
Abstract
The development of vectors that express a therapeutic transgene efficiently and specifically in hematopoietic cells (HCs) is an important goal for gene therapy of hematological disorders. We have previously shown that a 500-bp fragment from the proximal Was gene promoter in a lentiviral vector (LV) was sufficient to achieve more than 100-fold higher levels of Wiskott-Aldrich syndrome protein in HCs than in nonhematopoietic cells (non-HCs). We show now that this differential was reduced up to 10 times when the enhanced green fluorescent protein gene (eGFP) was expressed instead of Was in the same LV backbone. Insertion of Was cDNA sequences downstream of eGFP in these LVs had a negative effect on transgene expression. This effect varied in different cell types but, overall, Was cDNA sequences increased the hematopoietic specificity of Was promoter-driven LV. We have characterized the minimal fragment required to increase hematopoietic specificity and have demonstrated that the mechanism involves Was promoter regulation and RNA processing. In addition, we have shown that Was cDNA sequences interfere with the enhancer activity of the woodchuck posttranscriptional regulatory element. These results represent the first data showing the role of Was intragenic sequences in gene regulation.
Introduction
The development of efficient, stable, and regulated gene transfer vectors is a major goal for both basic science and gene therapy approaches. At present lentiviral vectors (LVs) are the most powerful of all integrative vector systems (Wiznerowicz and Trono, 2005). They not only transduce efficiently most cell types, including quiescent cells, but also express the transgene stably (Case et al., 1999). The development of self-inactivated LVs (Zufferey et al., 1998) improved their biosafety and regulatory properties because the expression of the transgene is directed only through internal promoters and not through the 5′ long terminal repeat (LTR). This property allows the use of various promoters (tissue-specific, drug-regulated, etc.) to achieve regulated expression and to reduce genotoxicity (Modlich et al., 2006; Zychlinski et al., 2008). The latest lentiviral generations allow drug-regulated and tissue-specific delivery of transgenes both in vitro and in vivo (Barde et al., 2006; Szulc et al., 2006; Chang and Sadelain, 2007; Frecha et al., 2008).
In most approaches, transcriptionally targeted LVs contain minimal promoter regions defined as accountable for transcriptional regulation of their genes. Using this strategy, several groups have obtained LVs that are finely regulated in liver (Oertel et al., 2003), hematopoietic cells (Martin et al., 2005), antigen-presenting cells (Cui et al., 2002), B cells (Lutzko et al., 2003; Moreau et al., 2004), CD4+ T cells (Marodon et al., 2003), erythroid cells (Moreau-Gaudry et al., 2001; Lotti et al., 2002; Han et al., 2007), and endothelial cells (Jager et al., 1999; revised in Goverdhana et al., 2005). However, the level of regulation of the transgene rarely mimics exactly the regulation of the gene in nature (Frecha et al., 2008). This can be easily understood because the final activity of a promoter is dependent on multiple factors (position of the promoter in the chromatin, long distant enhancers or silencers, epigenetic modifications, etc.) (Osborne et al., 2004) that are missing in the integrated vector. Several approaches can be used to attain improved regulated expression by integrative vectors such as the use of insulators (Hawley, 2001; Robert-Richard et al., 2007), or the combination of several elements that modulate promoter activity (Frecha et al., 2008).
The woodchuck posttranscriptional regulatory element (WPRE) is an enhancer element that affects gene expression at the posttranscriptional level (Gaszner and Felsenfeld, 2006), and its effect over retroviral vectors depends both on the inserted promoter and the target cell (Donello et al., 1998; Zufferey et al., 1999; Werner et al., 2004; Hlavaty et al., 2005; Mastroyiannopoulos et al., 2005). Moreau-Gaudry and co-workers demonstrated that inclusion of the WPRE in erythroid-specific LVs increased expression in erythroid cells without affecting expression in nontargeted cells (Moreau-Gaudry et al., 2001).
Although most of the studies concerning transcriptional regulation focus on regions upstream of the promoter, there are solid data demonstrating that downstream regions can also play important roles in regulating promoter activity (Levine and Tjian, 2003). In fact, elements that modulate transcriptional activity can be found inside introns and exons of genes (Sakonju et al., 1980; Sternberg et al., 1988; Xu et al., 1993; Cui et al., 1998; Seshasayee et al., 2000; Kobayashi et al., 2001; Lin and Tam, 2001; Saur et al., 2002; Wong et al., 2004; Abbasi et al., 2007). Therefore, in a retroviral backbone, if a gene contains regulatory elements inside exons, its expression will be modulated not only by the promoter used but also by these elements present in the cDNA.
The development of pan-hematopoietic specific LVs is of interest for both gene therapy approaches and basic research. We have developed a pan-hematopoietic specific Was promoter-driven LV that expresses the Wiskott-Aldrich syndrome (Was) gene more than 100 times more efficiently in hematopoietic cells than in nonhematopoietic cells (Martin et al., 2005).
In this paper we show that the hematopoietic specificity of Was promoter-driven LVs drops up to 10 times when eGFP is expressed instead of the Was gene. We demonstrate that Was cDNA sequences play an important role in the highly specific hematopoietic expression of Was promoter-driven LVs. We show that the effect of Was cDNA on hematopoietic specific expression is due to both transcriptional and posttranscriptional activity and that this effect is governed by a 5′ 900-bp region. We also show that the Was cDNA interferes with the enhancer activity of the WPRE element, reducing its activity.
Material and Methods
Cell lines and culture media
HVS-WAS/1, herpesvirus saimiri-immortalized T cells derived from a patient with WAS, and ALLO-WAS/1, a primary allospecific T cell line from a second patient with WAS (Gallego et al., 1997), were kindly provided by I. Molina (Granada University, Granada, Spain). ALLO-N/1 is a T cell line generated in our laboratory from a healthy donor (Martin et al., 2005). U937 (monocytic), HEL (erythroblastic), and K562 (erythroleukemic) cells lines were used as hematopoietic cells (HCs) of myeloid origin. All hematopoietic cells were cultured in RPMI 1640 supplemented with fetal calf serum (FCS; GIBCO-BRL, Middlesex, UK) to 10%, or fresh human serum (kindly provided by the Granada Regional Transfusion Center, Granada, Spain), glutamine, penicillin–streptomycin, and recombinant human interleukin-2 (rIL-2, 50 IU/ml) (Hoffman-LaRoche, Nutley, NJ; kindly supplied by the AIDS Research and Reference Reagent Program, National Institutes of Health, Rockville, MD); primary decidual stromal cells (DSCs) were kindly provided by E. Garcia-Olivares (Granada University) and obtained from human decidua as described previously (Garcia-Pacheco et al., 2001). Jurkat T cells were cultured in RPMI 1640 supplemented as described previously, without the addition of rIL-2. RKO (colon adenocarcinoma cells) and 293T (fetus kidney) cells were grown in Dulbecco's modified Eagle's medium, supplemented with 10% FCS, glutamine, and antibiotics as described previously. Human microvascular endothelial cells (HMECs) (ATCC, Molsheim, France) were grown in endothelial cell growth medium (PromoCell, Heidelberg, Germany).
Construction of vectors expressing Was, eGFP, and eGFP–Was
LVs were renamed according to the following letter code: W, Was gene endogenous promoter; WA, Was cDNA; S, spleen focus-forming virus LTR promoter; E, enhanced GFP cDNA; WP, woodchuck postregulatory element; EWA, eGFP–WASP (Wiskott-Aldrich syndrome protein) chimera. For simplicity, LVs previously described were renamed as indicated in Fig. 1A, Fig. 2A, and Fig. 5A. SEWP is the SE or HRSINCSGW vector in Demaison and colleagues (2002), SWA is the SW vector in Martin and colleagues (2005), WWA is the WW vector in Martin and colleagues (2005), and SEWAWP is the SEW vector in Toscano and colleagues (2008). All these plasmids share the self-inactivated (SIN) lentiviral backbone described by Zufferey and colleagues (1998). WEWAWP was obtained by replacing the SFFV-LTR promoter in the SEWAWP vector by the Was promoter, obtained by BamHI/NotI digestion of WWA. SEWA and WEWA vectors were obtained by excision of WPRE by ClaI digestion from SEWAWP and WEWAWP, respectively. The same strategy was used to obtain SE and WE from SEWP and WEWP vectors, respectively.
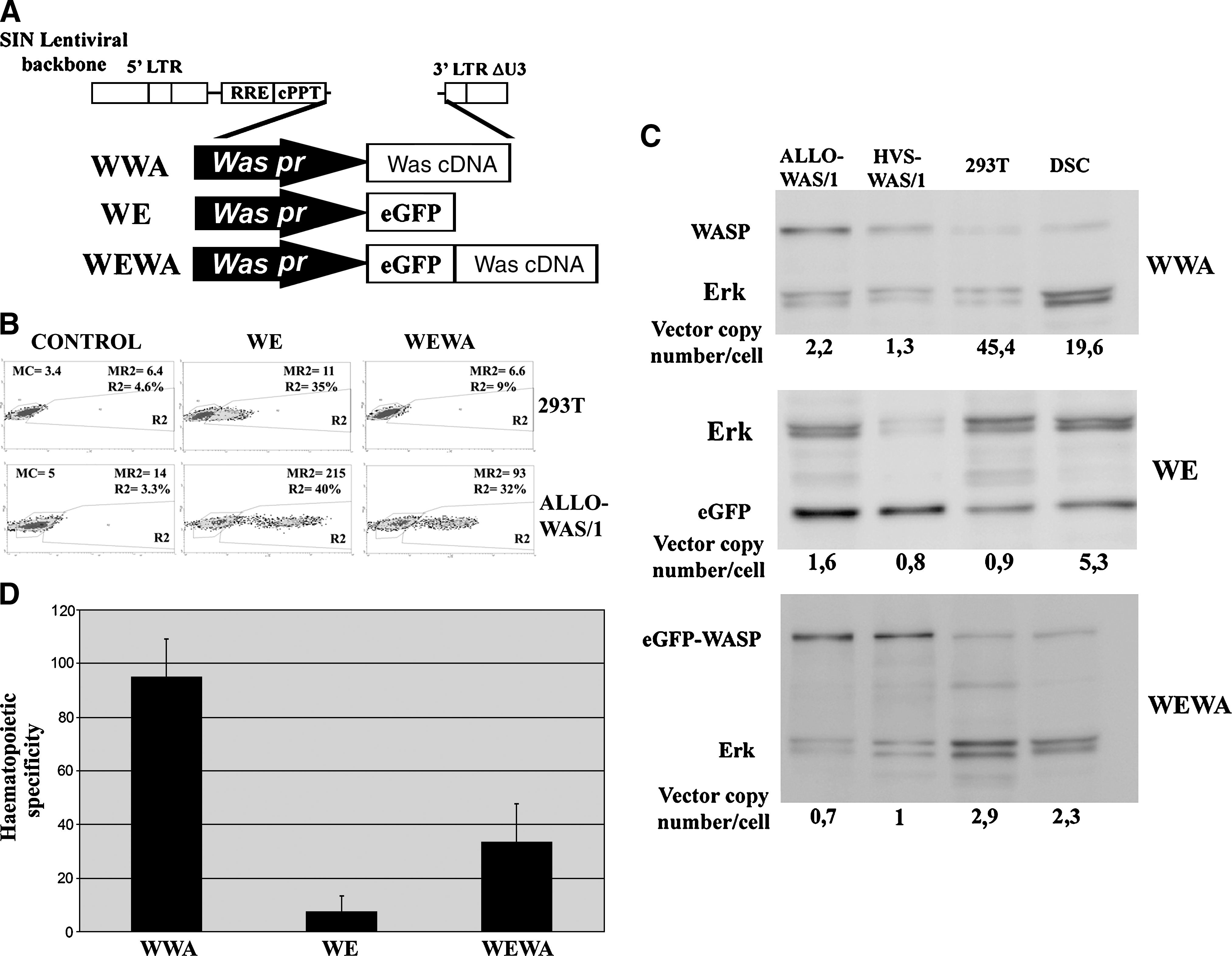
Hematopoietic specificity of Was promoter-driven lentiviral vectors (LVs) is enhanced by Was cDNA. (
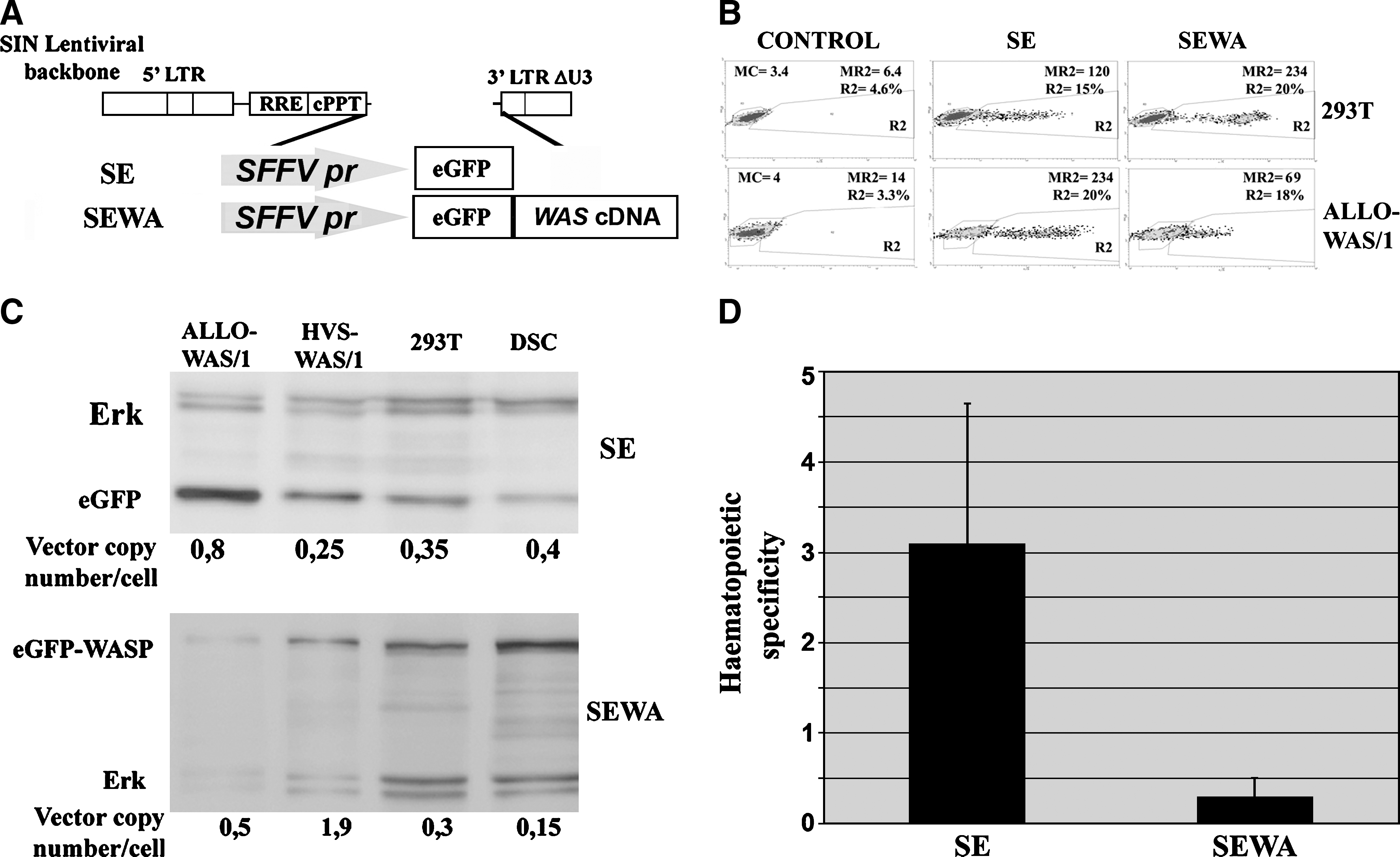
Hematopoietic cell-specificity improvement of LVs by Was cDNA sequences requires a Was promoter. (
Was cDNA fragments were obtained by digestions with BamHI/ClaI (BC fragment, 1613 bp), BamHI/XhoI (BX fragment, 960 bp), BamHI/ApaL1 (BA fragment, 719 bp), ApaL1/ClaI (AC fragment, 894 bp) and XhoI/XhoI (XX fragment, 680 bp) and cloned by blunt ends into the unique PstI site in the WE vector (right after the stop codon of eGFP cDNA). Of all the clones screened, only those inserted in the 5′ to 3′ orientation were selected. The resultant vectors were named WE-BC, WE-BX, WE-BA, WE-AC, and WE-XX (see Fig. 2A). The BC fragment was also cloned 5′ to 3′ into the SE and WEWP vectors, to obtain the SE-BC and WE-BC-WP vectors, using the PstI site that is located after the stop codon in both vectors.
Vector production
LVs were produced by cotransfection of 293T cells with three plasmids: (1) vector plasmid, (2) packaging plasmid pCMVΔR8.9, and (3) envelope plasmid pMD.G, as previously described (Neil et al., 2001). Briefly, 293T cells (6 × 106) were plated over a 10-cm tissue culture-grade Petri dish (Sarstedt, Newton, NC) the day before transfection to ensure exponential growth and more than 80% confluence. Vector plasmids, together with the packaging and envelope plasmids (total DNA, 27 μg; plasmid proportions of 3:2:1, respectively), were resuspended in 1.5 ml of Opti-MEM (GIBCO-BRL), mixed at room temperature for 20 min with 60 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA), and then diluted in 1.5 ml of Opti-MEM. The plasmid–Lipofectamine mixture was added to prewashed cells and then incubated for 6–8 hr. The producer cells were then washed and cultured for an additional 48 hr in 10 ml of Opti-MEM. Viral supernatants were collected and filtered through a 0.45-μm pore size filter (Nalgene, Rochester, NY), aliquoted, and immediately frozen at −80°C.
Cell transduction and vector titration
Exponentially growing target cells were washed in phosphate-buffered saline (PBS) and seeded onto 24-well plates at a concentration of 2 × 105 cells per well in 500 μl of the appropriate medium. Vector supernatants were added to the culture and incubated overnight. For expression analysis of eGFP or eGFP–WASP, cells were analyzed 72 hr later, or at other time points if indicated, in a BD FACScan flow cytometer (BD Biosciences, San Jose, CA). For analysis of Was-expressing vectors, the procedure was identical but the transduction efficiency was calculated by determining the vector copy number per cell by real-time polymerase chain reaction (PCR) (see later). Viral titers (transduction units [TU] per milliliter) were calculated on the basis of transduction of 293T cells by the various vectors. Each cell line was transduced at a predetermined multiplicity of infection (MOI) that allowed equivalent vector copy numbers among the different cells used in the panel, because the 293T cells or DSCs were four to six times more permissive than T cells (primary or HVS immortalized).
Quantitative Western blot
Cells were lysed with 1% Nonidet P-40 (NP-40) lysis buffer containing a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO), resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE; 10% polyacrylamide gels, reducing conditions), and electrotransferred to Hybond-P polyvinylidene difluoride (PVDF) membranes (GE Healthcare Life Sciences, Buckinghamshire, UK). Membranes were blocked with 5% nonfat milk and probed for 1 hr at room temperature with anti-WASP monoclonal antibody D-1 (2 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) or anti-GFP monoclonal antibody (Millipore, Billerica, MA) followed by incubation with horseradish peroxidase (HRP)-labeled goat anti-mouse antibody (1:10,000 dilution; Invitrogen Caltag). Quantitative analysis was performed with an ECL-Advance Western blotting detection system (GE Healthcare Life Sciences). Membranes were revealed after incubation for 1–5 min with ECL-Advance reagents. Quantification of light emission was done with a Molecular Imager ChemiDoc XRS system (Bio-Rad, Hercules, CA) and analyzed at 440 nm with Quantity One version 4.5.0 software (Bio-Rad). Contribution of each band was recorded and expressed as relative intensity per square millimeter. To determine WASP/eGFP expression in transduced cells, ERK (extracellular signal-regulated kinase) protein content was used as internal loading control. The ratio of WASP to ERK was calculated for each transduced cell and then normalized for vector copy number per cell.
The hematopoietic specificity of each vector was measured as follows: We first normalized transgene expression to protein loading (dividing by the ERK signal) and by the vector copy number (dividing this value again by the vector copy number) for each sample. The value obtained provided a relative measure of transgene expression per integrated vector (TEIV). To obtain the hematopoietic specific index of each vector we divided the TEIV obtained in HCs by the one obtained in non-HCs
Nucleic acid preparations and quantitative PCR and RT-PCR
Genomic DNA from cultured cells was isolated by adding 1 ml per 106 cells of SNET extraction buffer (20 mM Tris-HCl [pH 8], 5 mM EDTA [pH 8], 400 mM NaCl, 1% SDS; filtered and aliquoted) containing proteinase K (100 μg/ml; Sigma-Aldrich). DNA samples were incubated at 55°C for 2–18 hr, proteinase K was inactivated by a final heating at 95°C for 10 min, and RNase (1 μg/ml) was finally added for 30 min at 37°C.
Proteins were extracted twice with phenol–chloroform, and DNA was then precipitated and its concentration determined by spectrophotometry. Quantitative PCRs were performed in an ABI PRISM 7000 sequence detection system (Applied Biosystems, Foster City, CA). Samples were mixed with iTaq Supermix with ROX (Bio-Rad) containing dNTPs, iTaq DNA polymerase (50 U/ml), 40 mM Tris-HCl (pH 8.4), 100 mM KCl, 10 mM MgCl2, 1 μM ROX internal reference dye, and stabilizers. Specific primers were added at 400 nM. For SWA-, WWA-, SEWA-, and WEWA-transduced cells, a pair of primers comprising WAS exons 9 and 10 (forward, 5′-AGG CTG TGC GGC AGG AGA T-3′; reverse, 5′-CAG TGG ACC GAA CGA CCC TTG-3′) and a specific TaqMan probe (5′-FAM-CGC CAG GAG CCA CTT CCG CCG-TAMRA-3′) were used. The parameters for the PCR were as follows: 1 × (95°C for 2 min), 45 × (95°C for 30 sec, 58°C for 30 sec, and 72°C for 30 sec), 1 × (50°C for 2 min). For WE-, WE (B-C)-, and WE (B-X)-transduced cells, vector copy number determination was done with eGFP primers (forward, 5′-GCCCGACAACCACTACCT-3′; reverse, 5′-CGTCCATGCCGAGAGTGA-3′) and the TaqMan probe (5′-FAM-CGGCGGCGGTCACGAACTCCA-TAMRA-3′). The parameters for the PCR were as follows: 1 × (95°C for 2 min), 45 × (95°C for 30 sec, 61.4°C for 30 sec, and 72°C for 30 sec), 1 × (72°C 2 min). 293T cells (1 × 105) were mixed with 10-fold increasing amounts of plasmid DNA (from 102 to 1 × 107 copies) to determine the standard curve in each experiment.
For comparative quantitative RT-PCR, total RNA was extracted with an RNeasy Plus minikit (Qiagen, Hilden, Germany) and 1 μg was reverse transcribed into cDNA with a high-capacity cDNA reverse transcription kit (Applied Biosystems). One microliter of the final reaction was used to amplify GFP by PCR, using a QuantiTect SYBR green PCR kit (Qiagen) and GFP-specific primers (forward, 5′-GCCCGACAACCACTACCT-3′; and reverse, 5′-CGTCCATGCCGAGAGTGA-3′). The results were normalized by glyceraldehyde-3-phosphate dehydrogenase (GADPH) amplification of the same samples, using GADPH-specific primers (forward, 5′-GAAGGTGAAGGTCGGAGTC-3′; and reverse, 5′-GAAGATGGTGATGGGATTTC-3′). The PCR was done with a Stratagene Mx3005 P sequence detector (Stratagene, La Jolla, CA) and consisted of 40 cycles at 94°C (15 sec), then 60°C (30 sec) and 72°C (30 sec), and the results were analyzed with MxPro software (Stratagene).
Statistical analysis
Data are expressed as means ± SDEVP. Experiments were repeated two or three times with similar results. The analysis of significance was done using the Student t test (Microsoft Office Excel 2007; Microsoft, Redmond, WAS). p < 0.05 was consider significant.
Results
Hematopoietic specificity of Was promoter-driven LVs is influenced by Was cDNA
We have previously shown that the lentiviral vector (LV) WWA (Martin et al., 2005) expressing the Wiskott-Aldrich syndrome protein (WASP) is more than 100 times more efficient in hematopoietic cells (HCs) than in non-HCs. We explore here whether the same backbone, a Was promoter-driven self-inactivated LV, could express other genes such as eGFP in a similar hematopoietic cell-specific manner as the Was gene. We constructed the WE and WEWA vectors with the same backbone as the WWA vector but expressing eGFP or eGFP–Was, respectively (Fig. 1A). Figure 1B shows eGFP (from WE vectors) or eGFP–WASP chimera (from WEWA) expression levels in ALLO-WAS/1 (WASP-deficient T cell line) and 293T (epithelial cell line). Contrary to previous data for the expression of WASP, the Was promoter-driven LV expressed eGFP efficiently in 293T cells, although still performing better in T cells than in 293T cells (Fig. 1B). Interestingly, the Was promoter-driven LVs expressing the eGFP–WASP chimera (WEWA vectors) were efficient only in T cells (Fig. 1B) as we observed previously for the WWA vectors (Martin et al., 2005). Indeed, although WEWA-transduced 293T cells contained similar vector copy numbers per cell (vcn/c) as WE-transduced 293T cells (vcn/c = 0.4; data not shown), we could not detect any expression of the transgene (Fig. 1B, top right).
To quantify this effect in detail we performed quantitative Western blot analysis comparing transgene expression levels of WWA-, WE-, and WEWA-transduced T cells (ALLO-WAS/1 and HVS-WAS/1) and non-HCs (293T cells and DSCs) (Fig. 1C). We calculated the relative transgene expression per integrated vector (TEIV) for each cell line and vector (see Materials and Methods for detail). The TEIV values were used to estimate the hematopoietic specificity of each vector by dividing the TEIV of each HC line by the TEIV of each non-HC line. Figure 1D shows that the hematopoietic specificity of the WE vector is 5–10 times lower than the WWA vector (p < 0.001). This reduction in hematopoietic specificity is partially restored by the WEWA vectors (p < 0.02).
Was cDNA influences transgene expression at various levels in various cell types
The increased hematopoietic specificity of WASP-expressing LVs compared with eGFP-expressing LVs could be due to the presence of Was cDNA sequences in the vector (affecting RNA processing or Was promoter activity) or to the differences in WASP protein processing in HCs versus non-HCs. To determine the mechanism by which Was cDNA affects LV transgene expression, we first constructed the SE and SEWA vectors (Fig. 2A) expressing eGFP and eGFP–WASP proteins through the constitutive spleen focus-forming virus (SFFV) promoter. With these vectors we can determine whether the effect of Was cDNA implies RNA processing and/or protein stability. If that is the case, eGFP–WASP-expressing LVs will have reduced expression levels compared with eGFP-expressing LVs (as observed with WEWA vectors), independently of the promoter used to express the mRNA. Also, SEWA vectors should have improved hematopoietic specificity compared with SE vectors, as occurs for Was promoter-driven LVs. We therefore analyzed the expression pattern of SE and SEWA vectors in the 293T and ALLO-WAS/1 cell lines (Fig. 2B). Insertion of the Was cDNA into the SFFV-driven LV (SEWA vectors) did not downregulate transgene expression in 293T cells (see transgene expression of SE vs. SEWA vectors in Fig. 2B), indicating that the presence of Was cDNA sequence does not affect RNA processing or protein stability in this cell type. On the other hand, in ALLO-WAS/1 T cells we observed a substantial decrease in transgene expression of the SEWA vectors compared with SE vectors. These data indicate that, contrary to 293T cells, mRNA processing and/or protein stability (of eGFP vs. eGFP–WASP chimera) are important players in the reduced expression of Was-containing vectors (WEWA and SEWA) in T cells.
We further quantified the effect of Was cDNA sequences on SFFV-driven LVs by quantitative Western blot analysis, comparing transgene expression levels of SE- versus SEWA-transduced T cells (ALLO-WAS/1 and HVS-WAS/1) and non-HCs (293T and DSCs) (Fig. 2C). As in Fig. 1C, detail quantification was performed to compare transgene expression levels of the SE and SEWA vectors in the various cell lines. Interestingly, the hematopoietic specificity of SFFV-driven LVs harboring Was cDNA sequences was reduced (instead of increased as observed for Was-driven LVs) (Fig. 2D, SEWA vs. SE vector).
Part of the effect observed in WEWA and SEWA vectors compared with WE and SE vectors could be due in part to differences in protein processing (eGFP–WASP chimera vs. eGFP). To further investigate the mechanisms by which Was cDNA influences transgene expression we constructed a Was promoter-driven LV expressing eGFP harboring a full-length Was cDNA inserted downstream of the stop codon of the eGFP gene (Fig. 3A, WE-BC). Therefore, the WE-BC vector is identical to the WEWA vector but expresses eGFP instead of eGFP–WASP protein. We also constructed the SE-BC LV (same backbone as the WE-BC vector but driving expression through the SFFV promoter) to discriminate any effect due only to RNA processing (Fig. 3A, SE-BC). Six HC lines (HSV-WAS/1, ALLO-N/1 and ALLO-WAS/1, U937, HEL, and K562) and three non-HC lines (293T, RKO, and HMEC) were transduced with the SE, SE-BC, WE, and WE-BC vectors and analyzed by flow cytometry (Fig. 3B). As occurred with the WEWA vectors, the WE-BC vector expresses the transgene (eGFP) better in HCs (Fig. 3B, right: HCs, dark columns; non-HCs, light columns) and has a marked reduction in transgene expression in all cell lines compared with WE vectors. Interestingly, as also observed for WEWA vectors, this reduction is stronger in non-HCs than in HCs. On the other hand, the SE-BC vector (also harboring the Was cDNA sequence) maintains similar expression levels in non-HCs and in myeloid cells (U937 and K562) and therefore RNA processing is not affected by the presence of Was cDNA sequences in these cells. However, a strong reduction can be observed in T cells (as observed with the SEWA vectors), indicating that in T cells, the mRNAs containing Was cDNA sequences are either more instable or less well translated than the same RNA without this sequence.
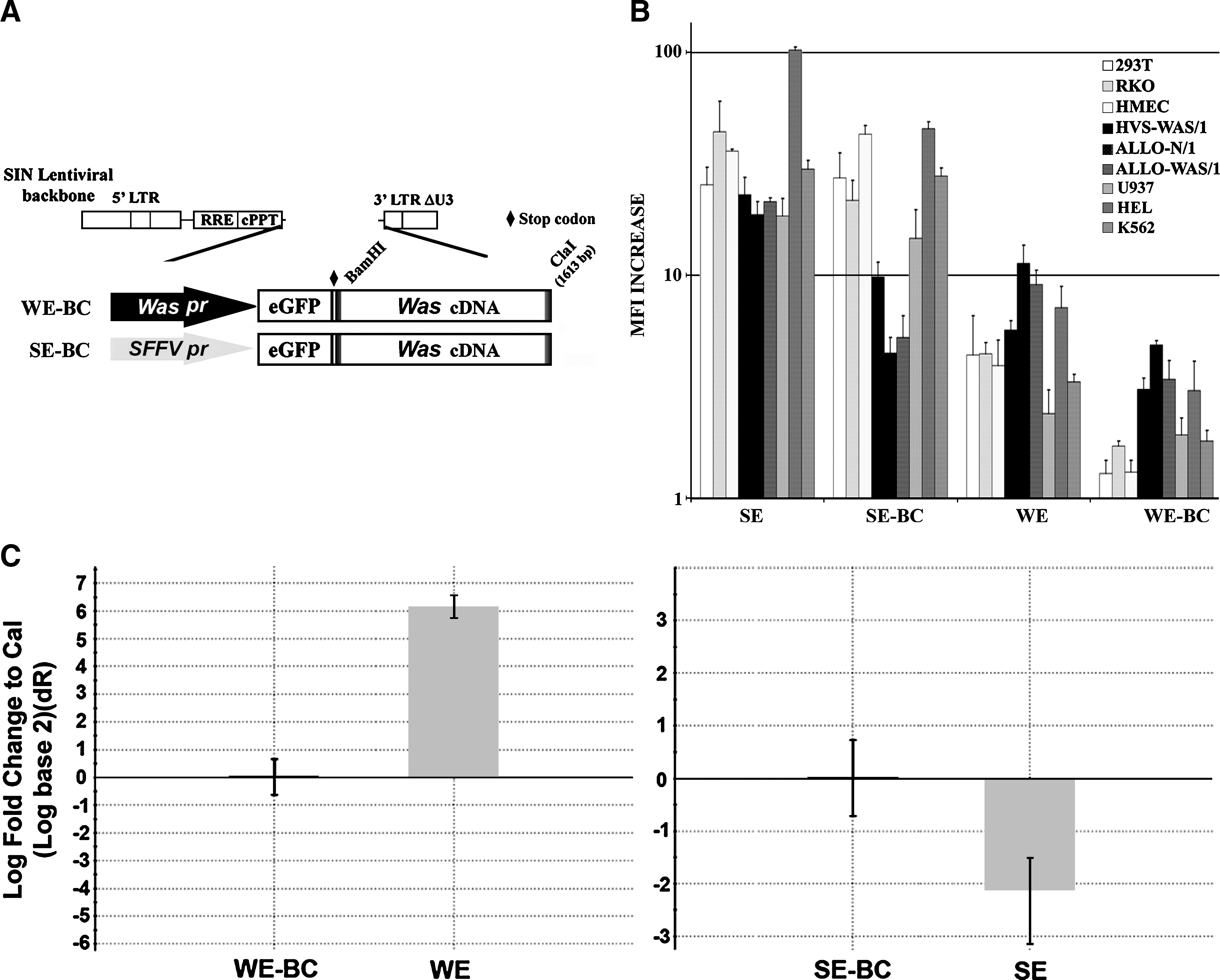
The effect of Was cDNA sequences on LV expression depends on the promoter and the cell type. (
Last, we quantified the effect of Was cDNA sequences on the transcriptional activity of LVs. We transduced 293T cells with Was- and SFFV promoter-driven LVs with or without the Was cDNA sequence and expressing eGFP (WE, WE-BC, SE, and SE-BC) and quantified relative eGFP mRNA levels by RT-PCR of transduced cells with identical vector copy numbers per cell. Figure 3C show the result of the comparative RT-PCR, using the BC-containing vector as reference. Was cDNA sequences dramatically reduced mRNA levels of Was promoter-driven LVs in 293T cells (WE-BC vectors expressed up to 60 times less RNA than WE vectors; Fig. 3C, left) but slightly increased mRNA levels of SFFV-driven LVs (Fig. 3C, right).
All these data together indicate that Was cDNA sequences have at least two different effects on LV transgene expression depending on the cell type. In T cells these sequences have a marked posttranscriptional effect, reducing either RNA stability and/or mRNA translation. In non-HCs such as 293T cells and HMECs, Was cDNA downregulates Was promoter activity but does not have any effect on the SFFV promoter or in posttranscriptional regulation.
A 900-bp fragment 5′ of the Was cDNA contains regulatory sequences that influence transcription and increase hematopoietic specificity
To further study the influence of the Was cDNA in Was promoter activity, we constructed a panel of LVs expressing eGFP and harboring different fragments of the Was cDNA downstream of the eGFP stop codon. We dissected the Was cDNA (BC, 1640 bp) into 5′ fragments (BA, 719 bp; and BX, 960 bp) and 3′ fragments (AC and XX) that included (BX and AC) or did not include (BA and XX) the 330-bp center of the Was cDNA (Fig. 4A). To study the minimal Was cDNA sequences that influence Was promoter activity we focused our studies on cells in which these sequences do not affect mRNA processing (non-HCs; 293T cells and HMECs). 293T cells and HMECs were transduced by the various vectors at high MOI (MOI of 5) to obtain detectable levels of eGFP expression with all vectors. Transduced cells were analyzed by fluorescence-activated cell sorting (FACS) 7 days posttransduction. The increments in fluorescence intensity of WE-, WE-BC-, WE-BA-, WE-AC-, WE-BX-, and WE-XX-transduced cells are plotted in Fig. 4B. All Was cDNA fragments but the 680-bp XX decreased significantly the eGFP expression of the Was promoter-driven LVs (Fig. 4B) in both cell lines. For instance, WE-transduced 293T cells expressed 10 times more eGFP than did WE-BC-transduced 293T cells (p < 0.01) and WE-transduced HMECs expressed 4 times more eGFP than did WE-BA-transduced HMECs (p < 0.05).
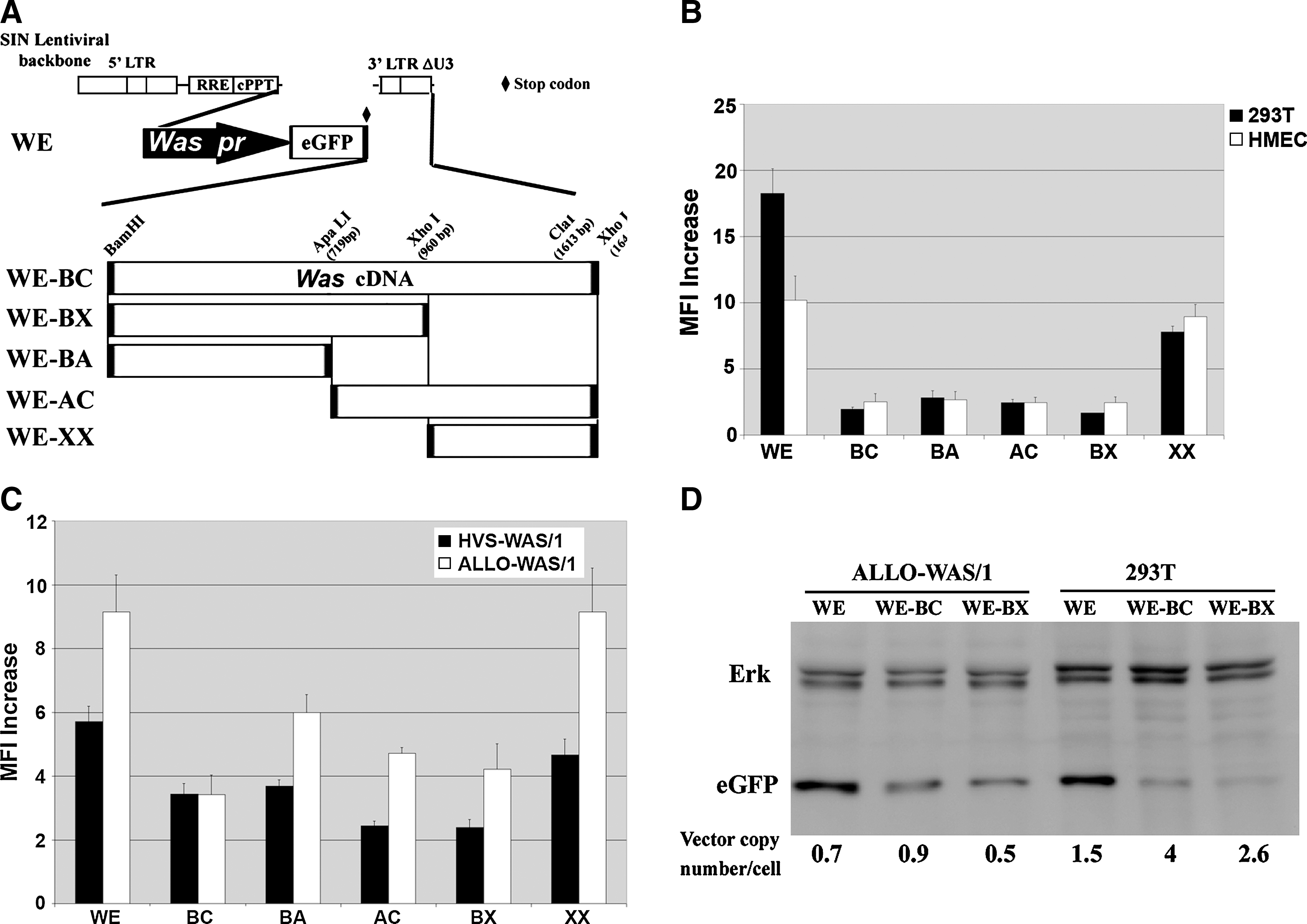
The first half of the Was cDNA is enough to downregulate promoter activity in non-HCs and to increase hematopoietic cell-specific expression of eGFP. (
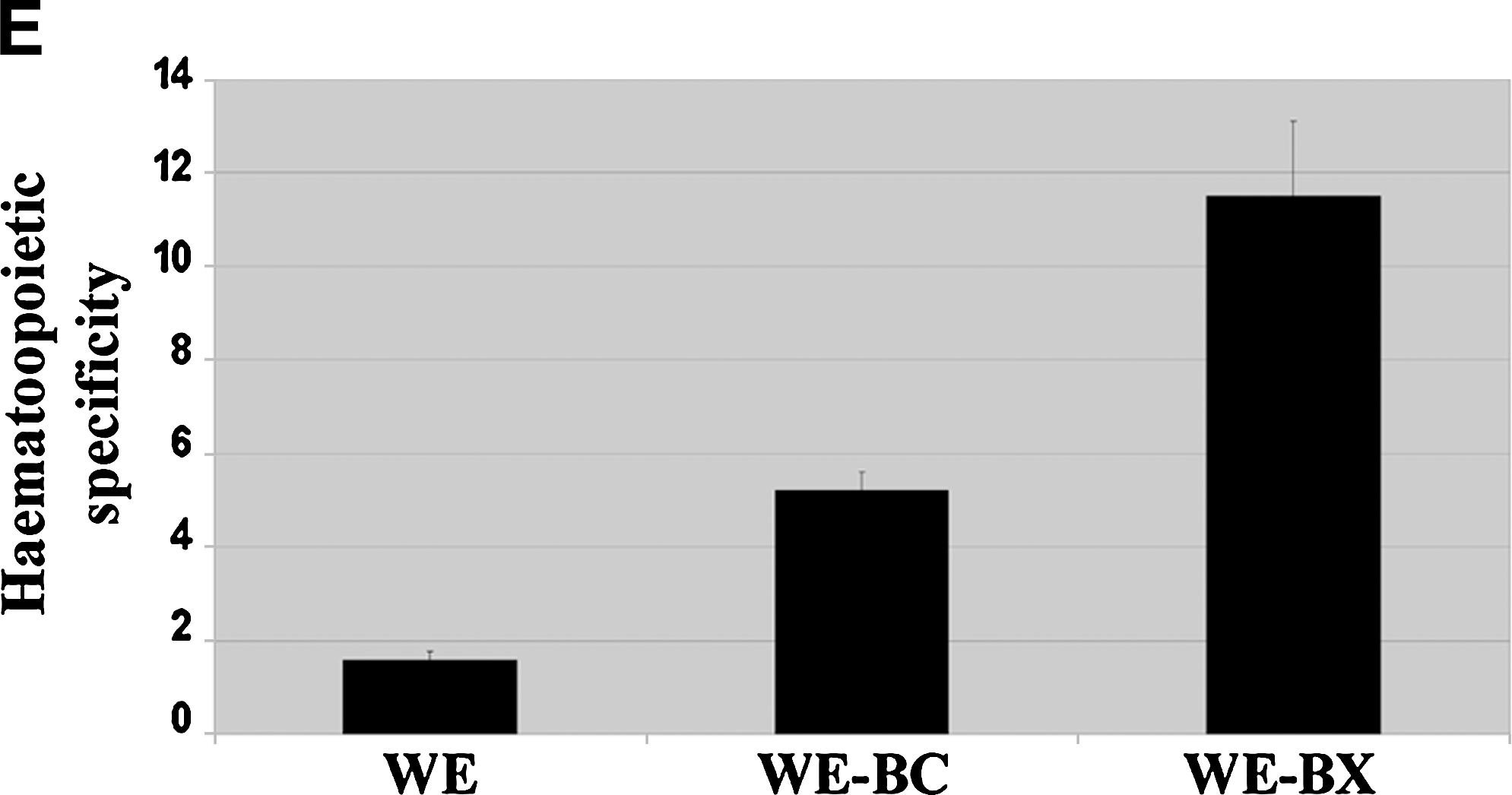
We next analyzed whether the presence of Was cDNA fragments resulted in improved hematopoietic specificity of eGFP-expressing vectors. We first performed an experiment similar to that described previously but comparing the effect of Was cDNA sequences in HCs. HVS-WAS/1 and ALLO-WAS/1 T cells were transduced with the various vectors at an MOI of 1–2 and analyzed for eGFP expression. The increments in fluorescence intensity of the various vectors are plotted in Fig. 4C. As for non-HCs, all vectors containing Was cDNA fragments, except for the 680-bp XX, showed a reduction in eGFP expression in both cell lines compared with WE vectors. However, this effect was less marked than for HCs. For instance, WE-transduced HVS-WAS/1 cells expressed 1.6 times more eGFP than did WE-BC-transduced HVS-WAS/1 cells (p < 0.01) and WE-transduced ALLO-WAS/1 cells expressed 2.6 times more eGFP than did WE-BC-transduced ALLO-WAS/1 cells (p < 0.05). To further study this observation we performed quantitative Western blot analysis comparing eGFP expression levels of WE-, WE-BC-, and WE-BX-transduced HCs (ALLO-WAS/1) and non-HCs (293T) (Fig. 4D). A detailed quantification of band intensities (relative to ERK protein levels and vector copy number per cell) and of vector copy numbers per cell allowed us to measure the relative transgene expression per integrated vector (TEIV) (see Materials and Methods) and to determine the hematopoietic specificity of the WE, WE-BC, and WE-BX LVs by comparing the TEIV of each vector in 293T cells versus ALLO-WAS/1 T cells. Figure 4E shows that WE-BC and WE-BX vectors have improved hematopoietic specificity (four to six times better; p < 0.05) than the WE vector (Fig. 4D). These results corroborate the involvement of Was cDNA in hematopoietic cell-specific expression and localize the area involved to the first 900 bp of the 5′ sequence.
Was cDNA partially neutralizes the enhancer effect of WPRE in most cell types
Moreau-Gaudry and colleagues have demonstrated the positive effect of the WPRE in erythroid-specific transgene expression (Moreau-Gaudry et al., 2001). On the basis of these results we decided to study whether the WPRE could also increase transgene expression of Was promoter-driven LVs harboring Was cDNA fragments. We transduced ALLO-WAS/1 and 293T cells with SE, WE, SE-BC, and WE-BC LVs and with their WPRE-containing counterparts (Fig. 5A; SEWP, WEWP, SE-BCWP, and WE-BCWP). Figure 5B shows the increment in eGFP expression levels (measured as the MFI increase with the WPRE+ vector divided by the MFI increase with its WPRE− counterpart) achieved by introduction of the WPRE into each vector analyzed. In general, the effect of the WPRE in transgene expression was higher in SFFV-driven than in Was-driven LVs. For instance, eGFP expression was 10 times higher with SEWP than with SE in 293T cells, and 4 times higher in ALLO-WAS/1 cells (Fig. 5B, solid columns). On the other hand, the presence of the WPRE in the WE backbone vector increased eGFP expression only four times in 293T cells and two times in ALLO-WAS/1 cells (Fig. 5B, open columns). Interestingly, the presence of Was cDNA in the SE and WE backbones reduced the effect of the WPRE in most cells, with a drastic effect in 293T cells. Indeed, in 293T cells, the WPRE increased the eGFP expression of SE vectors 10 times but only 2 times if incorporated into the SE-BC vector (Fig. 5B, solid columns vs. dashed columns; p < 0.03). A significant reduction was also observed in ALLO-WAS/1 cells (3-fold enhancement vs. no enhancement; p < 0.01). Similarly, the effect of the WPRE in the WE vector was lower when incorporated into Was cDNA-containing vectors (WE-BC): whereas the WPRE enhanced the eGFP expression of WE vectors four times in 293T cells and two times in ALLO-WAS/1 cells, the presence of Was sequences in the WE-BC vectors had almost no effect (Fig. 5B, open columns vs. gray columns; p < 0.05).
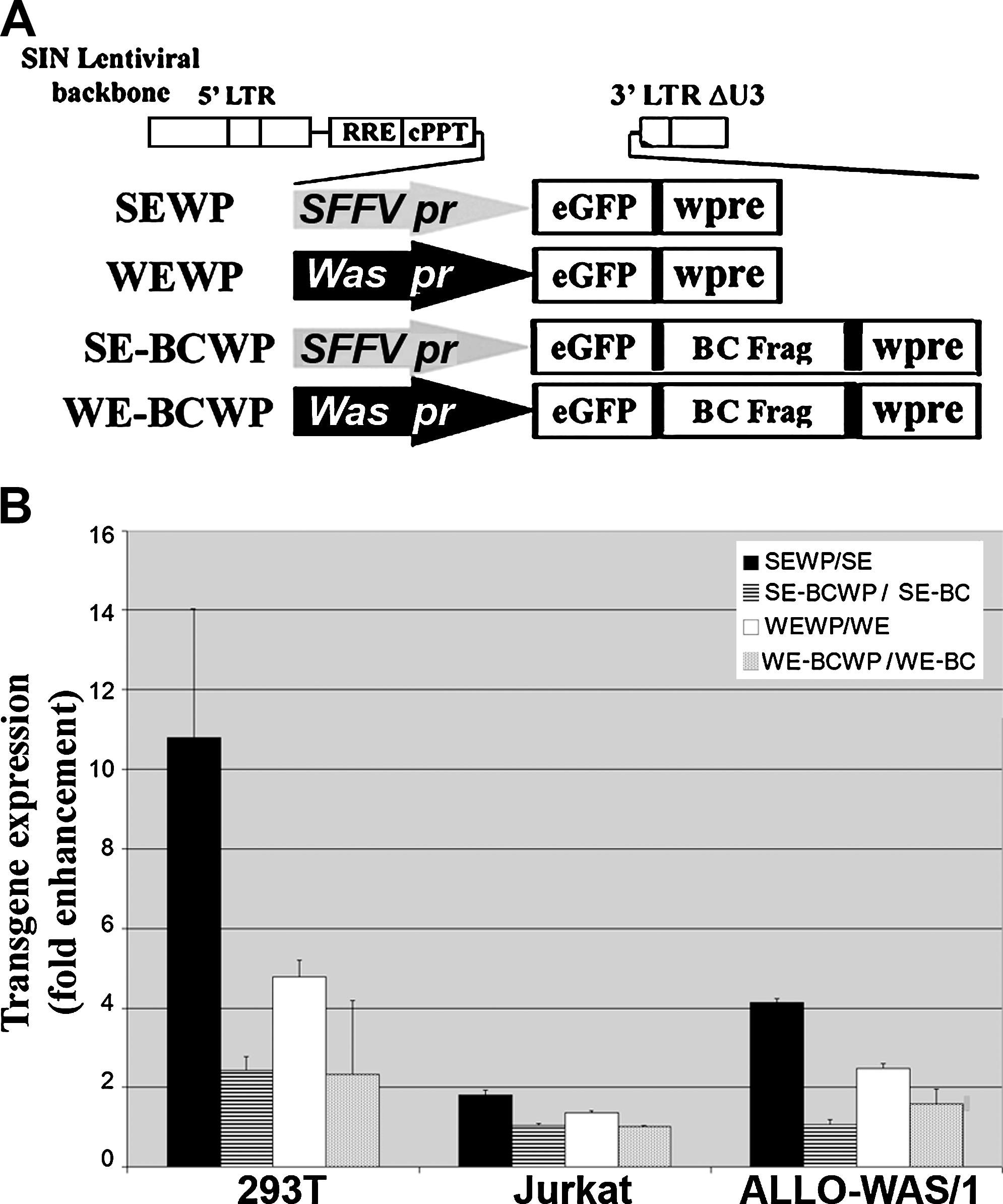
Inclusion of the woodchuck posttranscriptional regulatory element (WPRE) in Was promoter-driven and Was cDNA-containing LVs does not improve expression. (
Discussion
The development of vectors expressing a therapeutic transgene efficiently and specifically in hematopoietic cells (HCs) is an important goal for gene therapy of hematological disorders. We have previously shown that the WWA LV expressing the Was cDNA through a 500-bp fragment of the Was proximal promoter is more than 100 times more efficient in HCs than in non-HCs (Martin et al., 2005). In this paper we have shown that the same LV expressing eGFP (WE) partially loses hematopoietic cell-specific expression. We demonstrated that the enhanced hematopoietic cell specificity of Was-expressing vectors is due to a combined effect of both transcriptional and posttranscriptional effects of Was cDNA.
To study the mechanism by which the Was cDNA influences the hematopoietic specificity of Was promoter-driven LVs, we first investigated whether mRNA and/or protein stability was a major player. If posttranscriptional regulation was involved, SFFV-driven LVs (SW, SEWA, and SE-BC) should also have increased hematopoietic expression compared with SE LVs. However, we found no effect in this sense. In fact, the transgene expression of SFFV-driven vectors carrying Was cDNA sequences was drastically reduced in T cells, with no effect in myeloid cells and non-HCs compared with SE vectors. We further showed that Was cDNA sequences reduced mRNA levels of Was promoter-driven LVs in 293T cells up to 64 times compared with Was promoter-driven LVs carrying this sequence, an effect possible only by having a direct effect in blocking Was promoter activity, because SFFV-driven LVs expressing the same RNA were not affected or even had increased mRNA levels (Fig. 3C). These data indicate that the effect of Was cDNA sequences on LV transgene expression is variable depending on the cell type. In non-HCs, we assume that transcriptional modulation is the only process acting to regulate transgene expression of Was promoter-driven LVs. However, in T cells we have a more complicated scenario and we propose that probably both mRNA processing and promoter regulation play a role.
We further characterized the region of the Was cDNA involved in transgene regulation by incorporating various Was cDNAs fragments downstream of eGFP in the WE vector and studying eGFP expression in HCs and non-HCs, both by FACS and quantitative Western blotting. The results obtained corroborate the involvement of Was cDNA in hematopoietic cell-specific expression and localize the area involved to the first 900 bp of the 5′ sequence of the cDNA. All constructs but WE-XX decreased expression (compared with WE vectors) in all cell lines, with a stronger effect in non-HCs. The effect was independent of insert size because the WE-BA vector (719-bp insert size) was as efficient as the WE-BC vector (1640 bp) (Fig. 3B). In addition, the WE-XX vector (680-bp insert size) has an expression pattern similar to that of the WE vector. Transfac software analysis found potential inhibitory transcription factor (TF)-binding sites (CBF, WT1, GATA1, and C/EBPα) and potential activators (Sp1, Egr1, and Ap1) through the Was cDNA sequence; from BamHI to ApaLI we have Sp1, Egr1, C/EBPα, Ap1, and GATA1 TF-binding sites. From ApaLI to XhoI, we found GATA1, WT1, and Ap1 TF-binding sites; and from XhoI to the 3′ end we found Sp1, Egr1, H4TF1, CBF-1, and C/EBPα TF-binding sites. Taking together expression profiles of the various constructs and TF-binding sites, one could hypothesize that WT1 and GATA1 could play a role in transcriptional repression and that Sp1, Ap1, and Egr1 could play a role in increasing hematopoietic specificity. Our laboratory is actively working to determine whether any of these TFs is actually involved in active regulation of Was promoter-driven LVs and whether this mechanism is also involved in physiological expression of the Was gene.
The mechanism involved in the improved hematopoietic specificity is complex because Was sequences negatively affect mRNA stability and/or translation only in T cells and not in 293T cells or HMECs, where these sequences strongly decreased eGFP expression because of promoter downregulation. Therefore the improved hematopoietic specificity must be the result of transcriptional and posttranscriptional modulation in the different cell types. The combined effects result in a general downregulation of transgene expression but a gain in hematopoietic specificity.
The Was promoter-driven and Was cDNA-containing LVs express low protein levels, and this could be a good characteristic for some purposes by minimizing vector genotoxicity. However, it would also be of interest to have hematopoietic cell-specific vectors expressing higher transgene levels. On the basis of the work of Moreau-Gaudry and colleagues, in which they demonstrated that inclusion of the WPRE in erythroid cell-specific LVs increased expression in erythroid cells (Moreau-Gaudry et al., 2001), we decided to study whether the WPRE could improve the titer and transgene expression of our LVs. However, although we noticed an increase in vector titer (data not shown), the effect of WPRE in transgene expression was variable. In general, the effect of the WPRE in transgene expression was higher in SFFV-driven than in Was-driven LVs. Interestingly, the presence of Was cDNA in the LV backbone (SEWAWP, WEWAWP, and WE-BC vectors) blocks the enhancing effect of the WPRE in all cell lines and therefore partially inhibits its beneficial effect in our lentiviral backbone. Because Was sequences negatively modulate mRNA stability and/or translation, it is plausible that these sequences interfere with WPRE activity, partially neutralizing their effect on RNA stability.
In summary, this work demonstrated the effect of Was cDNA on LV transgene expression, showing an effect on Was promoter regulation and/or on mRNA processing, depending on the cell type. The global effect of these sequences on Was promoter-driven LVs is improved hematopoietic specificity, partially explaining the better performance when these vectors express WASP versus eGFP. We have characterized the minimal Was cDNA fragment required to increase hematopoietic specificity to the first 900 bp. Last, we have shown that inclusion of the WPRE in Was promoter-driven and Was cDNA-containing LVs does not improve expression of these vectors.
Footnotes
Acknowledgments
We are grateful to Dr. Didier Trono (University of Geneva, Geneva, Switzerland) for providing the HIV packaging pCMV.R8.91 and envelope pMD.G plasmids, Dr. A. Thrasher for providing the WEWP and SEWP vectors, and Dr. Ignacio Molina and Enrique Garcia Olivares (Granada University) for providing primary HVS-transformed T cell lines and DSCs, respectively. This work was supported by grant PI061035 to F.M.
Author Disclosure Statement
For all authors, no competing financial interests exist.