). On the other hand, the observation that treatment with BLs and US was not effective in the lung, kidney, and heart (Fig. 5) agrees in part with the study by Negishi et al. (2008) and Chen et al. (2006), who reported that gene transfection in the kidney was difficult in a treatment using microbubbles. Therefore, these observations support our finding that the combination method using mannosylated lipoplexes and BLs with US offers important advantages for gene transfection into mannose receptor-expressing cells in the RES.
In the experiments, we applied US externally using a comparatively large probe in mice (diameter, 20 mm) to evaluate the gene transfection efficiency in noninvasive conditions. Consequently, the organ specificity of the gene expression was characterized (Fig. 5). As the US-exposed organ-specific gene expression was observed by direct exposure of the organ to US (Fig. 8), more effective gene transfection into the targeted organ was also possible by this transfection method using mannosylated lipoplexes and BLs with US.
To date, nucleic acid introduction methods using microbubbles such as BLs have been applied in in vitro using not only naked pDNA but also naked siRNA and oligodeoxynucleotides (Haag et al., 2006; Negishi et al., 2008). However, it may be difficult to use these materials in in vivo because of the poor stability of naked nucleic acids in blood. The major feature of the combination method using mannosylated lipoplexes and BLs with US was to both improve the pDNA stability in the body and control the kinetics of pDNA by using lipoplexes. As the mannosylated liposomes used in this study can form complexes with different nucleic acids via electrostatic interaction, this transfection method can be applied to many types of diseases. We have reported that mannosylated liposomes/nuclear factor κB complexes could significantly suppress hepatitis (Higuchi et al., 2006; Huang et al., 2009). Therefore, in the future, the combination method using mannosylated liposomes/various types of nucleic acids and BLs with US would be widely applied using mannose receptor-expressing cells for a variety of genetic diseases.
In conclusion, we demonstrated that the gene transfection efficiency in macrophages and dendritic cells was significantly increased by the combination method using mannosylated lipoplexes and BLs with US. To date, various targeting ligands for cell-selective targeting, such as galactose (Fumoto et al., 2004; Letrou-Bonneval et al., 2008), folic acid (Zuber et al., 2003; Fernandes et al., 2008), anisamide (Li et al., 2008), hyaluronan (Eliaz and Szoka, 2001), RGD peptide (Anwer et al., 2004; Oba et al., 2008), IRQ peptide (Mudhakir et al., 2008), and anti-Her2 antibody (Chiu et al., 2004; Strehblow et al., 2005), have been reported. Therefore, these observations would be valuable for the development of cell-selective gene delivery systems by using various ligand-modified lipoplexes and BLs with US.
Footnotes
Acknowledgment
This work was supported in part by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by Health and Labour Sciences Research Grants for Research on Advanced Medical Technology from the Ministry of Health, Labour, and Welfare of Japan.
Disclosure Statement
No competing financial interests exist.